We first summarize simulation results from our earlier work on the shear viscosity of squalane at different rates and pressures relevant to the studies performed in later subsections. Next, we present the examination of the link between rheological properties and molecular structure using the intramolecular orientation tensors. This is followed by results that probe this link using the intermolecular orientation tensors.
3.1. Rheological Properties
Figure 3 shows the NEMD simulation results for the shear viscosity of squalane for different shear rates
and pressures
MPa at 293 K. Shear viscosity
is calculated as
, where
is the
component of the stress tensor. At all pressures, squalane exhibits a clear shear thinning behavior evident by a decreasing viscosity with increasing shear strain rate. Our previous papers [
17,
18] showed that phenomenological models and the associated molecular mechanisms that best describe the observed shear thinning behavior can be different at different pressures. For low pressures
100 MPa, where the Newtonian viscosity
is small, the viscosity of squalane is better fit by power-law models such as Carreau model and Ostwald-de Waele model [
53,
54], which predict a power-law shear thinning as a function of rate:
, where
is a characteristic shear rate, and the exponent
n is between 0 and 1. For low rates
,
reduces to the Newtonian viscosity
. For rates
,
shows power-law behavior. Our earlier work showed that the viscosity extracted from the Newtonian plateau reached in simulations and via the fit to the Carreau model is in excellent agreement with the Newtonian viscosity measured in experiments [
17,
18]. In such models, shear thinning is attributed to changes in molecular order. For example, an increase in the molecular alignment along the flow direction with increasing rate can reduce the rate of collisions between molecules, and thus decrease the fluid viscosity.
On the other hand, for high pressures
400 MPa, where
is large, the viscosity-rate scaling is better fit by Eyring model [
55,
56], which predicts a logarithmic rise in shear stress with rate, resulting in the limiting high-rate shear thinning behavior:
for
, where
is a characteristic rate related to the Eyring stress parameter
. Our previous papers showed that Eyring model fits to high-rate viscosity data from simulations were consistent with experimental measurements of viscosity at the same pressure that reach up to
and extend down into the Newtonian regime [
17,
18]. Eyring model assumes that shear flow is a stress-biased thermally activated process, where shear occurs through rare molecular rearrangements that require activation over a single potential energy barrier. In Eyring model, the reduction in viscosity with shear rate does not rely on a change of molecular order, but instead on a change of the competition between shear rate and thermal activation.
Given these alternate molecular mechanisms of shear thinning, one that relies on changes in molecular order and the other that does not, an accurate assessment of changes in molecular order with increasing shear rate for different pressures becomes important to probe the origins of the observed high-rate shear thinning behavior shown in
Figure 3. In the following, we examine the link between shear thinning and molecular order in squalane for different pressures by using the molecular structure information encoded in the intramolecular and intermolecular orientation tensors.
3.2. Intramolecular Orientation and Shear Thinning: Single Atom Pair Analysis
The intramolecular orientation tensor
(Equation (
7)) is defined for each (united-) atom pair
p associated with a squalane molecule. A squalane molecule has 30 united atoms, and therefore there are 435 intramolecular atom pairs. The intramolecular structure information is encoded in the associated 435 intramolecular orientation tensors. Each of these
tensors is a 3 × 3 symmetric matrix, where
=
. Therefore, 6 out of 9 components are non-trivial. The diagonal elements
measure the degree of atom pair alignment in the direction of the flow field (
x), velocity gradient (
y), and vorticity (
z), respectively. A value of 0 indicates no preference to align along the axis, i.e., the atom-pair distance vector assumes a random orientation. Positive and negative values imply the extent to which the atom-pair orientation is parallel or perpendicular to the axis, with 1 and −0.5 representing perfectly parallel and perpendicular alignments, respectively.
To illustrate,
Figure 4 shows the intramolecular orientation tensor for the atom pair 2−28 (see
Figure 1) for different rates and pressures. For pressures
100 MPa,
,
, and
(
Figure 4a–c) start at ≈0 at the lowest strain rates probed (
) and gradually increase or decrease with increasing rate, before plateauing to a non-zero value at the highest rates (
). This behavior signals a gradual shift in the orientation of the atom pair 2−28 from a random orientation to an ordered orientation as shear rate increases from
to
. The high-rate plateau values for
,
, and
are positive, negative, and negative, respectively, which indicates that the atom pair 2−28 orients along the flow direction while being perpendicular to the velocity gradient and vorticity directions.
In stark contrast, the diagonal components
,
, and
for pressures
400 MPa start at non-zero values at the lowest probed rates (
) and do not change noticeably as
increases to
. This behavior signals that the atom pair 2−28 does not have an appreciable change in the orientation even as squalane is sheared at increasingly higher rates.
Figure 4d shows the
component for all the pressures. A trend similar to
is observed:
increases from
to a saturated positive value with increasing
for
100 MPa, but is nearly constant at a positive saturated value for
400 MPa. For all pressures, the off-diagonal components
and
exhibit small fluctuations around zero for all rates.
For higher pressures, even though all the orientation tensor components associated with the 2−28 atom pair do not change significantly with increasing rate, the viscosity continues to steadily decrease. This shear thinning may result from either a change in a different intramolecular order parameter (e.g., orientation of a different atom pair), or from an entirely different molecular mechanism (e.g., thermal activation). Different atom pairs might contribute differently to the molecular order, and therefore the orientation tensor information associated with not just 1, but all 435 pairs needs to be examined. We next show the results of using dimension reduction methods to analyze and visualize this high-dimensional information.
3.3. Intramolecular Orientation and Shear Thinning: Dimension Reduction Results
We first classify the 30 united atoms of a squalane molecule into 3 types based on their positions (see
Figure 1): end atoms (atoms 1, 3, 29, and 30), side atoms (atoms 8, 13, 19, and 24), and backbone atoms (all the other atoms in
Figure 1). Using this classification, we separate the 435 atom pairs into 4 representative pair types: side-backbone pairs of a side atom and its bonded backbone atom (e.g., atom pair 7–8 in
Figure 1), end-backbone pairs of an end atom and its bonded backbone atom (e.g., atom pair 2–1 in
Figure 1), short backbone-backbone pairs of two backbone atoms directly connected by a covalent bond (e.g., atom pair 15–16 in
Figure 1), and long backbone-backbone pairs of two backbone atoms that are separated by more than one covalent bond (e.g., atom pair 2–28 in
Figure 1 which is separated by 21 bonds).
Each of the 435 atom pairs is associated with an intramolecular orientation tensor having 6 non-trivial components. This high-dimensional dataset of dimensions encapsulates the intramolecular structure information for each pressure and shear rate. In the following, we discuss the results of using PCA and t-SNE to reduce and visualize this high-dimensional information. In all dimension reduction tasks, each component of the orientation tensors is first centralized and normalized such that the mean and variance of the data points are 0 and 1 respectively.
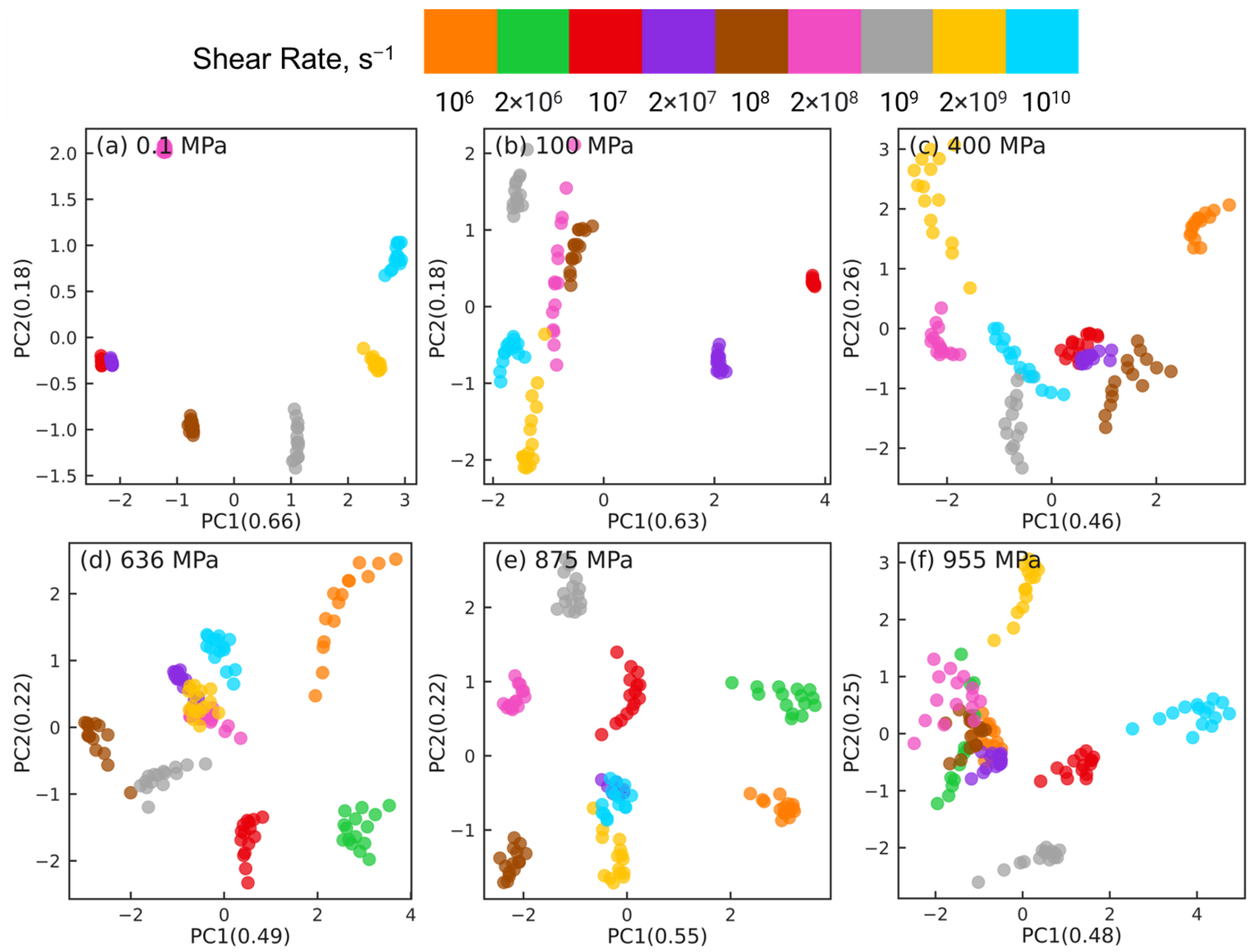
Figure 5 shows the PCA results for reducing the datasets of
dimensions to
dimensions at different pressures, where
indicates the number of shear rates associated with the corresponding pressure (
for 0.1 and 100 MPa,
for 400 MPa, and
for
MPa). For clarity, orientation tensors associated with the aforementioned 4 pair types (i.e., side-backbone, end-backbone, short backbone-backbone, long backbone-backbone pairs) are shown in
Figure 5. The sub-figures correspond to different pressures
0.1 MPa (a), 100 MPa (b), 400 MPa (c), 636 MPa (d), 875 MPa (e), and 955 MPa (f). In each sub-figure, the atom pairs are identified by the shear rate (symbol color) and the pair type: side-backbone, end-backbone, short backbone-backbone, long backbone-backbone (symbol type), as illustrated in the legend. The first and second principal components (PC1 and PC2) represent the components associated with the reduced dimensional space that, respectively, capture the largest and the second largest variance in the high-dimensional dataset. The numbers in the parentheses of PC1 and PC2 indicate the weight fraction of the information in the original data that is captured by the corresponding component. For example, in
Figure 5a, PC1 and PC2 capture 67% and 17% of the information embedded in the high-dimensional space, respectively. All the other components combined capture the remaining 16% of the information in the original data.
For all 6 pressures, by analyzing the eigenvectors associated with PC1 and PC2, we find that , , , and components contribute significantly to PC1, while PC2 is dominated by and components. This suggests that dimension reduction using PCA picks a low-dimensional axis that represents the shear-induced changes in the intramolecular orientation tensors as the first principal component. The and components that dominate PC2 are expected to be 0 on average, which suggests that the second most important dimension in the low-dimensional space represents the variations in the intramolecular orientation tensors that can be attributed to statistical fluctuations.
The PCA dimension reduction results for pressures
MPa (
Figure 5a,b) show that the atom pairs are better separated by strain rate (symbol color) compared to by pair type (symbol type). The datasets associated with the lowest rates, where squalane exhibits Newtonian flow, are not separable, consistent with our earlier work [
24]. Most atom pairs associated with the same shear rate are grouped together into clusters that are organized along the PC1 dimension by the strain rate, e.g., from left to right with increasing rate for 0.1 MPa. In stark contrast, PCA dimension reduction results for pressures
MPa (
Figure 5c–f) show that the atom pairs are better separated by pair type (symbol type) compared to by strain rate (symbol color). The side-backbone, end-backbone, short backbone-backbone, and long backbone-backbone pairs organize into distinct clusters well-separated along the PC1 axis. The large separation between clusters comprising side-backbone and long backbone-backbone pairs suggests that these pairs contribute to the variations in the intramolecular orientation tensors in contrasting roles.
The PCA results shown in
Figure 5 highlight that the changes in the intramolecular orientation tensors of all 435 atom pairs with increasing shear rate at low pressures (≤100 MPa) are dramatically different from the changes that occur when squalane is sheared over similar range of strain rates at high pressures. It is informative to examine if these differences persist when the intramolecular orientation tensors associated with atom pairs of only one specific type are examined using PCA.
Figure 6,
Figure 7,
Figure 8 and
Figure 9 show the PCA results for reducing the dimension of intramolecular orientation tensor datasets for side-backbone pairs, end-backbone pairs, short backbone-backbone pairs, and long backbone-backbone pairs, respectively, at different pressures. At low pressures
100 MPa, regardless of the pair type, the atom pairs are organized along the PC1 axis by strain rate. This indicates that rate-dependent shear-induced changes in the intramolecular orientation tensors dominate the overall intramolecular structure evolution at low pressures, and thus can be linked to the reduction in the viscosity of squalane. At high pressures
400 MPa, the side-backbone, end-backbone, short backbone-backbone, and long backbone-backbone atom pairs are dispersed and they do not organize in any systematic way. This signals an overall limited evolution of molecular orientation of these atom pairs with changes in shear rate. As a result, the shear thinning of squalane observed for these pressures can not be explained via the evolution in the intramolecular orientation tensors associated with atom pairs of these 4 pair types.
While the long backbone-backbone atom pairs associated with different strain rates are mixed together in
Figure 9, the overall large number of these pairs and the wide distribution of separation bonds (from 2 to 21) associated with these pairs might blur the information in the low-dimensional space. Therefore, we divide this group into 20 sub-groups where each group is characterized and distinguished by an integer bond separation number
representing the number of bonds separating an atom pair.
Figure 10 shows the same PCA dimension reduction results shown in
Figure 9, except, instead of strain rate, the symbols are colored with the number of bonds separating the atom pair (i.e., the bond separation number). A red-blue color gradient scale is used where red represents smaller bond separation numbers (e.g.,
) and blue represents higher bond separation numbers (e.g.,
). At low pressures
MPa, the red and blue symbols appear mixed with no clear organization along either principal components. Interestingly, unlike
Figure 9c–f, a pattern in the organization of atom pairs for higher pressures
MPa emerges in
Figure 10c–f: the atom pairs are organized along the PC1 axis in increasing or decreasing order by their bond separation number. In each case, the far-separated atom pairs with large
b values (blue symbols) are clearly separated from the near-separated atom pairs characterized with small
b values (red symbols).
The emergence of organization of atom pairs at higher pressures in the reduced dimensions motivates a further classification of the long backbone-backbone pairs into near pairs, which are atom pairs separated by more than 1 but fewer than 6 bonds (i.e., 1
), and far pairs, which are atom pairs separated by more than 16 but fewer than 22 bonds (
). We now repeat the dimension reduction tasks using PCA for these near and far long backbone-backbone pairs.
Figure 11 shows the PCA results for near pairs at different pressures. At low pressures
100 MPa, atom pairs associated with the same strain rate are grouped together into clusters that are organized along the PC1 axis by strain rate. However, the atom pairs associated with different rates mix together and are not separable at high pressures
400 MPa. Thus, similar to the side-backbone, end-backbone, and short backbone-backbone atom pairs, the near long backbone-backbone pairs are also unable to link the observed reduction in viscosity of squalane with increasing rate to the evolution of the associated intramolecular orientation tensors.
Figure 12 shows the PCA results for far pairs at different pressures. A distinct picture emerges: for all pressures, the far atom pairs are grouped together into clusters that are separable by shear rates. At low pressures (
100 MPa), the separation between these clusters is more significant, and the clusters are organized along the PC1 axis by strain rate, similar to all other pair types (e.g., side-backbone, end-backbone, short backbone-backbone, and the near long backbone-backbone pairs). On the other hand,
Figure 12c–f show that at high pressures
400 MPa, while the clusters of atom pairs associated with the same rate are still separable, their distribution in the low-dimensional space is random and not organized along either of the principal components. In other words, the clusters of atom pairs identified by the strain rate do not show a correlation with changes in the rate, as observed for low pressures in
Figure 12a,b, where atom pairs are organized along PC1 axis by increasing or decreasing shear rate.
We note that well-separated clusters do not necessarily imply a significant evolution in the intramolecular structure with rate. From the single-pair results shown in
Figure 4, we find that all the tensor components at high pressures are saturated and exhibit very small fluctuations with changes in strain rate. Because we normalize the data during the preprocessing stage, small fluctuations in these saturated values can get magnified and picked up by PCA during the dimension reduction. The enhanced separation between clusters of atom pairs can thus emerge from the differences between the magnified fluctuations associated with different rates. Therefore, in addition to the separation between clusters, it is also important to look at their organization in the reduced dimensions, which is random in
Figure 12 suggesting the lack of correlation between shear thinning and changes in molecular orientation with strain rate.
To cross-check the PCA dimension reduction results for near and far long backbone-backbone pairs, we use t-SNE to reduce the dimension of intramolecular orientation tensor datasets associated with these 2 types of pairs.
Figure 13 and
Figure 14 show the t-SNE dimension reduction results for near pairs and far pairs, respectively, at different pressures. For near pairs, the data points with different shear rates are separable at low pressures
P = 0.1, 100 MPa in
Figure 13a,b, the separation is better for 0.1 MPa compared to 100 MPa. The clusters of atom pairs are also organized with shear rate along the horizontal direction, similar to the PCA results in
Figure 11a,b. On the other hand, at high pressures
400 MPa in
Figure 13c–f, the atom pairs are not separable and are scattered all over the plot.
Figure 14a,b shows that at low pressures
P = 0.1, 100 MPa, the far long backbone-backbone atom pairs with different shear rates are clearly separated and well organized with increasing or decreasing shear rate along the diagonal of the plot (e.g., from bottom left to top right for
MPa). In contrast, while the far atom pairs associated with the same shear rate are grouped together into well-separated clusters for high pressures
400 MPa in
Figure 14c–f, these clusters are randomly distributed, and do not exhibit any organization as observed in
Figure 14a,b. Overall, t-SNE corroborates many of the PCA findings.
3.4. Intermolecular Orientation and Shear Thinning
The analysis based on the dimension reduction of the orientation tensors associated with intramolecular atom pairs does not consider effects arising from the interactions between the molecules on the viscosity of squalane. These intermolecular effects, such as the friction between a pair of molecules, can change with strain rate and pressure, thus altering the squalane viscosity. We now examine the relationship between intermolecular orientation and shear thinning. An intermolecular atom pair is defined for atoms belonging to two different squalane molecules. This atom pair is characterized with a separation distance parameter
d which is chosen between 3.9 Å (an approximate value of the united-atom diameter) and 10 Å (the LJ potential cut-off distance). Discretizing this distance interval with a step size of 0.1 Å, 61 intermolecular atom pairs are generated for each strain rate and pressure. An intermolecular orientation tensor
is computed for each pair
p belonging to the set of these 61 pairs using Equation (
7), where the ensemble average is taken over the number of atom pairs belonging to different squalane molecules that are associated with a given distance
d. These 61 orientation tensors encapsulate the intermolecular orientation information at each strain rate and pressure.
To illustrate,
Figure 15 shows the 61 intermolecular orientation tensors at a strain rate of
for pressures
MPa. The diagonal elements
,
, and
of the orientation tensor at a distance
4 Å increasingly deviate from zero as pressure
P increases from 0.1 MPa to 100 MPa, before plateauing for
MPa. As the distance
d between the intermolecular atom pair increases, the diagonal elements of the corresponding orientation tensors exhibit a non-monotonic trend. For example,
exhibits an oscillatory behavior with increasing
d, reaching a maximum value of 0 at
7−8 Å, and a minimum at ≈8−9 Å.
is nearly symmetric to
around the axis of zero orientation.
also oscillates with a relatively smaller amplitude and has a minimum near
7−8 Å. The off-diagonal elements do not change significantly with either pressure or the pair distance.
exhibits mild variations within the range of ≈0.15−0.2, and
and
are ≈0 for all
d and
P.
Figure 16 shows the 61 intermolecular orientation tensors at a high pressure
MPa for different strain rates
. The 6 sub-figures show the 6 tensor components:
,
,
,
,
, and
.
Figure 16a–c show the variation of the diagonal elements
,
, and
as the separation distance
d between the atoms is increased. For small
, these components are different for different rates and assume non-zero values; the deviation from 0 is larger for higher strain rates. As
d increases,
,
, and
approach 0 in a non-monotonic manner. Variations in these components with rate diminishes with increasing
d such that for
,
,
, and
are nearly the same for all rates. The off-diagonal components
,
, and
do not change significantly with either rate or
d.
Figure 17 shows the results for reducing the intermolecular orientation tensor datasets of
dimensions to
dimensions using PCA at different pressures, where
indicates the number of shear rates associated with the corresponding pressure (
for 0.1 and 100 MPa,
for 400 MPa, and
for
MPa.) The sub-figures correspond to different pressures
0.1 MPa (a), 100 MPa (b), 400 MPa (c), 636 MPa (d), 875 MPa (e), and 955 MPa (f). In each sub-figure, different colors are used to label the atom pairs associated with different shear rates. For all 6 pressures, by analyzing the eigenvectors associated with PC1 and PC2, we find that the non-zero components:
,
,
, and
contribute significantly to PC1, while PC2 is dominated by
and
components, which are nearly zero.
Figure 17a,b show the dimension reduction results at low pressures
MPa. These plots look distinct from
Figure 17c–f which show the dimension reduction results at higher pressures
MPa. For low pressures, the atom pairs associated with smaller shear rates (
for 0.1 MPa and
for 100 MPa) cluster together into compact groups that are not easily separable; for 0.1 MPa, these groups are vertically aligned. On the other hand, the atom pairs for higher shear rates are dispersed and spread out horizontally. For high pressures
400 MPa, all dimension reduction plots look similar. In each case, the atom pairs associated with all rates share the same overall pattern: they are spread out horizontally, and are somewhat separable by the PC2 dimension. This separation, however, does not correlate in any significant way with changes in shear rate. These observations are similar to the ones made for the dimension reduction of intramolecular far long backbone-backbone pairs in
Figure 12. Far long backbone-backbone pairs were also able to separate the orientation tensor datasets, but the well-separated clusters were not organized along strain rate. Overall, the PCA results show that shear thinning at high pressures is not related to changes in the orientation order associated with the intermolecular atom pairs.
Figure 18 shows the t-SNE dimension reduction results for the intermolecular atom pairs at different pressures. Unlike PCA, t-SNE is able to separate the atom pairs associated with different strain rates for all rates at the low pressures
MPa. For all pressures, the atom pairs associated with the same strain rate distribute in a linear manner across the two-dimensional t-SNE plot. At low pressures, there is a degree of organization in the separation of different “strands” of atom pairs associated with the same shear rate. However, at high pressures, there is no consistent pattern in the organization of these strands, which is similar to the linear dimension reduction results obtained via PCA.
The PCA and t-SNE results suggest the saturation of the molecular orientation order associated with squalane sheared under different strain rates at high pressures. Motivated by this, in order to quantify the degree of evolution in the molecular orientation with shear rate for different pressures, we extract two angles,
and
, associated with the intramolecular and intermolecular orientation, respectively. We choose a far long backbone-backbone atom pair, 2–28, to compute the angle
between the squalane molecule axis and the flow direction (
).
Figure 19a shows that at low pressure
100 MPa,
decreases slowly from ∼
at strain rate
to ∼10
at
, indicating that the squalane molecules gradually align along the flow direction with increasing shear rate. In contrast, for high pressures
400 MPa,
is ≈
regardless of the strain rate, indicating that the squalane molecules are already well-aligned and the system exhibits a saturated intramolecular orientation order. These trends are consistent with the PCA and t-SNE dimension reduction results shown in
Figure 12 and
Figure 14.
Figure 19b shows the angle
between two neighboring squalane molecules, which is computed as an average using the distance vectors associated with the 2–28 atom pair belonging to two neighboring squalane molecules. The average is taken over all pairs of squalane molecules whose center of masses are separated by a distance less than
. Similar to the single molecule results,
decreases with increasing
at low
MPa, varying from ∼
to ∼
. On the other hand, at high
MPa,
exhibits a much smaller variation with
, oscillating around ∼
. Overall, these results indicate that shear thinning at low pressures is correlated with molecular alignment, however, at high pressures, it occurs with little change in the molecular structure as encapsulated by the intramolecular and intermolecular orientation tensors.





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}