
Ferrocene-Modified Polyacrylonitrile-Containing Block Copolymers as Preceramic Materials


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Instrumentations
2.3. General Synthesis of PAN Macroinitiator
2.4. General Synthesis of PAN-b-PMMA Block Copolymers
2.5. Post-Modification of the PAN-b-PMMA Block Copolymers Using 3-Ferrocenyl Propylamine
2.6. Stabilization and Pyrolysis of the Block Copolymer Films
3. Results and Discussion
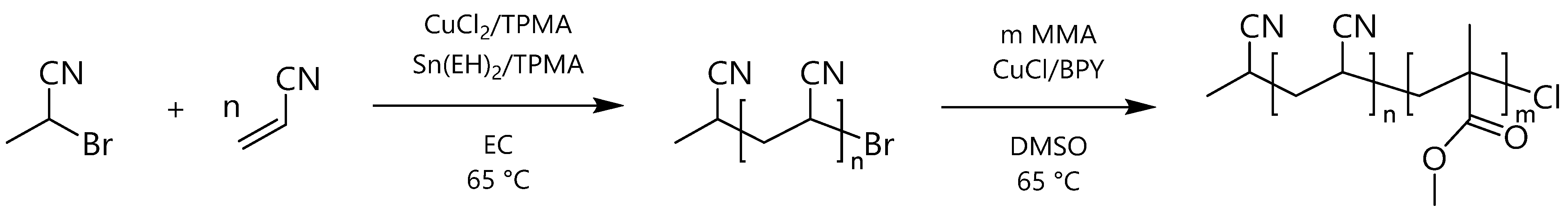
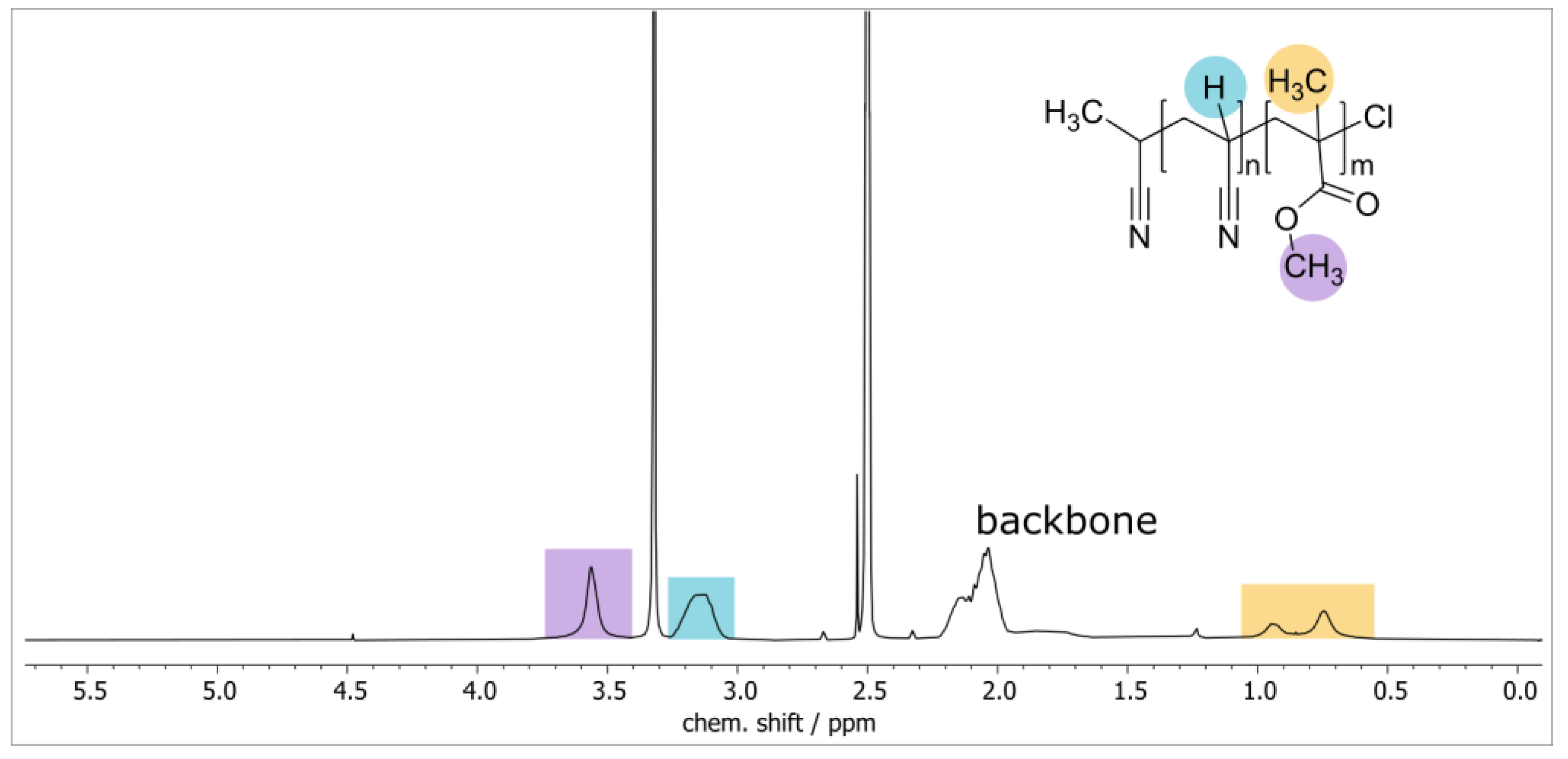
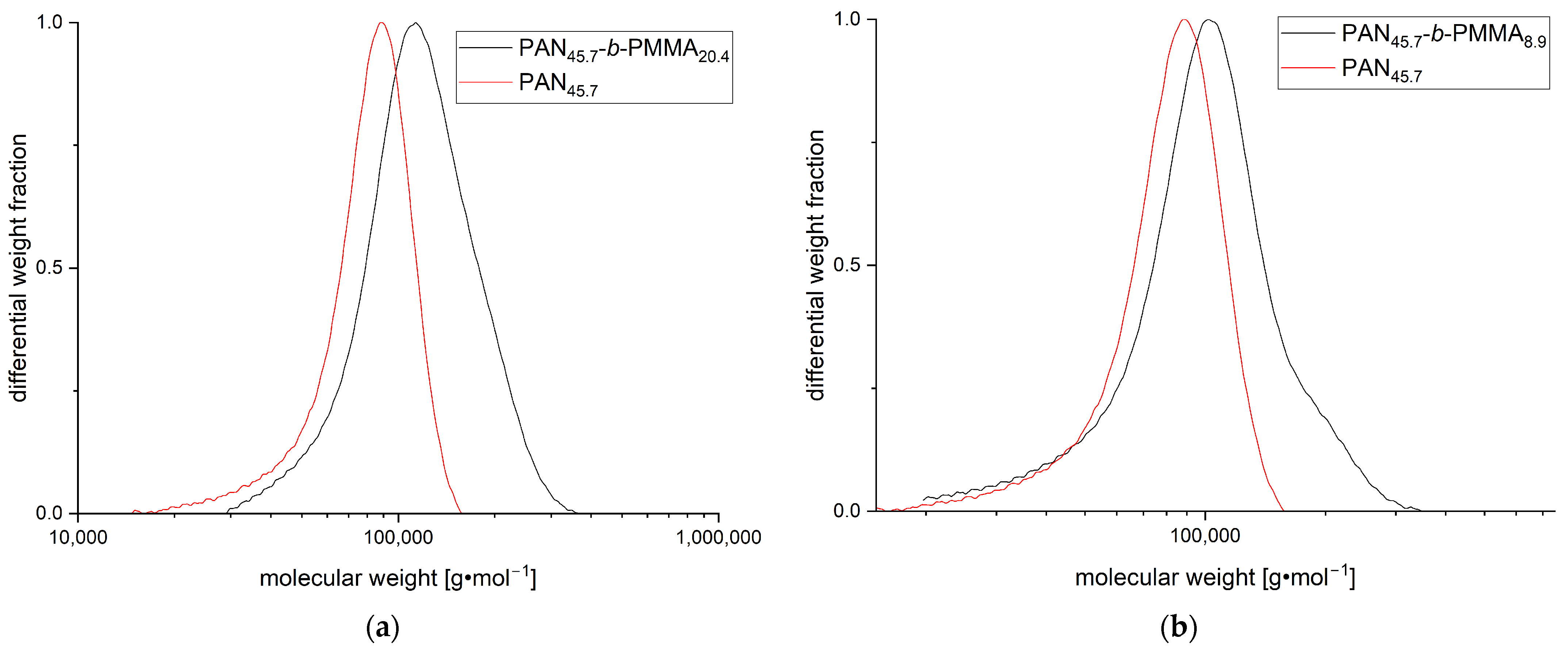
3.1. Synthesis and Characterization of PAN-b-PMMA Block Copolymers
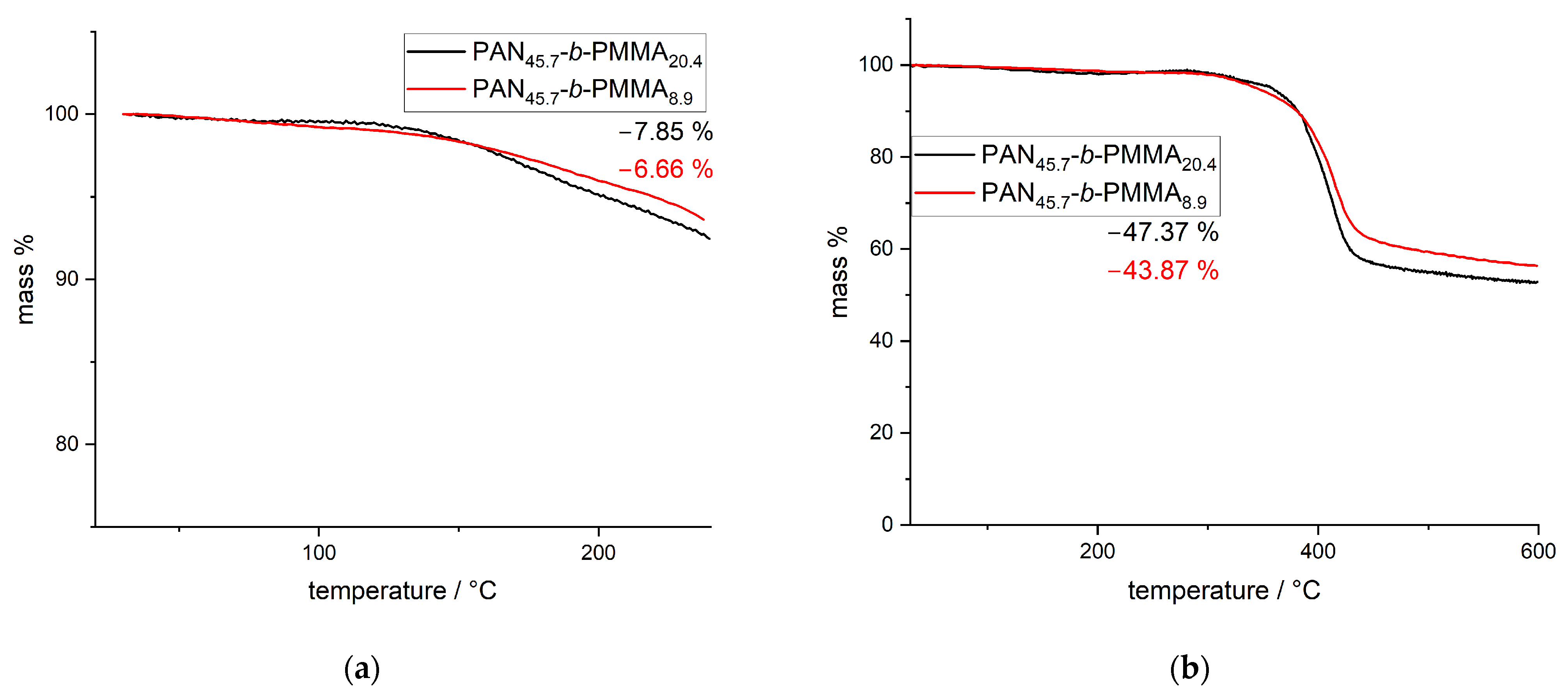
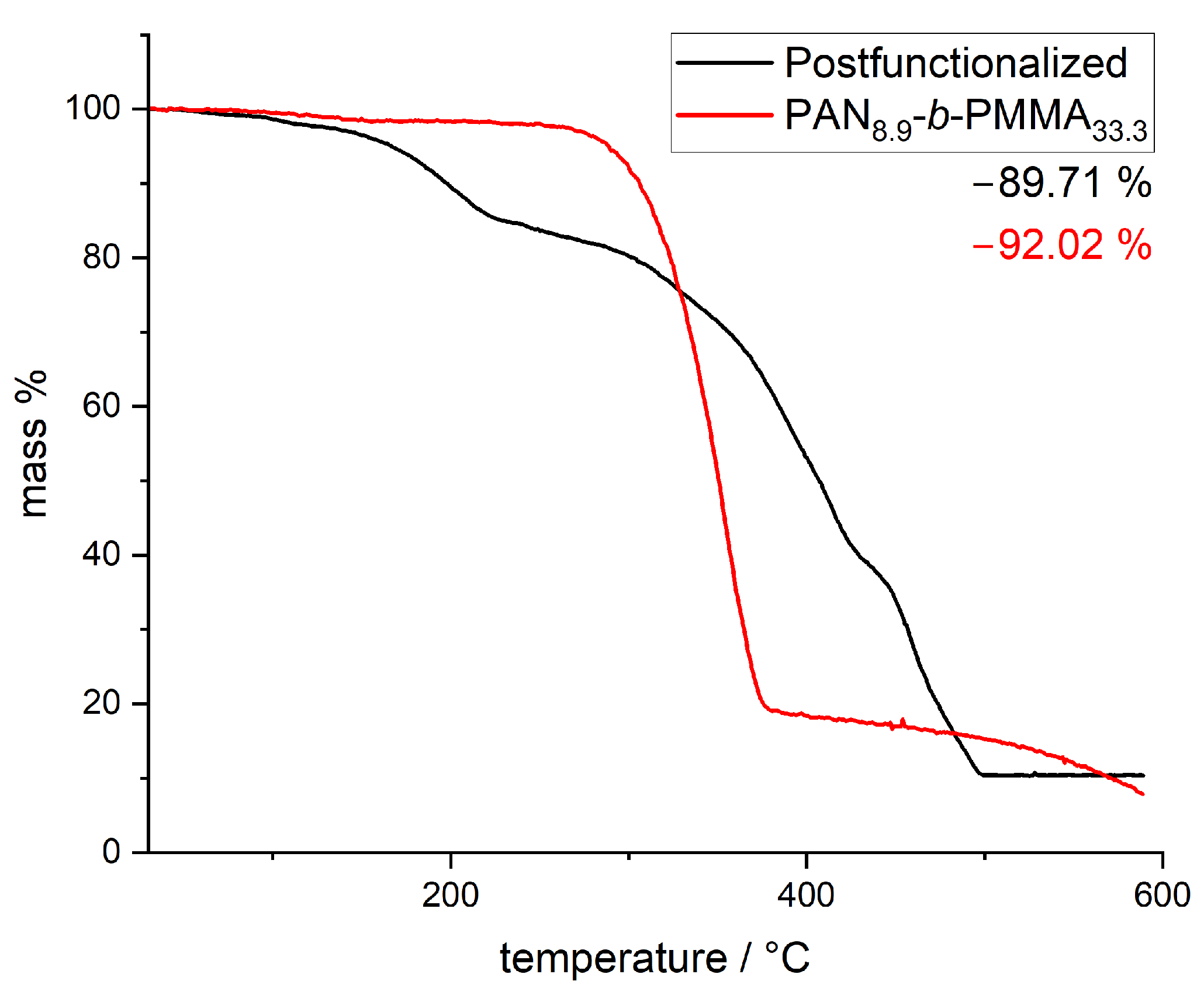
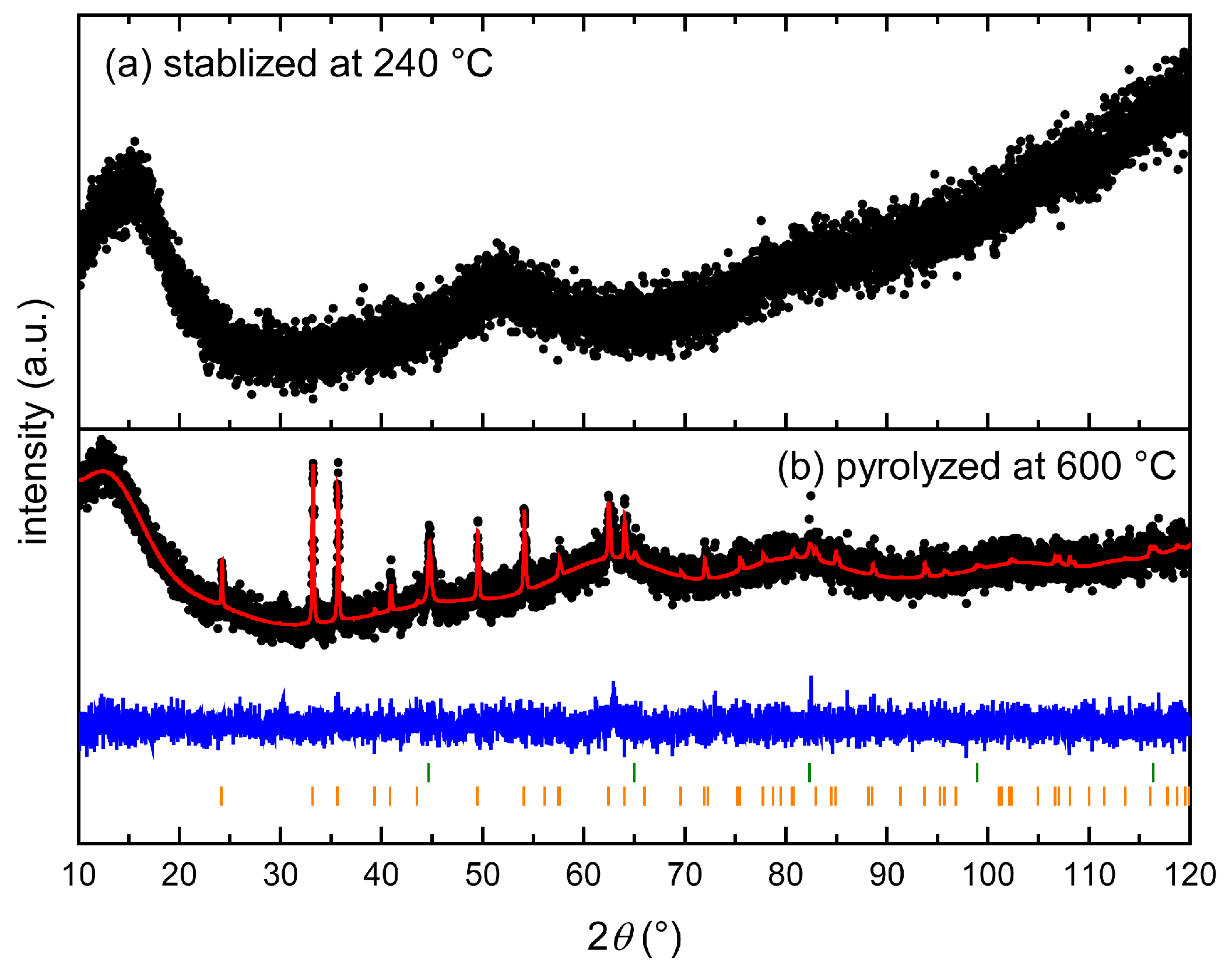
3.2. Thermal Stabilization and Pyrolysis of the Block Copolymer Films
3.3. Post-Modification Using 3-Ferrocenyl Propylamine
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feng, H.; Lu, X.; Wang, W.; Kang, N.-G.; Mays, J. Block Copolymers: Synthesis, Self-Assembly, and Applications. Polymers 2017, 9, 494. [Google Scholar] [CrossRef]
- Perumal, S.; Atchudan, R.; Lee, W. A Review of Polymeric Micelles and Their Applications. Polymers 2022, 14, 2510. [Google Scholar] [CrossRef]
- Stefik, M.; Guldin, S.; Vignolini, S.; Wiesner, U.; Steiner, U. Block copolymer self-assembly for nanophotonics. Chem. Soc. Rev. 2015, 44, 5076–5091. [Google Scholar] [CrossRef]
- Lazzari, M.; López-Quintela, M.A. Block copolymers as a tool for nanomaterial fabrication. Adv. Mater. 2003, 15, 1583–1594. [Google Scholar] [CrossRef]
- Kim, H.C.; Park, S.M.; Hinsberg, W.D. Block copolymer based nanostructures: Materials, processes, and applications to electronics. Chem. Rev. 2010, 110, 146–177. [Google Scholar] [CrossRef] [PubMed]
- Zhulina, E.B.; Borisov, O.V. Theory of Block Polymer Micelles: Recent Advances and Current Challenges. Macromolecules 2012, 45, 4429–4440. [Google Scholar] [CrossRef]
- Hamley, I.W. The Physics of Block Copolymers; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Lazzari, M.; Liu, G.; Lecommandoux, S. Block Copolymers in Nanoscience; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Hayward, R.C.; Pochan, D.J. Tailored Assemblies of Block Copolymers in Solution: It Is All about the Process. Macromolecules 2010, 43, 3577–3584. [Google Scholar] [CrossRef]
- Schacher, F.H.; Rupar, P.A.; Manners, I. Functional Block Copolymers: Nanostructured Materials with Emerging Applications. Angew. Chem. Int. Ed. 2012, 51, 7898–7921. [Google Scholar] [CrossRef] [PubMed]
- Brendel, J.C.; Schacher, F.H. Block Copolymer Self-Assembly in Solution—Quo Vadis? Chem. Asian J. 2018, 13, 230–239. [Google Scholar] [CrossRef]
- Shao, L.; Ma, J.; Prelesnik, J.L.; Zhou, Y.; Nguyen, M.; Zhao, M.; Jenekhe, S.A.; Kalinin, S.V.; Ferguson, A.L.; Pfaendtner, J.; et al. Hierarchical Materials from High Information Content Macromolecular Building Blocks: Construction, Dynamic Interventions, and Prediction. Chem. Rev. 2022, 122, 17397–17478. [Google Scholar] [CrossRef]
- Schüth, F.; Schmidt, W. Microporous and Mesoporous Materials. Adv. Mater. 2002, 14, 629–638. [Google Scholar] [CrossRef]
- Stein, A. Advances in microporous and mesoporous solids—Highlights of recent progress. Adv. Mater. 2003, 15, 763–775. [Google Scholar] [CrossRef]
- Thomas, A.; Goettmann, F.; Antonietti, M. Hard templates for soft materials: Creating nanostructured organic materials. Chem. Mater. 2008, 20, 738–755. [Google Scholar] [CrossRef]
- Yang, L.; Wang, Z.; Ji, Y.; Wang, J.; Xue, G. Highly Ordered 3D Graphene-Based Polymer Composite Materials Fabricated by “Particle-Constructing” Method and Their Outstanding Conductivity. Macromolecules 2014, 47, 1749–1756. [Google Scholar] [CrossRef]
- Nasir, S.; Hussein, M.Z.; Zainal, Z.; Yusof, N.A. Carbon-Based Nanomaterials/Allotropes: A Glimpse of Their Synthesis, Properties and Some Applications. Materials 2018, 11, 295. [Google Scholar] [CrossRef]
- Renschler, C.L.; Sylwester, A.P.; Salgado, L.V. Carbon-Films from Polyacrylonitrile. J. Mater. Res. 1989, 4, 452–457. [Google Scholar] [CrossRef]
- Zhong, M.; Tang, C.; Kim, E.K.; Kruk, M.; Celer, E.B.; Jaroniec, M.; Matyjaszewski, K.; Kowalewski, T. Preparation of porous nanocarbons with tunable morphology and pore size from copolymer templated precursors. Mater. Horiz. 2014, 1, 121–124. [Google Scholar] [CrossRef]
- Einert, M.; Wessel, C.; Badaczewski, F.; Leichtweiß, T.; Eufinger, C.; Janek, J.; Yuan, J.; Antonietti, M.; Smarsly, B.M. Nitrogen-Doped Carbon Electrodes: Influence of Microstructure and Nitrogen Configuration on the Electrical Conductivity of Carbonized Polyacrylonitrile and Poly(ionic liquid) Blends. Macromol. Chem. Phys. 2015, 216, 1930–1944. [Google Scholar] [CrossRef]
- Kruk, M.; Dufour, B.; Celer, E.B.; Kowalewski, T.; Jaroniec, M.; Matyjaszewski, K. Synthesis of mesoporous carbons using ordered and disordered mesoporous silica templates and polyacrylonitrile as carbon precursor. J. Phys. Chem. B 2005, 109, 9216–9225. [Google Scholar] [CrossRef]
- Lu, A.H.; Kiefer, A.; Schmidt, W.; Schüth, F. Synthesis of polyacrylonitrile-based ordered mesoporous carbon with tunable pore structures. Chem. Mater. 2004, 16, 100–103. [Google Scholar] [CrossRef]
- Kowalewski, T.; Tsarevsky, N.V.; Matyjaszewski, K. Nanostructured carbon arrays from block copolymers of polyacrylonitrile. J. Am. Chem. Soc. 2002, 124, 10632–10633. [Google Scholar] [CrossRef] [PubMed]
- Vowinkel, S.; Schäfer, C.G.; Cherkashinin, G.; Fasel, C.; Roth, F.; Liu, N.; Dietz, C.; Ionescu, E.; Gallei, M. 3D-ordered carbon materials by melt-shear organization for tailor-made hybrid core–shell polymer particle architectures. J. Mater. Chem. C 2016, 4, 3976–3986. [Google Scholar] [CrossRef]
- Schäfer, C.G.; Vowinkel, S.; Hellmann, G.P.; Herdt, T.; Contiu, C.; Schneider, J.J.; Gallei, M. A polymer based and template-directed approach towards functional multidimensional microstructured organic/inorganic hybrid materials. J. Mater. Chem. C 2014, 2, 7960–7975. [Google Scholar] [CrossRef]
- Zheng, B.; Lin, X.; Zhang, X.; Wu, D.; Matyjaszewski, K. Emerging Functional Porous Polymeric and Carbonaceous Materials for Environmental Treatment and Energy Storage. Adv. Funct. Mater. 2019, 30, 1907006. [Google Scholar] [CrossRef]
- Duranoğlu, D.; Trochimczuk, A.W.; Beker, U. Kinetics and Thermodynamics of Hexavalent Chromium Adsorption onto Activated Carbon Derived from Acrylonitrile-Divinylbenzene Copolymer. Chem. Eng. J. 2012, 187, 193–202. [Google Scholar] [CrossRef]
- Wu, D.; Dong, H.; Pietrasik, J.; Kim, E.K.; Hui, C.M.; Zhong, M.; Jaroniec, M.; Kowalewski, T.; Matyjaszewski, K. Novel Nanoporous Carbons from Well-Defined Poly(styrene-co-acrylonitrile)-Grafted Silica Nanoparticles. Chem. Mater. 2011, 23, 2024–2026. [Google Scholar] [CrossRef]
- Fanous, J.; Wegner, M.; Grimminger, J.; Andresen, Ä.; Buchmeiser, M.R. Structure-Related Electrochemistry of Sulfur-Poly(acrylonitrile) Composite Cathode Materials for Rechargeable Lithium Batteries. Chem. Mater. 2011, 23, 5024–5028. [Google Scholar] [CrossRef]
- Zhong, M.; Kim, E.K.; McGann, J.P.; Chun, S.-E.; Whitacre, J.F.; Jaroniec, M.; Matyjaszewski, K.; Kowalewski, T. Electrochemically Active Nitrogen-Enriched Nanocarbons with Well-Defined Morphology Synthesized by Pyrolysis of Self-Assembled Block Copolymer. J. Am. Chem. Soc. 2012, 134, 14846–14857. [Google Scholar] [CrossRef]
- Schlander, A.M.; Gallei, M. Temperature-Induced Colouration and Interface Shell Cross-Linking for the Preparation of Polymer-Based Opal Films. ACS Appl. Mater. Interf. 2019, 47, 44764–44773. [Google Scholar] [CrossRef]
- Wu, M.M. Acrylonitirle and Acrylonitrile Polymers. Encycl. Polym. Sci. Technol. 2003, 1, 143–144. [Google Scholar]
- Bandrup, J.; Immergut, E.H.; Grulke, E.A. (Eds.) Polymer Handbook, 4th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1999. [Google Scholar]
- Gemmer, L.; Niebuur, B.-J.; Dietz, C.; Rauber, D.; Plank, M.; Frieß, F.V.; Presser, V.; Stark, R.W.; Kraus, T.; Gallei, M. Polyacrylonitrile-containing amphiphilic block copolymers: Self-assembly and porous membrane formation. Polym. Chem. 2023, 14, 4825–4837. [Google Scholar] [CrossRef]
- Fukuda, T.; Terauchi, T.; Goto, A.; Tsujii, Y.; Miyamoto, T.; Shimizu, Y. Well-Defined Block Copolymers Comprising Styrene−Acrylonitrile Random Copolymer Sequences Synthesized by “Living” Radical Polymerization. Macromolecules 1996, 29, 3050–3052. [Google Scholar] [CrossRef]
- Detrembleur, C.; Sciannamea, V.; Koulic, C.; Claes, M.; Hoebeke, M.; Jérôme, R. Controlled Nitroxide-Mediated Radical Polymerization of Styrene, Styrene/Acrylonitrile Mixtures, and Dienes Using a Nitrone. Macromolecules 2002, 35, 7214–7223. [Google Scholar] [CrossRef]
- Sciannamea, V.; Jerome, R.; Detrembleur, C. In-situ nitroxide-mediated radical polymerization (NMP) processes: Their understanding and optimization. Chem. Rev. 2008, 108, 1104–1126. [Google Scholar] [CrossRef] [PubMed]
- Chiefari, J.; Chong, Y.K.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T.P.T.; Mayadunne, R.T.A.; Meijs, G.F.; Moad, C.L.; Moad, G.; et al. Living Free-Radical Polymerization by Reversible Addition−Fragmentation Chain Transfer: The RAFT Process. Macromolecules 1998, 31, 5559–5562. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, G. Controlling the Pore Size of Mesoporous Carbon Thin Films through Thermal and Solvent Annealing. Small 2017, 13, 1603107. [Google Scholar] [CrossRef]
- Keddie, D.J. A guide to the synthesis of block copolymers using reversible-addition fragmentation chain transfer (RAFT) polymerization. Chem. Soc. Rev. 2014, 43, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Perrier, S. 50th Anniversary Perspective: RAFT Polymerization—A User Guide. Macromolecules 2017, 50, 7433–7447. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Jo, S.M.; Paik, H.-j.; Shipp, D.A. An Investigation into the CuX/2,2′-Bipyridine (X = Br or Cl) Mediated Atom Transfer Radical Polymerization of Acrylonitrile. Macromolecules 1999, 32, 6431–6438. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Atom Transfer Radical Polymerization (ATRP): Current Status and Future Perspectives. Macromolecules 2012, 45, 4015–4039. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Advanced Materials by Atom Transfer Radical Polymerization. Adv. Mater. 2018, 30, e1706441. [Google Scholar] [CrossRef] [PubMed]
- Matyjaszewski, K.; Davis, K.; Patten, T.E.; Wei, M. Observation and analysis of a slow termination process in the atom transfer radical polymerization of styrene. Tetrahedron 1997, 53, 15321–15329. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Mu Jo, S.; Paik, H.-j.; Gaynor, S.G. Synthesis of Well-Defined Polyacrylonitrile by Atom Transfer Radical Polymerization. Macromolecules 1997, 30, 6398–6400. [Google Scholar] [CrossRef]
- Tsarevsky, N.V.; Sarbu, T.; Göbelt, B.; Matyjaszewski, K. Synthesis of Styrene−Acrylonitrile Copolymers and Related Block Copolymers by Atom Transfer Radical Polymerization. Macromolecules 2002, 35, 6142–6148. [Google Scholar] [CrossRef]
- Tsarevsky, N.V.; Bernaerts, K.V.; Dufour, B.; Du Prez, F.E.; Matyjaszewski, K. Well-Defined (Co)polymers with 5-Vinyltetrazole Units via Combination of Atom Transfer Radical (Co)polymerization of Acrylonitrile and “Click Chemistry”-Type Postpolymerization Modification. Macromolecules 2004, 37, 9308–9313. [Google Scholar] [CrossRef]
- Manners, I. Synthetic Metal-Containing Polymers; VCH: Weinheim, Germany, 2004. [Google Scholar]
- Whittell, G.R.; Hager, M.D.; Schubert, U.S.; Manners, I. Functional soft materials from metallopolymers and metallosupramolecular polymers. Nat. Mater. 2011, 10, 176–188. [Google Scholar] [CrossRef]
- Abd-El-Aziz, A.S.; Strohm, E.A. Transition metal-containing macromolecules: En route to new functional materials. Polymer 2012, 53, 4879–4921. [Google Scholar] [CrossRef]
- Gallei, M. The Renaissance of Side-Chain Ferrocene-Containing Polymers: Scope and Limitations of Vinylferrocene and Ferrocenyl Methacrylates. Macromol. Chem. Phys. 2014, 215, 699–704. [Google Scholar] [CrossRef]
- Abd-El-Aziz, A.S.; Agatemor, C.; Etkin, N. Sandwich complex-containing macromolecules: Property tunability through versatile synthesis. Macromol. Rapid Commun. 2014, 35, 513–559. [Google Scholar] [CrossRef]
- Beladi-Mousavi, S.M.; Sadaf, S.; Walder, L.; Gallei, M.; Rüttiger, C.; Eigler, S.; Halbig, C.E. Poly(vinylferrocene)-Reduced Graphene Oxide as a High Power/High Capacity Cathodic Battery Material. Adv. Energy Mater. 2016, 6, 1600108. [Google Scholar] [CrossRef]
- Su, X.; Tan, K.-J.; Elbert, J.; Rüttiger, C.; Gallei, M.; Jamison, T.F.; Hatton, T.A. Asymmetric Faradaic systems for selective electrochemical separations. Energy Environ. Sci. 2017, 10, 1272–1283. [Google Scholar] [CrossRef]
- Gallei, M.; Rüttiger, C. Recent Trends in Metallopolymer Design: Redox-Controlled Surfaces, Porous Membranes, and Switchable Optical Materials Using Ferrocene-Containing Polymers. Chem. Eur. J. 2018, 24, 10006–10021. [Google Scholar] [CrossRef] [PubMed]
- Rohland, P.; Schröter, E.; Nolte, O.; Newkome, G.R.; Hager, M.D.; Schubert, U.S. Redox-active polymers: The magic key towards energy storage—A polymer design guideline progress in polymer science. Prog. Polym. Sci. 2022, 125, 101474. [Google Scholar] [CrossRef]
- Hailes, R.L.; Oliver, A.M.; Gwyther, J.; Whittell, G.R.; Manners, I. Polyferrocenylsilanes: Synthesis, properties, and applications. Chem. Soc. Rev. 2016, 45, 5358–5407. [Google Scholar] [CrossRef]
- Zhou, J.W.; Whittell, G.R.; Manners, I. Metalloblock Copolymers: New Functional Nanomaterials. Macromolecules 2014, 47, 3529–3543. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, J.; Ren, L.; Tang, C. Metal-containing and related polymers for biomedical applications. Chem. Soc. Rev. 2016, 45, 5232–5263. [Google Scholar] [CrossRef]
- Hardy, C.G.; Zhang, J.Y.; Yan, Y.; Ren, L.X.; Tang, C.B. Metallopolymers with transition metals in the side-chain by living and controlled polymerization techniques. Prog. Polym. Sci. 2014, 39, 1742–1796. [Google Scholar] [CrossRef]
- Pietschnig, R. Polymers with pendant ferrocenes. Chem. Soc. Rev. 2016, 45, 5216–5231. [Google Scholar] [CrossRef] [PubMed]
- Mera, G.; Gallei, M.; Bernard, S.; Ionescu, E. Ceramic Nanocomposites from Tailor-Made Preceramic Polymers. Nanomaterials 2015, 5, 468–540. [Google Scholar] [CrossRef]
- Kaur, S.; Gallei, M.; Ionescu, E. Polymer-Ceramic Nanohybrid Materials. Org.-Inorg. Hybrid Nanomater. 2015, 267, 143–185. [Google Scholar] [CrossRef]
- Scheid, D.; Cherkashinin, G.; Ionescu, E.; Gallei, M. Single-Source Magnetic Nanorattles By Using Convenient Emulsion Polymerization Protocols. Langmuir 2014, 30, 1204–1209. [Google Scholar] [CrossRef]
- Schöttner, S.; Hossain, R.; Rüttiger, C.; Gallei, M. Ferrocene-Modified Block Copolymers for the Preparation of Smart Porous Membranes. Polymers 2017, 9, 491. [Google Scholar] [CrossRef]
- Mazurowski, M.; Gallei, M.; Li, J.; Didzoleit, H.; Stühn, B.; Rehahn, M. Redox-responsive polymer brushes grafted from polystyrene nanoparticles by means of surface initiated atom transfer radical polymerization. Macromolecules 2012, 45, 8970–8981. [Google Scholar] [CrossRef]
- Dong, H.; Tang, W.; Matyjaszewski, K. Well-Defined High-Molecular-Weight Polyacrylonitrile via Activators Regenerated by Electron Transfer ATRP. Macromolecules 2007, 40, 2974–2977. [Google Scholar] [CrossRef]
- Vapnik, H.; Elbert, J.; Su, X. Redox-copolymers for the recovery of rare earth elements by electrochemically regenerated ion-exchange. J. Mater. Chem. A 2021, 9, 20068–20077. [Google Scholar] [CrossRef]
- Lamson, M.; Kopeć, M.; Ding, H.; Zhong, M.; Matyjaszewski, K. Synthesis of well-defined polyacrylonitrile by ICARATRP with low concentrations of catalyst. J. Polym. Sci. A Polym. Chem. 2016, 54, 1961–1968. [Google Scholar] [CrossRef]
- Seo, Y.; Park, S.Y.; Lee, J.; Kim, J.K.; Duan, C.; Li, W. Inverted Cylindrical Microdomains by Blending Star-Shaped and Linear Block Copolymers. Macromolecules 2021, 54, 629–636. [Google Scholar] [CrossRef]
- Karbownik, I.; Fiedot, M.; Rac, O.; Suchorska-Woźniak, P.; Rybicki, T.; Teterycz, H. Effect of doping polyacrylonitrile fibers on their structural and mechanical properties. Polymer 2015, 75, 97–108. [Google Scholar] [CrossRef]
- Houtz, R.C. “Orlon” Acrylic Fiber: Chemistry and Properties. Text. Res. J. 1950, 20, 786–801. [Google Scholar] [CrossRef]
- Grassie, N.; McGuchan, R. Pyrolysis of polyacrylonitrile and related polymers—III. Thermal analysis of preheated polymers. Eur. Polym. J. 1971, 7, 1357–1371. [Google Scholar] [CrossRef]
- Fitzer, E. Pan-based carbon fibers—Present state and trend of the technology from the viewpoint of possibilities and limits to influence and to control the fiber properties by the process parameters. Carbon 1989, 27, 621–645. [Google Scholar] [CrossRef]
- Korobeinichev, O.P.; Paletsky, A.A.; Gonchikzhapov, M.B.; Glaznev, R.K.; Gerasimov, I.E.; Naganovsky, Y.K.; Shundrina, I.K.; Snegirev, A.Y.; Vinu, R. Kinetics of thermal decomposition of PMMA at different heating rates and in a wide temperature range. Thermochim. Acta 2019, 671, 17–25. [Google Scholar] [CrossRef]
- Hull, A.W. A New Method of X-Ray Crystal Analysis. Phys. Rev. 1917, 10, 661–696. [Google Scholar] [CrossRef]
- Pauling, L.; Hendricks, S.B. The crystal structures of hematite and corundum. J. Am. Chem. Soc. 1925, 47, 781–790. [Google Scholar] [CrossRef]
- Rietveld, H.M. Line profiles of neutron powder-diffraction peaks for structure refinement. Acta Crystallogr. 1967, 22, 151–152. [Google Scholar] [CrossRef]
- Rietveld, H.M. A profile refinement method for nuclear and magnetic structures. J. Appl. Crystallogr. 1969, 2, 65–71. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mn/kg mol−1 (vs. PMMA) | Ð (vs. PMMA) | φPMMA/% (NMR) | |
|---|---|---|---|
| PAN45.7 | 74.4 | 1.13 | / |
| PAN11.1 | 19.2 | 1.24 | / |
| PAN58.4 | 80.77 | 1.34 | / |
| PAN45.7-b-PMMA20.4 | 105.9 | 1.19 | 31 |
| PAN45.7-b-PMMA8.9 | 87.4 | 1.22 | 16 |
| PAN11.1-b-PMMA33.3 | 53.4 | 1.50 | 75 |
| PAN58.4-b-PMMA24.0 | 152.8 | 1.38 | 43 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heinz, S.; Gemmer, L.; Janka, O.; Gallei, M. Ferrocene-Modified Polyacrylonitrile-Containing Block Copolymers as Preceramic Materials. Polymers 2024, 16, 2142. https://doi.org/10.3390/polym16152142
Heinz S, Gemmer L, Janka O, Gallei M. Ferrocene-Modified Polyacrylonitrile-Containing Block Copolymers as Preceramic Materials. Polymers. 2024; 16(15):2142. https://doi.org/10.3390/polym16152142
Chicago/Turabian StyleHeinz, Sebastian, Lea Gemmer, Oliver Janka, and Markus Gallei. 2024. "Ferrocene-Modified Polyacrylonitrile-Containing Block Copolymers as Preceramic Materials" Polymers 16, no. 15: 2142. https://doi.org/10.3390/polym16152142
APA StyleHeinz, S., Gemmer, L., Janka, O., & Gallei, M. (2024). Ferrocene-Modified Polyacrylonitrile-Containing Block Copolymers as Preceramic Materials. Polymers, 16(15), 2142. https://doi.org/10.3390/polym16152142