
Block Copolymers of Poly(N-Vinyl Pyrrolidone) and Poly(Vinyl Esters) Bearing n-alkyl Side Groups via Reversible Addition-Fragmentation Chain-Transfer Polymerization: Synthesis, Characterization, and Thermal Properties




Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of PNVP-b-PVE Block Copolymers via RAFT Polymerization
2.3. Characterization Techniques
3. Results and Discussion
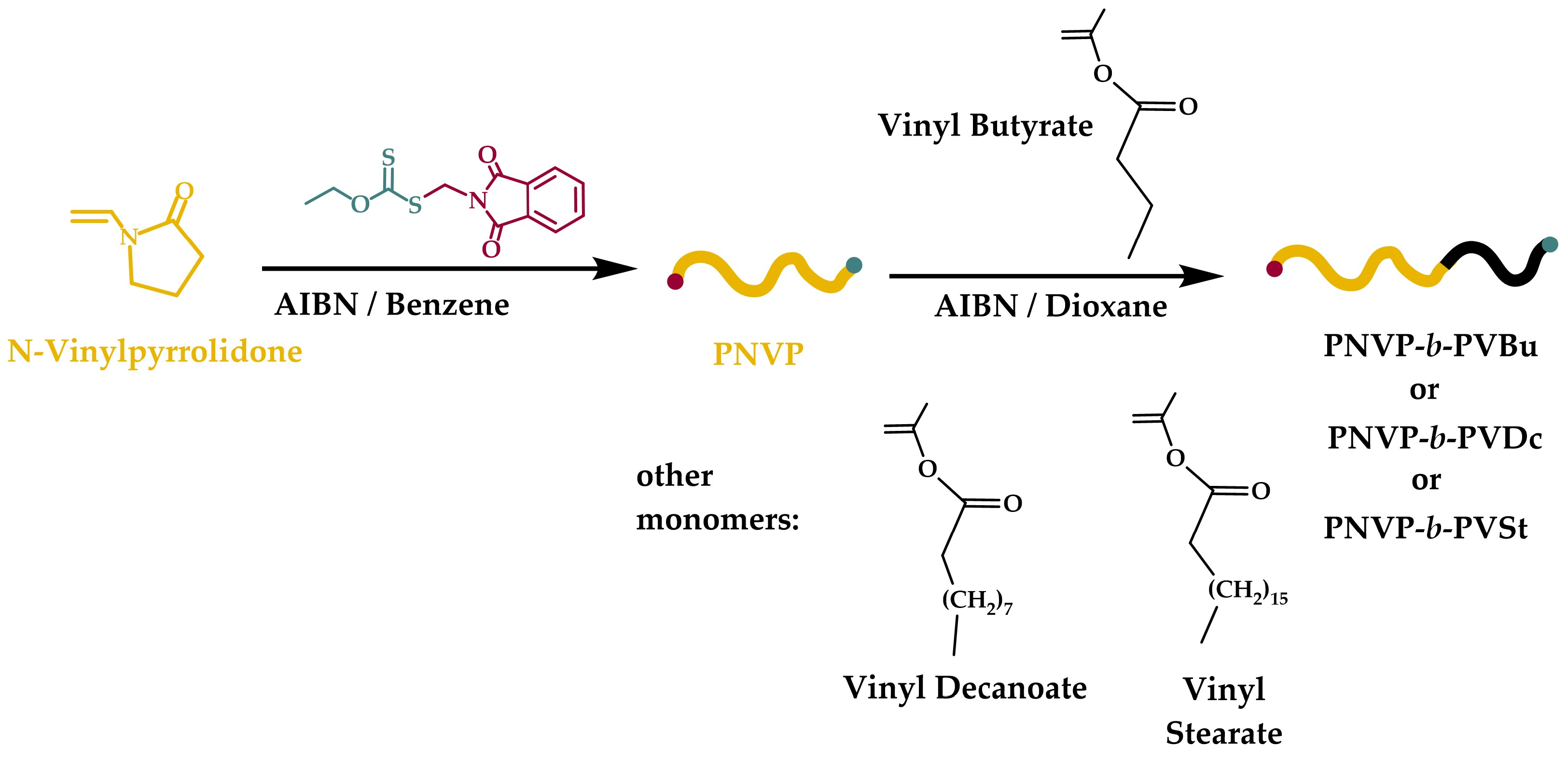
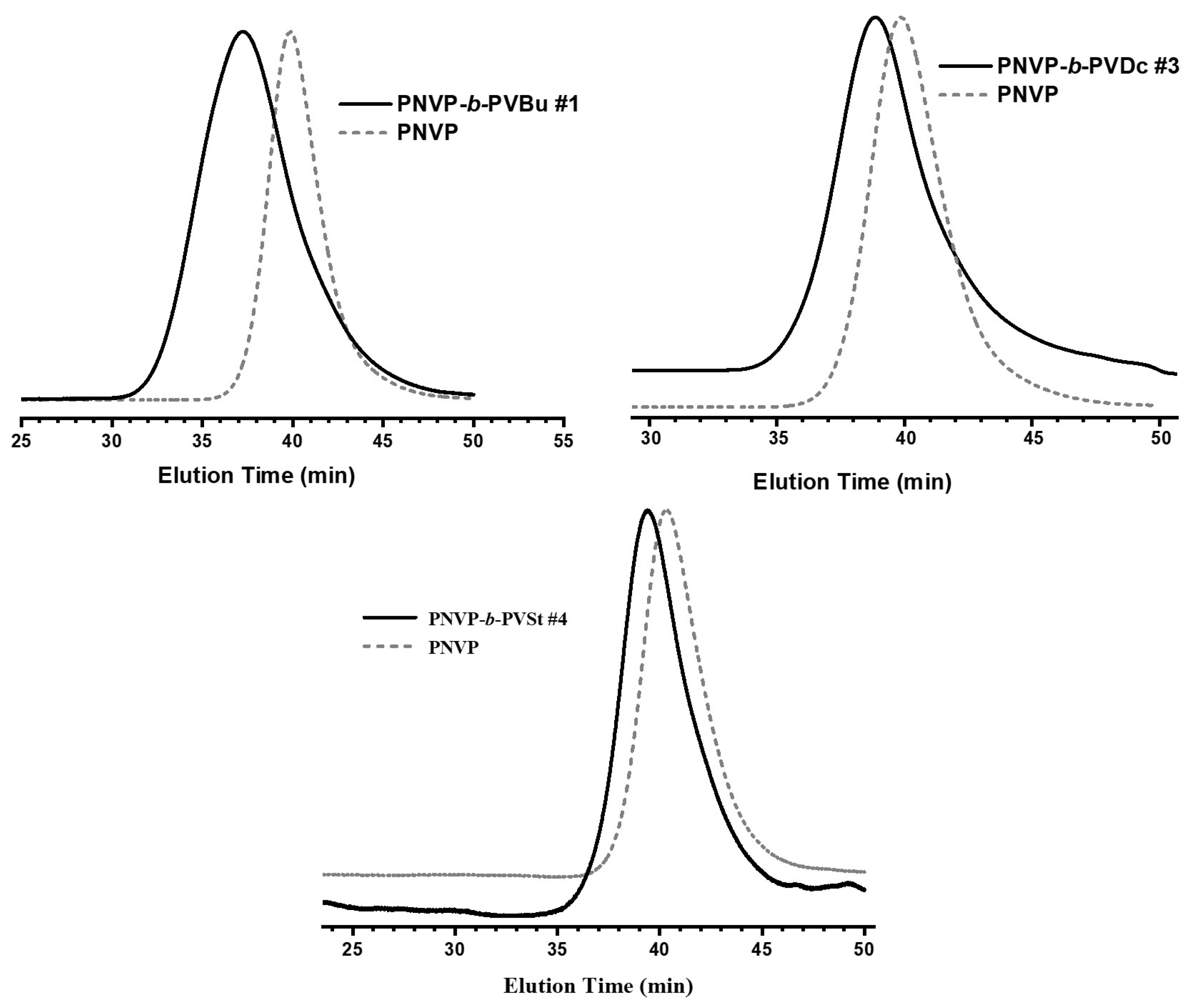
3.1. Synthesis of the Block Copolymers of NVP and VEs
3.2. Thermal Properties
3.2.1. DSC Analysis
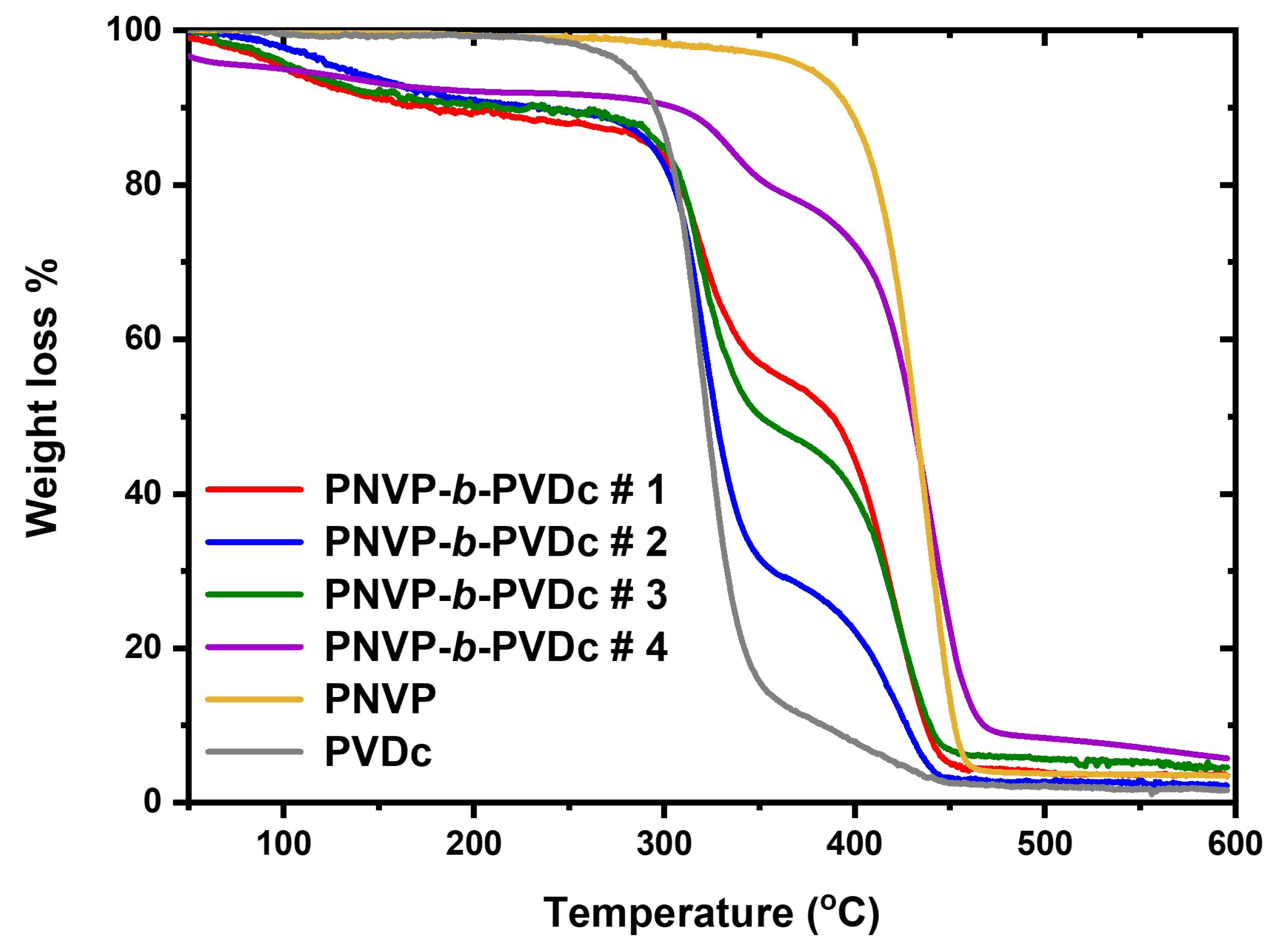
3.2.2. TGA Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rinno, H. Poly(vinyl esters). In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000; Volume 28, pp. 469–479. [Google Scholar]
- Ljungberg, N.; Wesslén, B. Preparation and properties of plasticized poly(lactic acid) films. Biomacromolecules 2005, 6, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, N.; Kawasumi, M.; Kato, M.; Usuki, A.; Okada, A. Preparation and mechanical properties of polypropylene–clay hybrids using a maleic anhydride-modified polypropylene oligomers. J. Appl. Polym. Sci. 1998, 67, 87–92. [Google Scholar] [CrossRef]
- Mark, H.F.; Bikales, N.; Overberger, C.G.; Menges, G.; Kroschwitz, J.I. Encyclopedia of Polymer Science and Engineering, 2nd ed.; Wiley: New York, NY, USA, 1989; Volume 17, p. 420. [Google Scholar]
- Ahmad, H.; Hasan, M.K.; Miah, M.A.J.; Ali, A.M.I.; Tauer, K. Solvent effect on the emulsion copolymerization of MMA and LMA in aqueous media. Polymer 2011, 52, 3925–3932. [Google Scholar] [CrossRef]
- Yang, D.J.; Fu, X.K.; Gong, Y.F. Study on the preparation and performances of P(VAc-MMA) polymer electrolytes for lithium ion battery. J. Polym. Sci. 2008, 26, 375–380. [Google Scholar] [CrossRef]
- Baily, N.; Pound-Lana, G.; Klumperman, B. Synthesis, characterization and self-assembly of poly(N-vinylpyrrolidone)-block-poly(vinyl acetate). Aust. J. Chem. 2012, 65, 1124–1131. [Google Scholar] [CrossRef]
- Braun, D.; Leiss, J.; Bergmann, M.J.; Hellmann, G.P. Miscibility behavior of various polymers made of alkyl and carboxyl groups. Eur. Polym. J. 1993, 29, 225–230. [Google Scholar] [CrossRef]
- Saikia, P.J.; Baruah, S.D. Structural and thermal behavior of comb-like polymer having n-octadecyl side chains. J. Appl. Polym. Sci. 2007, 104, 1226–1231. [Google Scholar] [CrossRef]
- Reimschuessel, H.K. Glass-transition temperature of comblike polymers-effects of side chain length and backbone chain structure. J. Polym. Sci. Polym. Chem. Ed. 1979, 17, 2447–2457. [Google Scholar] [CrossRef]
- Inomata, K.; Sakamaki, Y.; Nose, T.; Sasaki, S. Solid-state structure of comb-like polymers having n-octadecyl side chains. 2. Crystalline-amorphous layered structure. Polym. J. 1996, 28, 992–999. [Google Scholar] [CrossRef]
- Morossof, N.; Morawetz, H.; Post, B. Polymerization in crystalline state. 7. A crystallographic study of radiation-initiated polymerization in single crystals of vinyl stearate. J. Am. Chem. Soc. 1965, 87, 3035–3036. [Google Scholar] [CrossRef]
- Platé, N.A.; Shibaev, V.P. Comb-like polymers. Structure and properties. J. Polym. Sci. Macromol. Rev. 1974, 8, 117–253. [Google Scholar] [CrossRef]
- Jordan, E.F.; Feldeisen, D.W.; Wrigsley, A.N. Side-chain crystallinity. 1. Heats of fusion and melting transitions on selected homopolymers having long side chains. J. Polym. Sci. Part A-1 1971, 9, 1835–1852. [Google Scholar] [CrossRef]
- Shibasaki, Y.; Fukuda, K. Thermal behavior of vinyl esters of long-chain fatty-acids and their comblike polymers. Thermochim. Acta 1985, 88, 211–216. [Google Scholar] [CrossRef]
- Hsieh, H.W.S.; Post, B.; Morawetz, H. Crystallographic study of polymers exhibiting side-chain crystallization. J. Polym. Sci. Polym. Phys. Ed. 1976, 14, 1241–1255. [Google Scholar] [CrossRef]
- Greenberg, S.A.; Alfrey, T. Side chain crystallization of normal-alkyl polymethacrylates and polyacrylates. J. Am. Chem. Soc. 1954, 76, 6280–6285. [Google Scholar] [CrossRef]
- Port, W.S.; Hansen, J.E.; Jordan, E.F., Jr.; Dietz, T.J.; Swern, D. Polymerizable derivatives of long-chain fatty acids. 4. Vinyl esters. J. Polym. Sci. 1951, 7, 207–220. [Google Scholar] [CrossRef]
- De Merlis, C.C.; Schoneker, D.R. Review of the oral toxicity of polyvinyl alcohol (PVA). Food Chem. Toxicol. 2003, 41, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Tadavarthy, S.M.; Moller, J.H.; Amplatz, K. Polyvinyl alcohol (Ivalon)—A new embolic material. Am. J. Roentgenol. Radium Ther. Nucl. Med. 1975, 125, 609–616. [Google Scholar] [CrossRef]
- Davidson, G.S.; Terbrugge, K.G. Histologic long-term follow-up after embolization with polyvinyl alcohol particles. Am. J. Neuroradiol. 1995, 16, 843–846. [Google Scholar]
- Kojima, Y.; Maeda, H. Evaluation of poly(vinyl alcohol) for protein tailoring: Improvements in pharmacokinetic properties of superoxide dismutase. J. Bioact. Compat. Polym. 1993, 8, 115–131. [Google Scholar] [CrossRef]
- Hassan, C.M.; Peppas, N.A. Structure and applications of poly(vinyl alcohol) hydrogels produced by conventional crosslinking or by freezing/thawing methods. Adv. Polym. Sci. 2000, 153, 37–65. [Google Scholar]
- Bunck, D.N.; Sorenson, G.P.; Mahanthapp, M.K. Cobalt-mediated radical polymerization routes to poly (vinyl ester) block copolymers. J. Polym. Sci. Polym. Part A Chem. Ed. 2011, 49, 242–249. [Google Scholar] [CrossRef]
- Yamada, K.; Nakano, T.; Okamoto, Y. Synthesis of syndiotactic poly(vinyl alcohol) from fluorine-containing vinyl esters. Polym. J. 1998, 42, 9679–9686. [Google Scholar] [CrossRef]
- Murray, R.E.; Lincoln, D.M. Catalytic route to vinyl esters. Catal. Today 1992, 13, 93–102. [Google Scholar] [CrossRef]
- Charmot, D.; Corpart, P.; Adam, H.; Zard, S.Z.; Biadatti, T.; Bouhadir, G. Controlled radical polymerization in dispersed media. Macromol. Symp. 2000, 150, 23–32. [Google Scholar] [CrossRef]
- Matioszek, D.; Brusylovets, O.; Wilson, D.J.; Mazières, S.; Destarac, M. Reversible addition-fragmentation chain-transfer polymerization of vinyl monomers with N,N-dimethylsdiselenocarbamates. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 4361–4368. [Google Scholar] [CrossRef]
- Benard, J.; Favier, A.; Zhang, L.; Nilararoya, A.; Davis, T.P.; Barner-Kowollik, C.; Stenzel, M.H. Poly(vinyl ester) star polymers via xanthate-mediated living radical polymerization: From poly(vinyl alcohol) to glycopolymer stars. Macromolecules 2005, 38, 5475–5484. [Google Scholar] [CrossRef]
- Gu, Y.; He, J.; Li, C.; Zhou, C.; Song, S.; Yang, Y. Block copolymerization of vinyl acetate and vinyl neo-decanoate mediated by dithionosulfide. Macromolecules 2010, 43, 4500–4510. [Google Scholar] [CrossRef]
- Lipscomb, C.E.; Mahanthappa, M.K. Poly(vinyl ester) block copolymers synthesized by reversible addition-fragmentation chain transfer polymerizations. Macromolecules 2009, 42, 4571–4579. [Google Scholar] [CrossRef]
- Lipscomb, C.E.; Mahanthappa, M.K. Microphase separation mode-dependent mechanical response in poly(vinyl ester)/PEO triblock copolymers. Macromolecules 2011, 44, 4401–4409. [Google Scholar] [CrossRef]
- Odian, G. Principles of Polymerization; Wiley Interscience: Hoboken, NJ, USA, 2004. [Google Scholar]
- Britton, D.; Heatley, F.; Lovell, P.A. Chain transfer to polymer in free-radical bulk and emulsion polymerization of vinyl acetate studied by NMR spectroscopy. Macromolecules 1998, 31, 2828–2837. [Google Scholar] [CrossRef]
- Lindemann, M.K. The Mechanism of Vinyl Acetate in Vinyl Polymerization; Ham, G.E., Ed.; Dekker: New York, NY, USA, 1967; pp. 252–255. [Google Scholar]
- Kwak, Y.; Goto, A.; Fukuda, T.; Kobayashi, Y.; Yamago, S. A systematic study on activation process in organotellurium-mediated living radical polymerization of styrene, methyl methacrylate, methyl acrylate and vinyl acetate. Macromolecules 2006, 39, 4671–4679. [Google Scholar] [CrossRef]
- Flory, P.J.; Leutner, F.S. Occurrence of head-to-head arrangements of structural units in polyvinyl alcohol. J. Polym. Sci. 1948, 3, 880–890. [Google Scholar] [CrossRef]
- Iovu, M.C.; Matyjaszewski, K. Controlled/living radical polymerization of vinyl acetate by degenerative transfer with alkyl iodides. Macromolecules 2003, 36, 9346–9354. [Google Scholar] [CrossRef]
- Borkar, S.; Sen, A. Controlled copolymerization of vinyl acetate with 1-alkenes and their fluoro derivatives by degenerative transfer. J. Polym. Sci. Polym. Part A Chem. Ed. 2005, 43, 3728–3736. [Google Scholar] [CrossRef]
- Koumura, K.; Satoh, K.; Kamigaito, M.; Okamoto, Y. Iodine transfer radical polymerization of vinyl acetate in fluoroalcohols for simultaneous control of molecular weight, stereospecificity, and regiospecificity. Macromolecules 2006, 39, 4054–4061. [Google Scholar] [CrossRef]
- Yamago, S. Development of organotellurium-mediated and organostibine-mediated living radical polymerization reactions. J. Polym. Sci. Polym. Part A Chem. Ed. 2006, 44, 1–12. [Google Scholar] [CrossRef]
- Shaver, M.P.; Hanhan, M.F.; Jones, M.R. Controlled radical polymerization of vinyl acetate mediated by a vanadium complex. Chem. Commun. 2010, 46, 2127–2128. [Google Scholar] [CrossRef]
- Debuigne, A.; Caillie, J.-R.; Jérôme, R. Highly Efficient cobalt-mediated radical polymerization of vinyl acetate. Angew. Chem. 2005, 44, 1101–1104. [Google Scholar]
- Debuigne, A.; Poli, R.; Jérôme, C.; Jérôme, R.; Detrembleur, C. Overview of cobalt-mediated radical polymerization: Roots, state of the art and future prospects. Prog. Polym. Sci. 2009, 34, 211–239. [Google Scholar] [CrossRef]
- Bryaskova, R.; Detrembleur, C.; Debuigne, A.; Jérôme, R. Cobalt-mediated radical polymerization (CMRP) of vinyl acetate initiated by redox systems: Toward the scale-up of CMRP. Macromolecules 2006, 39, 8263–8268. [Google Scholar] [CrossRef]
- Roka, N.; Kokkorogianni, O.; Kontoes-Georgoudakis, P.; Choinopoulos, I.; Pitsikalis, M. Recent advances in the synthesis of complex macromolecular architectures based on poly(N-vinyl pyrrolidone) and the RAFT polymerization technique. Polymers 2022, 14, 701. [Google Scholar] [CrossRef]
- Tilottama, B.; Manojkumar, K.; Haribabu, P.M.; Vijayakrishna, K. A short review on RAFT polymerization of less activated monomers. J. Macromol. Sci. Part A Pure Appl. Chem. 2022, 59, 180–201. [Google Scholar] [CrossRef]
- Teodorescu, M.; Bercea, M. Poly(vinylpyrrolidone)—A Versatile Polymer for Biomedical and Beyond Medical Applications. Polym.-Plast. Technol. Eng. 2015, 54, 923–943. [Google Scholar] [CrossRef]
- Franco, F.; De Marco, I. The Use of Poly(N-vinyl pyrrolidone) in the Delivery of Drugs: A Review. Polymers 2020, 12, 1114. [Google Scholar] [CrossRef] [PubMed]
- Moulay, S. Molecular iodine/polymer complexes. J. Polym. Eng. 2013, 33, 389–443. [Google Scholar] [CrossRef]
- Kurakula, M.; Rao, G.S.N.K. Pharmaceutical assessment of polyvinylpyrrolidone (PVP): As excipient from conventional to controlled delivery systems with a spotlight on COVID-19 inhibition. J. Drug Deliv. Sci. Technol. 2020, 60, 102046. [Google Scholar] [CrossRef]
- Turner, D.T.; Schwartz, A. The glass transition temperature of poly(N-vinyl pyrrolidone) by differential scanning calorimetry. Polymer 1985, 26, 757–762. [Google Scholar] [CrossRef]
- Buera, M.D.P.; Levi, G.; Karel, M. Glass transition in poly(vinylpyrrolidone): Effect of molecular weight and diluents. Biotechnol. Prog. 1992, 8, 144–148. [Google Scholar] [CrossRef]
- Wang, H.; Kolodka, E.; Tande, B.M. Thermomechanical and rheological studies of copolymers of methyl methacrylate with a series of vinyl esters. Ind. Eng. Chem. Res. 2013, 52, 5111–5119. [Google Scholar] [CrossRef]
- Nzé, R.-P.; Colombani, O.; Nicol, E. Synthesis of poly(vinyl laurate)-b-poly(vinyl stearate) deblock copolymers by cobalt-mediated radical polymerization in solution. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 4046–4054. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, C.; Tian, J.; Xie, Y.; Zhang, K. A comparative study of self-assembled superstructures from cellulose stearoyl ester and poly(vinyl stearate). Macromol. Chem. Phys. 2018, 219, 1800229. [Google Scholar] [CrossRef]
- Perrier, S. 50th Anniversary Perspective: RAFT Polymerization—A User Guide. Macromolecules 2017, 50, 7433–7447. [Google Scholar] [CrossRef]
- Stenzel, M.H.; Cummins, L.; Roberts, G.E.; Davis, T.R.; Vana, P. Barner-Kowollik Xanthate mediated living polymerization of vinyl acetate: A systematic variation in MADIX/RAFT agent structure. Macromol. Chem. Phys. 2003, 204, 1160–1168. [Google Scholar] [CrossRef]
- Favier, A.; Barner-Kowollik, C.; Davis, T.P.; Stenzel, M.H. A Detailed On-line FT/NIR and 1 H NMR spectroscopic investigation into factors causing inhibition in xanthate-mediated vinyl acetate polymerization. Macromol. Chem. Phys. 2004, 205, 925–936. [Google Scholar] [CrossRef]
- Tong, Y.-Y.; Dong, Y.-Q.; Du, F.-S.; Li, Z.-C. Synthesis of well-defined poly(vinyl acetate)-b-polystyrene by combination of ATRP and RAFT polymerization. Macromolecules 2008, 41, 7339–7346. [Google Scholar] [CrossRef]
- Wan, D.; Satoh, K.; Kamigaito, M.; Okamoto, Y. Xanthate-mediated radical polymerization of N-vinylpyrrolidone in fluoroalcohols for simultaneous control of molecular weight and tacticity. Macromolecules 2005, 38, 10397–10405. [Google Scholar] [CrossRef]
- Postma, A.; Davis, T.P.; Li, G.; Moad, G.; O’Shea, M.S. RAFT polymerization with phthalimidomethyl trithiocarbonates or xanthates. On the origin of bimodal molecular weight distributions in living radical polymerization. Macromolecules 2006, 39, 5307–5318. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Iatrou, H.; Pispas, S.; Pitsikalis, M. Anionic polymerization: High vacuum techniques. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 3211–3234. [Google Scholar] [CrossRef]
- Uhrig, D.; Mays, J.W. Experimental techniques in high-vacuum anionic polymerization. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 6179–6222. [Google Scholar] [CrossRef]
- Nakabayashi, K.; Mori, H. Recent progress in controlled radical polymerization of N-vinyl monomers. Eur. Polym. J. 2013, 49, 2808–2838. [Google Scholar] [CrossRef]
- Daniel, J.K. A guide to the synthesis of block copolymers using reversible-addition fragmentation chain transfer (RAFT) polymerization. Chem. Soc. Rev. 2014, 43, 495–505. [Google Scholar]
- Renjith, D.; Raveendra, L.B.; Redouane, B.; Nathalie, M.; Yves, G. Controlled Radical Polymerization of N-Vinylpyrrolidone by Reversible Addition-Fragmentation Chain Tranfer Process. Macromol. Symp. 2005, 229, 8–17. [Google Scholar]
- Zard, S.Z. Discovery of the RAFT/MADIX Process: Mechanistic Insights and Polymer Chemistry Implications. Macromolecules 2020, 53, 8144–8159. [Google Scholar] [CrossRef]
- Smith, A.A.A.; Hussmann, T.; Elich, J.; Postma, A.; Alves, M.-H. Macromolecular design of poly(vinyl alcohol) by RAFT polymerization). Polym. Chem. 2012, 3, 85–88. [Google Scholar] [CrossRef]
- Roka, N.; Pitsikalis, M. Statistical Copolymers of N-Vinylpyrrolidone and Benzyl Methacrylate via RAFT: Monomer Reactivity Ratios, Thermal Properties and Kinetics of Thermal Decomposition. J. Macromol. Sci. Part A Pure Appl. Chem. 2018, 55, 222–230. [Google Scholar] [CrossRef]
- Roka, N.; Pitsikalis, M. Synthesis and micellization behavior of amphiphilic block copolymers of poly(N-Vinylpyrrolidone) and poly(Benzyl Methacrylate): Block vs. statistical copolymers. Polymers 2023, 15, 2225. [Google Scholar] [CrossRef]
- Barrales-Rienda, J.M.; Chaves, M.S.; Mazòn-Arechederra, J.M. Polymer precursors of polyacetylene. Thermal degradation of poly(vinyl esters). Part II-Effect of the n-acyl chain length on the autocatalytic thermal degradation of a homologous series of poly(vinyl n-alkyl esters) (PV-n-AEs). Polym. Degrad. Stab. 1989, 23, 279–298. [Google Scholar] [CrossRef]
- Hamley, I.W. The Physics of Block Copolymers; Oxford Science Publications: New York, NY, USA, 1998; Chapter 2; p. 24. [Google Scholar]
- Rogers, S.S.; Mandelkern, I. Glass formation in polymers. 1. The glass transitions of the poly(n-alkyl methacrylates). J. Phys. Chem. 1957, 61, 985–990. [Google Scholar] [CrossRef]
- Rehberg, C.E.; Fischer, C.H. Properties of monomeric and polymeric alkyl acrylates and methacrylates. Ind. Eng. Chem. 1948, 40, 1429–1433. [Google Scholar] [CrossRef]
- Wiley, R.H.; Brauer, G.M. Refractometric determination of 2nd-order transition temperatures in polymers. 3. Acrylates and methacrylates. J. Polym. Sci. 1948, 3, 647–651. [Google Scholar] [CrossRef]
- Rehberg, C.E.; Fischer, C.H. Preparation and properties of the n-alkyl acrylates. J. Am. Chem. Soc. 1944, 66, 1203–1207. [Google Scholar] [CrossRef]
- Flory, P.J. Thermodynamics of crystallization in high polymers. IV. A theory of crystalline states and fusion in polymers, copolymers, and their mixtures with diluents. J. Chem. Phys. 1949, 17, 223–240. [Google Scholar] [CrossRef]
- Ruzette, A.-V.G.; Banerjee, P.; Mayes, A.M.; Pollard, M.; Russell, T.P.; Jerome, R.; Slawecki, T.; Hjelm, R.; Thiyagarajan, P. Phase behavior of diblock copolymers between styrene and n-alkyl methacrylates. Macromolecules 1998, 31, 8509–8516. [Google Scholar] [CrossRef]
- Roka, N.; Kokkorogianni, O.; Pitsikalis, M. Statistical copolymers of N-vinylpyrrolidone and 2-(dimethylamino)ethyl methacrylate via RAFT: Monomer reactivity ratios, thermal properties, and kinetics of thermal decomposition. J. Polym. Sci. Polym. Chem. Ed. 2017, 15, 3776–3787. [Google Scholar] [CrossRef]
- Šimon, P.; Rybár, M. Kinetics of polymer degradation involving the splitting off of small molecules: Part 8. Thermal degradation of polyvinyl esters. Polym. Degrad. Stab. 1992, 38, 255–259. [Google Scholar] [CrossRef]
- Gilbert, J.B.; Kipling, J.J.; McEnaney, B.; Sherwood, J.N. Carbonization of polymers. I-Thermogravimetric analysis. Polymer 1962, 3, 1–10. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | PNVP (g) | AIBN (g) | Vinyl Ester Monomer (mL) | Dioxane (mL) | Yield % a | Overall Yield % |
|---|---|---|---|---|---|---|
| PNVP-b-PVBu #1 | 2 | 0.0410 | 6 | 4 | 40 | 33 |
| PNVP-b-PVBu #2 | 2 | 0.0410 | 1 | 4 | 33 | 26 |
| PNVP-b-PVBu #3 | 2 | 0.0410 | 7 | 4 | 32 | 26 |
| PNVP-b-PVBu #4 | 2 | 0.0410 | 3 | 4 | 38 | 31 |
| PNVP-b-PVDc #1 | 2 | 0.0410 | 1.5 | 4 | 70 | 56 |
| PNVP-b-PVDc #2 | 2 | 0.0410 | 7 | 4 | 68 | 54 |
| PNVP-b-PVDc #3 | 2 | 0.0410 | 2.5 | 4 | 48 | 38 |
| PNVP-b-PVDc #4 | 3 | 0.0615 | 1 | 4 | 73 | 58 |
| PNVP-b-PVSt #1 | 3 | 0.0615 | 2.25 | 4 | 75 | 60 |
| PNVP-b-PVSt #2 | 3 | 0.0615 | 3.75 | 4 | 52 | 41 |
| PNVP-b-PVSt #3 | 3 | 0.0615 | 4.30 | 7 | 46 | 36 |
| PNVP-b-PVSt #4 | 3 | 0.0615 | 6.00 | 9 | 43 | 34 |
| Macro-CTA (PNVP) a | Block Copolymers a | NVP | Vinyl Ester | |||
|---|---|---|---|---|---|---|
| Sample | Mn 103 (Daltons) | Ð | Mn 103 (Daltons) | Ð | % mol b | % mol b |
| PNVP-b-PVBu #1 | 8.5 | 1.30 | 16.0 | 1.90 | 22 | 78 |
| PNVP-b-PVBu #2 | 28.0 | 1.27 | 32.0 | 1.32 | 84 | 16 |
| PNVP-b-PVBu #3 | 8.9 | 1.35 | 17.5 | 1.40 | 57 | 43 |
| PNVP-b-PVBu #4 | 8.9 | 1.35 | 15.5 | 1.54 | 48 | 52 |
| PNVP-b-PVDc #1 | 8.5 | 1.30 | 12.5 | 1.31 | 63 | 37 |
| PNVP-b-PVDc #2 | 5.5 | 1.47 | 12.5 | 1.60 | 38 | 62 |
| PNVP-b-PVDc #3 | 8.5 | 1.30 | 11.0 | 1.45 | 56 | 44 |
| PNVP-b-PVDc #4 | 9.5 | 1.36 | 10.5 | 1.36 | 93 | 7 |
| PNVP-b-PVSt #1 | 8.5 | 1.30 | 10.5 | 1.44 | 78 | 22 |
| PNVP-b-PVSt #2 | 7.5 | 1.30 | 10.4 | 1.51 | 61 | 39 |
| PNVP-b-PVSt #3 | 8.1 | 1.30 | 10.9 | 1.37 | 85 | 15 |
| PNVP-b-PVSt #4 | 8.1 | 1.30 | 12.5 | 1.22 | 83 | 17 |
| Sample | Tg Experimental (°C) | ||
|---|---|---|---|
| PNVP-b-PVBu #1 | −9.1 | 80.3 | 126.6 |
| PNVP-b-PVBu #2 | - | 84.0 | 168.7 |
| PNVP-b-PVBu #3 | −8.9 | 93.6 | 153.3 |
| PNVP-b-PVBu #4 | −2.0 | 88.7 | 155.2 |
| PNVP | - | - | 187.1 |
| PVBu | −8.5 | - | - |
| Sample | Tg Experimental (°C) | ||
|---|---|---|---|
| PNVP-b-PVDc #1 | −39.3 | 62.8 | 113.3 |
| PNVP-b-PVDc #2 | −39.7 | 63.3 | 100.8 |
| PNVP-b-PVDc #3 | −52.5 | 66.0 | 112.2 |
| PNVP-b-PVDc #4 | 2.9 | 56.3 | 137.8 |
| PNVP | - | - | 187.1 |
| PVDc | −45.0 | - | - |
| Sample | WVSt % | Tm (°C) | ΔH (j/g VSt) | Xc % | Tg1 (°C) | Tg2 (°C) |
|---|---|---|---|---|---|---|
| PNVP-b-PVSt #1 | 43.6 | 33.6 | 24.7 | 11.2 | 77.4 | 148.6 |
| PNVP-b-PVSt #2 | 64.2 | 38.2 | 23.0 | 10.4 | 70.2 | 133.3 |
| PNVP-b-PVSt #3 | 33.0 | 36.4 | 36.9 | 16.8 | 64.2 | 116.1 |
| PNVP-b-PVSt #4 | 36.4 | 35.4 | 35.9 | 16.3 | 62.9 | 112.9 |
| PNVP | - | - | - | - | - | 187.1 |
| PVSt | - | 43.9 | 87.7 | 39.9 | - | - |
| Sample | Start | End | Max1 (°C) | Max2 (°C) |
|---|---|---|---|---|
| PNVP-b-PVBu #1 | 266.7 | 475.7 | 321.5 | 429.4 (broad) |
| PNVP-b-PVBu #2 | 284.3 | 459.1 | 321.7 | 423.9 |
| PNVP-b-PVBu #3 | 280.2 | 492.7 | 335.0 | 442.4 |
| PNVP-b-PVBu #4 | 283.6 | 499.7 | 335.8 | 445.7 |
| PNVP | 347.6 | 484.1 | 437.5 | |
| PVBu | 256.2 | 457.6 | 319.5 | 416.1 |
| Sample | Start1 | End1 | Max1 (°C) | Max2 (°C) |
|---|---|---|---|---|
| PNVP-b-PVDc #1 | 266.7 | 463.6 | 320.7 | 421.6 |
| PNVP-b-PVDc #2 | 247.6 | 453.9 | 319.5 | 420.1 |
| PNVP-b-PVDc #3 | 257.7 | 467.9 | 322.2 | 425.3 |
| PNVP-b-PVDc #4 | 286.5 | 483.3 | 335.2 | 439.7 |
| PNVP | 347.63 | 484.06 | - | 437.5 |
| PVDc | 238.6 | 450.8 | 322.7 | 417.8 |
| Sample | Start | End | Max1 (°C) | Max2 (°C) | Max3 (°C) |
|---|---|---|---|---|---|
| PNVP-b-PVSt #1 | 116.2 | 479.6 | 200.8 | 319.1 | 422.9 |
| PNVP-b-PVSt #2 | 128.9 | 465.9 | 196.4 | 322.1 | 419.4 |
| PNVP-b-PVSt #3 | 100.9 | 502.3 | 206.1 | 334.6 | 441.2 |
| PNVP-b-PVSt #4 | 135.0 | 494.7 | 212.5 | 338.8 | 439.8 |
| PNVP | 347.6 | 484.0 | - | 437.5 | |
| PVSt | 113.5 | 412.2 | 194.2 | 320.9 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roka, N.; Papazoglou, T.-P.; Pitsikalis, M. Block Copolymers of Poly(N-Vinyl Pyrrolidone) and Poly(Vinyl Esters) Bearing n-alkyl Side Groups via Reversible Addition-Fragmentation Chain-Transfer Polymerization: Synthesis, Characterization, and Thermal Properties. Polymers 2024, 16, 2447. https://doi.org/10.3390/polym16172447
Roka N, Papazoglou T-P, Pitsikalis M. Block Copolymers of Poly(N-Vinyl Pyrrolidone) and Poly(Vinyl Esters) Bearing n-alkyl Side Groups via Reversible Addition-Fragmentation Chain-Transfer Polymerization: Synthesis, Characterization, and Thermal Properties. Polymers. 2024; 16(17):2447. https://doi.org/10.3390/polym16172447
Chicago/Turabian StyleRoka, Nikoletta, Theodosia-Panagiota Papazoglou, and Marinos Pitsikalis. 2024. "Block Copolymers of Poly(N-Vinyl Pyrrolidone) and Poly(Vinyl Esters) Bearing n-alkyl Side Groups via Reversible Addition-Fragmentation Chain-Transfer Polymerization: Synthesis, Characterization, and Thermal Properties" Polymers 16, no. 17: 2447. https://doi.org/10.3390/polym16172447
APA StyleRoka, N., Papazoglou, T.-P., & Pitsikalis, M. (2024). Block Copolymers of Poly(N-Vinyl Pyrrolidone) and Poly(Vinyl Esters) Bearing n-alkyl Side Groups via Reversible Addition-Fragmentation Chain-Transfer Polymerization: Synthesis, Characterization, and Thermal Properties. Polymers, 16(17), 2447. https://doi.org/10.3390/polym16172447