
Polyesters and Polyester Nano- and Microcarriers for Drug Delivery


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Polyesters for Fabrication of Drug Carriers
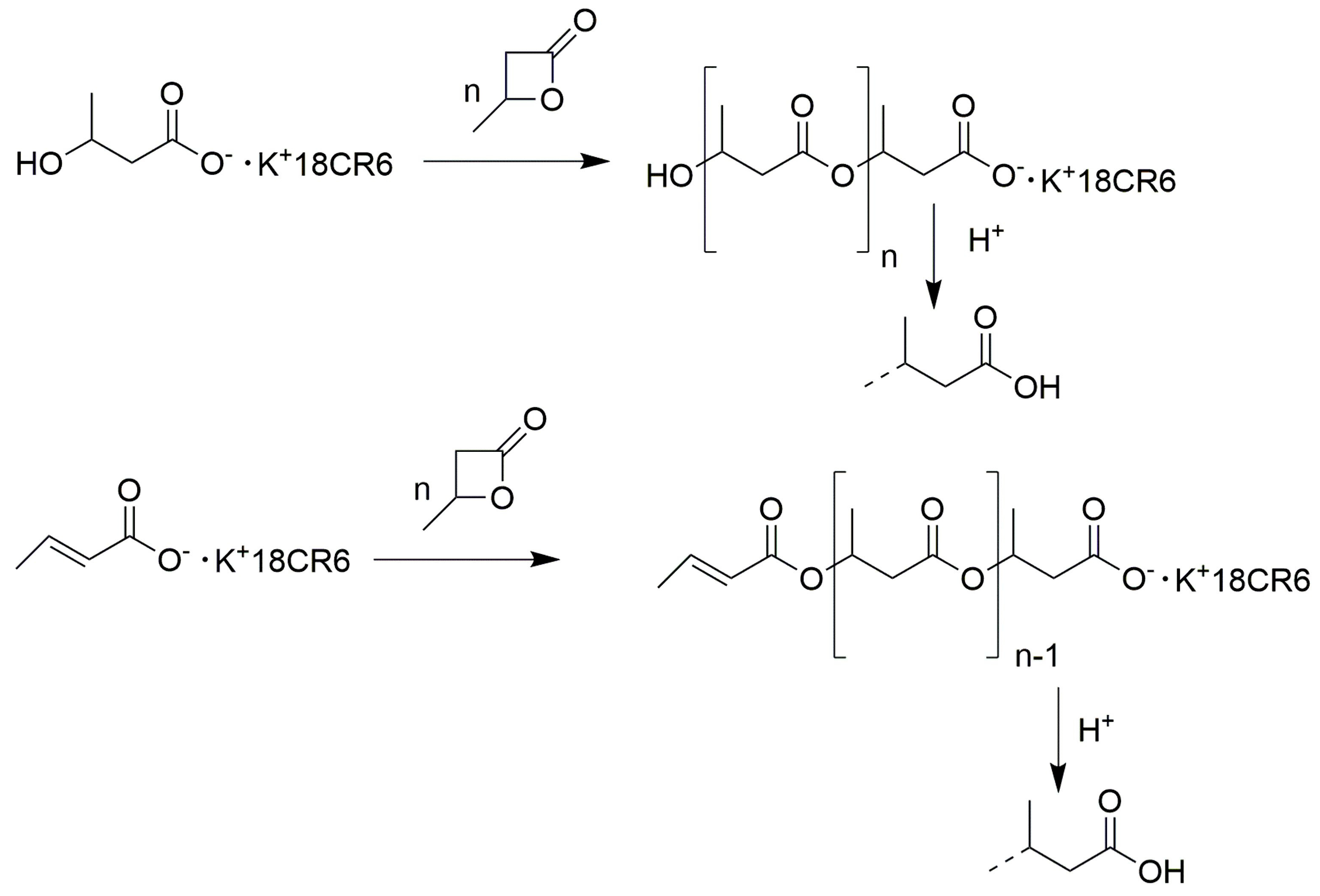
2.1. Synthesis of Poly(β-Butyrolactone) (Other Name Poly(Hydroxybutyrate))
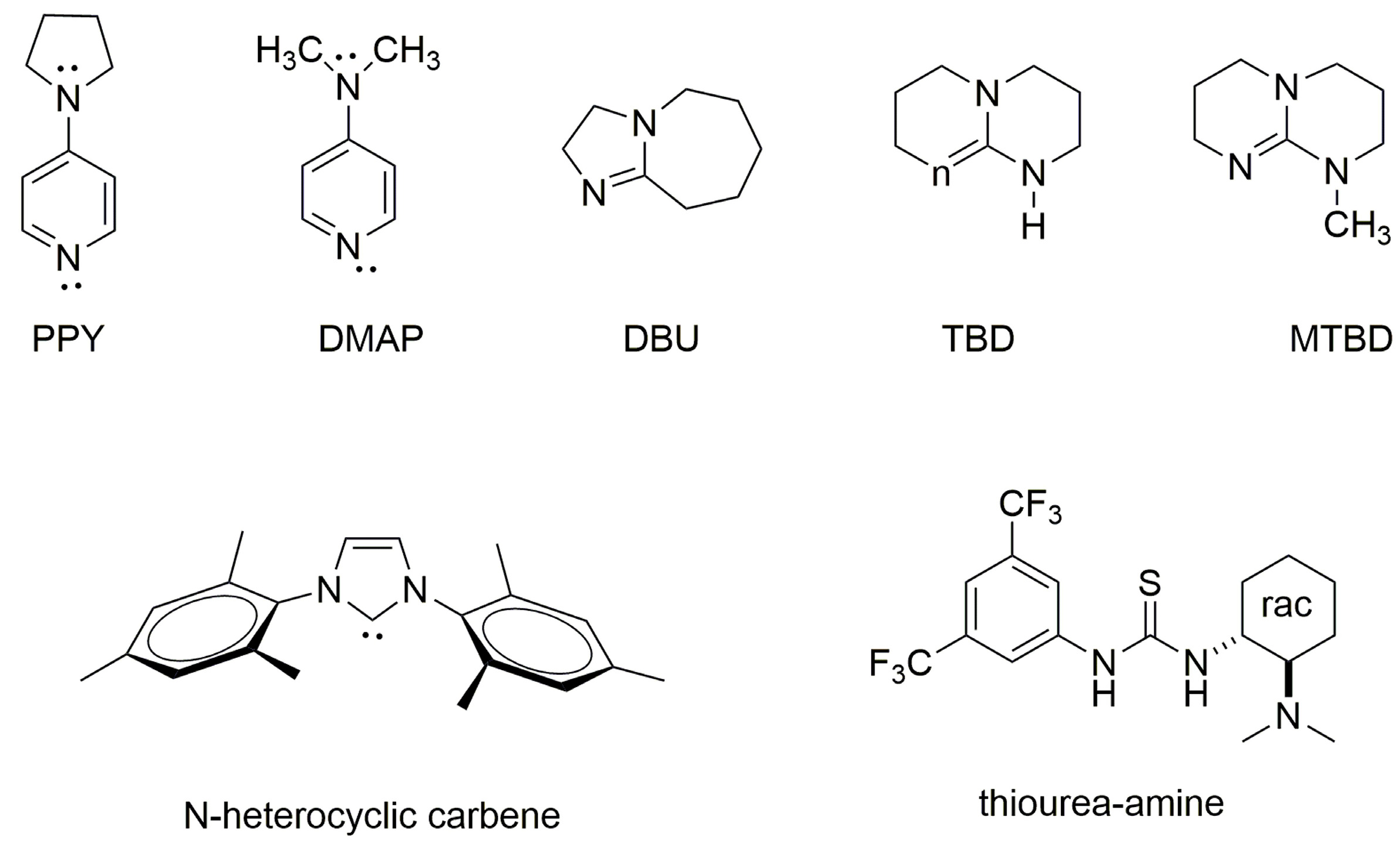
2.2. Polylactides
2.3. Polyglycolic Acid (Other Name: Polyglycolide)
2.4. Poly(ε-Caprolactone) and Macrolactones
3. Preparation of Polyester Drug Carriers
3.1. Drug Carriers by Oil-in-Water (O/W) Method
3.2. Drug Carriers by Water-in-Oil-in-Water (W1/O/W2) Method
3.3. Drug Carriers by Nanoprecipitation
3.4. Preparation of Drug Carriers Using Spray-Drying Equipment and Chips Used in Microfluidic Techniques
3.5. Direct Synthesis of Polyester Nano- and Microparticles by Ring-Opening Dispersion Polymerization of Lactides and ε-Caprolactone
3.6. Preparation of the Directly Synthesized Polyester Nano- and Microparticles Loaded with Drugs or Drug Models
4. Targeted Drug Delivery to the Brain Based on Polyester Nanoparticles
5. Preclinical and Clinical Studies of Polyester Nanocarriers
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, B.; Yang, J.-Z.; Wang, L.-F.; Zhang, Y.-J.; Lin, X.-J. Ifosfamide-loaded poly (lactic-co-glycolic acid) PLGA-dextran polymeric nanoparticles to improve the antitumor efficacy in osteosarcoma. BMC Cancer 2015, 15, 752. [Google Scholar] [CrossRef] [PubMed]
- Lilienthal, I.; Herold, N. Targeting molecular mechanisms underlying treatment efficacy and resistance in osteosarcoma: A review of current and future strategies. Int. J. Mol. Sci. 2020, 21, 6885. [Google Scholar] [CrossRef]
- Si, M.; Xia, Y.; Cong, M.; Wang, D.; Hou, Y.; Ma, H. In situ co-delivery of doxorubicin and cisplatin by injectable thermosensitive hydrogels for enhanced osteosarcoma treatment. Int. J. Nanomed. 2022, 17, 1309–1322. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Zhu, Q. Recent advances of PLGA micro/nanoparticles for the delivery of biomacromolecular therapeutics. Mater. Sci. Eng. C 2018, 92, 1041–1060. [Google Scholar] [CrossRef] [PubMed]
- Piperno, A.; Sciortino, M.T.; Giusto, E.; Montesi, M.; Panseri, S.; Scala, A. Recent advances and challenges in gene delivery mediated by polyester-based nanoparticles. Int. J. Nanomed. 2021, 16, 5981–6002. [Google Scholar] [CrossRef]
- Conte, C.; Monteiro, P.F.; Gurnani, P.; Stolnik, S.; Ungaro, F.; Quaglia, F.; Clarke, P.; Grabowska, A.; Kavallaris, M.; Alexander, C. Multi-component bioresponsive nanoparticles for synchronous delivery of docetaxel and TUBB3 siRNA to lung cancer cells. Nanoscale 2021, 13, 11414–11426. [Google Scholar] [CrossRef]
- Thangudu, S.; Cheng, F.-Y.; Su, C.-H. Advancements in the blood-brain barrier penetrating nanoplatforms for brain related disease diagnostics and therapeutic applications. Polymers 2020, 12, 3055. [Google Scholar] [CrossRef]
- Maher, R.; Moreno-Borrallo, A.; Jindal, D.; Mai, B.T.; Ruiz-Hernandez, E.; Harkin, A. Intranasal polymeric and lipid-based nanocarriers for CNS drug delivery. Pharmaceutics 2023, 15, 746. [Google Scholar] [CrossRef]
- Shen, K.; Sun, G.; Chan, L.; He, L.; Li, X.; Yang, S.; Wang, B.; Zhang, H.; Huang, J.; Chang, M.; et al. Anti-inflammatory nanotherapeutics by targeting matrix metalloproteinases for immunotherapy spinal cord injury. Small 2021, 17, 2102102. [Google Scholar] [CrossRef]
- Sun, Y.; Zabihi, M.; Li, Q.; Li, X.; Kim, B.J.; Ubogu, E.E.; Raja, S.N.; Wesselmann, U.; Zhao, C. Drug permeability: From the blood–brain barrier to the peripheral nerve barriers. Adv. Therap. 2023, 6, 2200150. [Google Scholar] [CrossRef]
- Zhao, M.; Zhu, T.; Chen, J.; Cui, Y.; Zhang, X.; Lee, R.J.; Sun, F.; Li, Y.; Teng, L. PLGA/PCADK composite microspheres containing hyaluronic acid–chitosan siRNA nanoparticles: A rational design for rheumatoid arthritis therapy. Int. J. Pharm. 2021, 596, 120204. [Google Scholar] [CrossRef] [PubMed]
- Sunoqrot, S.; Niazi, M.; Al-Natour, M.A.; Jaber, M.; Abu-Qatouseh, L. Loading of coal tar in polymeric nanoparticles as a potential therapeutic modality for psoriasis. ACS Omega 2022, 7, 7333–7340. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.; Chandra, S.; Dodson, K.; Shaheen, F.; Wiltz, K.; Ireland, S.; Syed, M.; Dash, S.; Wiese, T.; Mandal, T.; et al. Aptamer-functionalized hybrid nanoparticle for the treatment of breast cancer. Europ. J. Pharm. Biopharm. 2017, 114, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Jothar, L.; Sofia Doulkeridou, S.; Raymond, M.; Schiffelers, R.M.; Javier Sastre Torano, J.S.; Oliveira, S.; van Nostrum, C.E.; Hennink, W.E. Insights into maleimide-thiol conjugation chemistry: Conditions for efficient surface functionalization of nanoparticles for receptor targeting. J. Control. Release 2018, 282, 101–109. [Google Scholar] [CrossRef]
- Farran, B.; Montenegro, R.C.; Kasa, P.; Pavitra, E.; Yun Suk Huh, Y.S.; Han, Y.-K.; Kamal, M.A.; Nagarajua, G.P.; Rajue, G.S.R. Folate-conjugated nanovehicles: Strategies for cancer therapy. Mater. Sci. Eng. C 2020, 107, 110341. [Google Scholar] [CrossRef]
- Lee, S.E.; Lee, C.M.; Won, J.F.; Jang, G.-Y.; Lee, J.H.; Park, S.H.; Kang, T.H.; Han, H.D.; Park, Y.-M. Enhancement of anticancer immunity by immunomodulation of apoptotic tumor cells using annexin A5 protein-labeled nanocarrier system. Biomaterials 2022, 288, 121677. [Google Scholar] [CrossRef]
- Sanjanwala, D.; Vandana Patravale, V. Aptamers and nanobodies as alternatives to antibodies for ligand-targeted drug delivery in cancer. Drug Discov. Today 2023, 28, 103550. [Google Scholar] [CrossRef]
- Pelosi, C.; Tinè, M.R.; Wurm, F.R. Main-chain water-soluble polyphosphoesters: Multi-functional polymers as degradable PEG-alternatives for biomedical applications. Eur. Polym. J. 2020, 141, 110079. [Google Scholar] [CrossRef]
- Panyue Wen, P.; Ke, W.; Dirisala, A.; Toh, K.; Tanaka, M.; Li, J. Stealth and pseudo-stealth nanocarriers. Adv. Drug Deliv. Rev. 2023, 198, 114895. [Google Scholar] [CrossRef]
- Bona, B.L.; Lagarrigue, P.; Chirizzi, C.; Espinoza, M.I.M.; Pipino, C.; Metrangolo, P.; Cellesi, F.; Bombelli, F.B. Design of fluorinated stealth poly(ε-caprolactone) nanocarriers. Colloids Surf. B Biointerfaces 2024, 234, 113730. [Google Scholar] [CrossRef]
- Chountoulesi, M.; Naziris, N.; Pippa, N.; Pispas, S.; Demetzos, D. Stimuli-responsive nanocarriers for drug delivery. In Nanomaterials for Clinical Applications, Case Studies in Nanomedicines a Volume in Micro and Nano Technologies; Pippa, N., Demetzos, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar] [CrossRef]
- Guo, Q.; Liu, J.; Yang, H.; Lei, Z. Synthesis of photo, oxidation, reduction triple-stimuli-responsive interface-cross-linked polymer micelles as nanocarriers for controlled release. Macromol. Chem. Phys. 2021, 222, 2000365. [Google Scholar] [CrossRef]
- Sun, Y.; Davis, E. Nanoplatforms for targeted stimuli-responsive drug delivery: A review of platform materials and stimuli-responsive release and targeting mechanisms. Nanomaterials 2021, 11, 746. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Sharma, Y.; Dheer, D. Stimuli responsiveness of recent biomacromolecular systems (concept to market): A review. Int. J. Biol. Macromol. 2024, 261, 129901. [Google Scholar] [CrossRef]
- Kiamohammadi, L.; Asadi, L.; Shirvalilou, S.; Khoei, S.; Khoee, S.; Soleymani, M.; Minaei, S.E. Physical and biological properties of 5-fluorouracil polymer-coated magnetite nanographene oxide as a new thermosensitizer for alternative magnetic hyperthermia and a magnetic resonance imaging contrast agent: In vitro and in vivo study. ACS Omega 2021, 6, 20192–20204. [Google Scholar] [CrossRef] [PubMed]
- Trotsenko, Y.A.; Belova, L.L. Biosynthesis of poly(3-hydroxybutyrate) and poly(3-hydroxybutyrate-co-3-hydroxyvalerate) and its regulation in bacteria. Microbiology 2000, 6, 635–645. [Google Scholar] [CrossRef]
- Verlinden, R.A.J.; Hill, D.J.; Kenward, M.A.; Williams, C.D.; Radecka, I. Bacterial synthesis of biodegradablepolyhydroxyalkanoates. J. Appl. Microbiol. 2007, 102, 1437–1449. [Google Scholar] [CrossRef] [PubMed]
- Jendrossek, D.; Pfeiffer, D. New insights in the formation of polyhydroxyalkanoate granules (carbonosomes) and novel functions of poly(3-hydroxybutyrate). Environment. Microbiol. 2014, 16, 2357–2373. [Google Scholar] [CrossRef]
- Nagarajan, D.; Aristya, G.R.; Lin, Y.-J.; Chang, J.-J.; Yen, H.-W.; Chang, J.-S. Microbial cell factories for the production of polyhydroxyalkanoates. Essays Biochem. 2021, 65, 337–353. [Google Scholar] [CrossRef]
- Zhou, W.; Bergsma, S.; Colpa, D.I.; Euverink, G.-J.W.; Krooneman, J. Polyhydroxyalkanoates (PHAs) synthesis and degradation by microbes and applications towards a circular economy. J. Environ. Manag. 2023, 341, 118033. [Google Scholar] [CrossRef]
- Koller, M. A review on stability replacing shed and emerging fermentation schemes for microbial production of polyhydroxyalkanoate (PHA) biopolyesters. Fermentation 2018, 4, 30. [Google Scholar] [CrossRef]
- Lee, S.Y. Bacterial polyhydroxyalkanoates. Biotechnol. Bioeng. 1996, 49, 1–14. [Google Scholar] [CrossRef]
- Hirt, T.D.; Neuenschwander, P.; Suter, U.W. Telechelic diols from poly[®-3-hydroxybutyric acid] and poly([®-3-hydroxybutyric acid]-co-[®-3-hydroxyvaleric acid]. Macromol. Chem. Phys. 1996, 197, 1609–1614. [Google Scholar] [CrossRef]
- Andrade, A.P.; Witholt, B.; Hany, R.; Egli, T.; Li, Z. Preparation and characterization of enantiomerically pure telechelic diols from mcl-Poly[®-3-hydroxyalkanoates]. Macromolecules 2002, 35, 684–689. [Google Scholar] [CrossRef]
- Saruul, P.; Srienc, F.; Somers, D.A.; Samac, D.A. Production of a biodegradable plastic polymer, poly-β-hydroxybutyrate, in transgenic alfalfa. Crop Sci. 2002, 42, 919–927. [Google Scholar] [CrossRef]
- Menzel, G.; Harloff, H.-J.; Jung, C. Expression of bacterial poly(3-hydroxybutyrate) synthesis genes in hairy roots of sugar beet (Beta vulgaris L.). Appl. Microbiol. Biotechnol. 2003, 60, 571–576. [Google Scholar] [CrossRef]
- Wróbel, M.; Zebrowski, J.; Szopa, J. Polyhydroxybutyrate synthesis in transgenic flax. J. Biotechnol. 2004, 107, 41–54. [Google Scholar] [CrossRef]
- Parveez, G.K.A.; Bahariah, B.; Ayub, M.H.; Masani, M.Y.A.; Rasid, O.A.; Tarmizi, A.H.; Ishak, Z. Production of polyhydroxybutyrate in oil palm (Elaeis guineensis Jacq.) mediated, by microprojectile bombardment of PHB biosynthesis genes into embryogenic calli. Front. Plant Sci. 2015, 6, 598. [Google Scholar] [CrossRef]
- McQualter, R.B.; Somleva, M.N.; Gebbie, L.K.; Li, X.; Petrasovits, L.A.; Snell, K.D.; Nielsen, L.K.; Brumbley, S.M. Factors affecting polyhydroxybutyrate accumulation in mesophyll cells of sugarcane and switchgrass. BMC Biotechnol. 2014, 14, 83. [Google Scholar] [CrossRef]
- Malik, M.R.; Yang, W.; Patterson, N.; Tang, J.; Wellinghoff, R.L.; Preuss, M.L.; Burkitt, C.; Sharma, N.; Ji, Y.; Jez, J.M.; et al. Production of high levels of poly-3-hydroxybutyrate in plastids of Camelina sativa seeds. Plant Biotechnol. J. 2015, 13, 675–688. [Google Scholar] [CrossRef]
- Agostini, D.E.; Lando, J.B.; Shelton, J.R.J. Synthesis and characterization of poly-β-Hydroxybutyrate. I. Synthesis of crystalline DL-poly-β-hydroxybutyrate from DL-β-Butyrolactone. J. Polym. Sci. A-1 1971, 9, 2775–2787. [Google Scholar] [CrossRef]
- Shelton, J.R.; Agostini, D.E.; Lando, J.B. Synthesis and characterization of poly-β-hydroxybutyrate. II. Synthesis of D-poly-β-hydroxybutyrate and the mechanism of ring-opening polymerization of β-butyrolactone. J. Polym. Sci. A-1 1971, 9, 2789–2799. [Google Scholar] [CrossRef]
- Teranishi, K.; Iida, M.; Araki, T.; Yamashita, S.; Tani, H. Stereospecific polymerization of β-alkyl-β-propiolactone. Macromolecules 1974, 7, 421. [Google Scholar] [CrossRef]
- Iida, M.; Araki, T.; Teranishi, K.; Tani, H. Effect of substituents on stereospecific polymerization of /β-alkyl- and β-chloroalkyl-β-propiolactones. Macromolecules 1977, 10, 275–284. [Google Scholar] [CrossRef]
- Le Borgne, A.; Spassky, N. Stereoelective polymerization of β-butyrolactone. Polymer 1989, 30, 2312–2319. [Google Scholar] [CrossRef]
- Jedlinski, Z.; Kurcok, P.; Lenz, R.W. First facile synthesis of biomimetic poly-®-3-hydroxybutyrate via regioselective anionic polymerization of (S)-β-butyrolactone. Macromolecules 1998, 31, 6718–6720. [Google Scholar] [CrossRef]
- Kurcok, P.; Kowalczuk, M.; Hennek, K.; Jedlinski, Z. Anionic polymerization of beta-lactones initiated with alkali-metal alkoxides: Reinvestigation of the polymerization mechanism. Macromolecules 1992, 25, 2017–2020. [Google Scholar] [CrossRef]
- Zhang, Y.; Gross, R.A.; Lenz, R.W. Stereochemistry of the ring-opening polymerization of (S)-S-Butyrolactone. Macromolecules 1990, 23, 3206–3212. [Google Scholar] [CrossRef]
- Philip, S.; Keshavarz, T.; Roy, I. Polyhydroxyalkanoates: Biodegradable polymers with a range of applications. J. Chem. Technol. Biotechnol. 2007, 82, 233–247. [Google Scholar] [CrossRef]
- Khanna, S.; Srivastava, A. Recent advances in microbial polyhydroxyalkanoates. Process Biochem. 2005, 40, 607–619. [Google Scholar] [CrossRef]
- Domínguez-Díaz, M.; Meneses-Acosta, A.; Romo-Uribe, A.; Peña, C.; Segura, D.; Espin, G. Thermo-mechanical properties, microstructure and biocompatibility in poly-b-hydroxybutyrates (PHB) produced by OP and OPN strains of Azotobacter vinelandii. Eur. Polym. J. 2015, 63, 101–112. [Google Scholar] [CrossRef]
- Hong, S.-G.; Gau, T.-K.; Huang, S.-C. Enhancement of the crystallization and thermal stability of polyhydroxybutyrate by polymeric additives. J. Therm. Anal. Calorim. 2011, 103, 967–975. [Google Scholar] [CrossRef]
- Arcana, M.; Giani-Beaune, O.; Schue, F.; Amass, W.; Amass, A. Structure and morphology of poly(b-hydroxybutyrate) synthesized by ring-opening polymerization of racemic (R,S)-b-butyrolactone with distannoxane derivatives. Polym. Int. 2000, 49, 1348–1355. [Google Scholar] [CrossRef]
- Tang, X.; Westlie, A.H.; Watson, E.M.; Chen, E.Y.-X. Stereosequenced crystalline polyhydroxyalkanoates from diastereomeric monomer mixtures. Science 2019, 366, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Gay-Lussac, H.J.; Pelouze, H. Über die Milchsäure. Ann. Phys. 1833, 105, 108. [Google Scholar] [CrossRef]
- Carothers, W.H.; Dorough, G.L.; Van Natta, F.J. Studies of polymerization and ring formation. x. the reversible polymerization of cyclic esters. J. Am. Chem. Soc. 1932, 54, 761–772. [Google Scholar] [CrossRef]
- Garlotta, D. A literature review of poly(lactic acid). J. Polym. Environ. 2001, 9, 63–83. [Google Scholar] [CrossRef]
- Duda, A. ROP of Cyclic Esters. Mechanisms of Ionic and Coordination Processes. Polym. Sci. 2012, IV, 213–246. [Google Scholar] [CrossRef]
- Penczek, S.; Cypryk, M.; Duda, A.; Kubisa, P.; Slomkowski, S. Living ring-opening polymerizations of heterocyclic monomers. Prog. Polym. Sci. 2007, 32, 247–282. [Google Scholar] [CrossRef]
- Slomkowski, S.; Penczek, S.; Duda, A. Polylactides-an overview. Polym. Adv. Technol. 2014, 25, 436–447. [Google Scholar] [CrossRef]
- Pretula, J.; Slomkowski, S.; Penczek, S. Polylactides—Methods of synthesis and characterization. Adv. Drug Deliv. Rev. 2016, 107, 3–16. [Google Scholar] [CrossRef]
- Vert, M.; Chen, J.; Hellwich, K.-H.; Hodge, P.; Nakano, T.; Scholz, C.; Slomkowski, S.; Vohlidal, J. Nomenclature and terminology for linear lactic acid-based polymers (IUPAC Recommendations 2019). Pure Appl. Chem. 2020, 92, 193–211. [Google Scholar] [CrossRef]
- Matsutani, K.; Kimura, Y. PLA synthesis. Rrom the monomer to the polymer. In Poly(Lactic Acid) Science and Technology: Processing, Properties, Additives and Applications; Jiménez, A., Peltzer, M., Ruseckaite, R., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2015. [Google Scholar]
- Okano, K.; Hama, S.; Kihara, M.; Noda, H.; Tsutomu Tanaka, T.; Kondo, A. Production of optically pure D-lactic acid from brown rice using metabolically engineered Lactobacillus plantarum. Appl. Microbiol. Biotechnol. 2017, 101, 1869–1875. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, R.; Tadi, S.R.R.; Sivaprakasam, S.; Rajaram, S. Optimization of acid and enzymatic hydrolysis of kodo millet (Paspalum scrobiculatum) bran residue to obtain fermentable sugars for the production of optically pure D (−) lactic acid. Ind. Crops Prod. 2018, 111, 731–742. [Google Scholar] [CrossRef]
- Zaini, N.A.M.; Chatzifragkou, A.; Tverezovskiy, A.; Charalampopoulos, D. Purification and polymerisation of microbial D-lactic acid from DDGS hydrolysates fermentation. Biochem. Eng. J. 2019, 150, 107265. [Google Scholar] [CrossRef]
- Din, N.A.S.; Lim, S.J.; Maskat, M.Y.; Zaini, N.A.M. Microbial D-lactic acid production, In Situ separation and recovery from mature and young coconut husk hydrolysate fermentation broth. Biochem. Eng. J. 2022, 188, 108680. [Google Scholar] [CrossRef]
- Abedi, E.; Hashemi, S.M.B. Lactic acid production—Producing microorganisms and substrates sources-state of art. Heliyon 2020, 6, e04974. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, Y.; Shang, N.; Li, P. Microbial fermentation processes of lactic acid: Challenges, solutions, and future prospects. Foods 2023, 12, 2311. [Google Scholar] [CrossRef]
- Yankov, D. Fermentative lactic acid production from lignocellulosic feedstocks: From source to purified product. Front. Chem. 2022, 10, 823005. [Google Scholar] [CrossRef]
- Ajioka, M.; Enomoto, K.; Suzuki, K.; Yamaguchi, A. Basic properties of polylactic acid produced by the direct condensation polymerization of lactic-acid. Bull. Chem. Soc. Jpn. 1995, 68, 2125–2131. [Google Scholar] [CrossRef]
- Moon, S.-I.; Taniguchi, I.; Miyamoto, M.; Kimura, Y.; Lee, C.-W. Synthesis and properties of high-molecular-weight poly(L-lactic acid) by melt/solid polycondensation under different reaction conditions. High Perform. Polym. 2001, 13, S189–S196. [Google Scholar] [CrossRef]
- Moon, S.-I.; Lee, C.-W.; Taniguchi, I.; Miyamoto, M.; Kimura, Y. Melt/solid polycondensation of l-lactic acid: An alternative route to poly(l-lactic acid) with high molecular weight. Polymer 2001, 42, 5059–5062. [Google Scholar] [CrossRef]
- Kulkarni, R.K.; Pani, K.; Neuman, C.; Leonard, F. Lactic acid for surgical implants. Arch. Surg. 1966, 93, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Kohn, F.E.; van Den Berg, J.W.A.; van de Ridder, G.; Feijen, J. The ring-opening polymerization of D,L-lactide in the melt initiated with tetraphenyltin. J. Appl. Polym. Sci. 1984, 29, 4265–4277. [Google Scholar] [CrossRef]
- Lin, Z.; Guo, X.; He, Z.; Liang, X.; Wang, M.; Jin, G. Thermal degradation kinetics study of molten polylactide based on Raman spectroscopy. Polym. Eng. Sci. 2021, 61, 201–210. [Google Scholar] [CrossRef]
- Duda, A.; Penczek, S. Polymerization of ε-caprolactone initiated by aluminum isopropoxide trimer and/or tetramer. Macromolecules 1995, 28, 5981–5992. [Google Scholar] [CrossRef]
- Duda, A.; Penczek, S. On the difference of reactivities of various aggregated forms of aluminium triisopropoxide in initiating ring-opening polymerizations. Macromol. Rapid Commun. 1995, 16, 67–76. [Google Scholar] [CrossRef]
- Kowalski, A.; Libiszowski, J.; Majerska, K.; Duda, A.; Penczek, S. Kinetics and mechanism of ε-caprolactone and L,L-lactide polymerization coinitiated with zinc octoate or aluminum acetylacetonate: The next proofs for the general alkoxide mechanism and synthetic applications. Polymer 2007, 48, 3952–3960. [Google Scholar] [CrossRef]
- Gadomska-Gajadhur, A.; Ruśkowski, P. Biocompatible catalysts for lactide polymerization—Catalyst activity, racemization effect, and optimization of the polymerization based on design of experiments. Org. Process Res. Dev. 2020, 24, 1435–1442. [Google Scholar] [CrossRef]
- Kowalski, A.; Duda, A.; Penczek, S. Kinetics and mechanism of cyclic esters polymerization initiated with Tin(II) octoate. 3. Polymerization of L,L-dilactide. Macromolecules 2000, 33, 7359–7370. [Google Scholar] [CrossRef]
- Majerska, K.; Duda, A.; Penczek, S. Kinetics and mechanism of cyclic esters polymerization initiated with tin(II) octoate, 4—Influence of proton trapping agents on the kinetics of epsilon-caprolactone and L,L-dilactide polymerization. Macromol. Rapid Commun. 2000, 21, 1327–1332. [Google Scholar] [CrossRef]
- Duda, A.; Penczek, S. Thermodynamics of L-Lactide polymerization. equilibrium monomer concentration. Macromolecules 1990, 23, 1636–1639. [Google Scholar] [CrossRef]
- Shinno, K.; Miyamoto, M.; Kimura, Y.; Hirai, Y.; Yoshitome, H. Solid-state postpolymerization of l-lactide promoted by crystallization of product polymer: An effective method for reduction of remaining monomer. Macromolecules 1997, 30, 6438–6444. [Google Scholar] [CrossRef]
- Mosnacek, J.; Duda, A.; Libiszowski, J.; Penczek, S. Copolymerization of LL-lactide at its living polymer-monomer equilibrium with ε-caprolactone as comonomer. Macromolecules 2005, 38, 2027–2029. [Google Scholar] [CrossRef]
- Degee, P.; Dubois, P.; Jerome, R. Bulk polymerization of lactides initiated by aluminium isopropoxide.3. Thermal stability and viscoelastic properties. Macromol. Chem. Phys. 1997, 198, 1973–1984. [Google Scholar] [CrossRef]
- Kowalski, A.; Libiszowski, J.; Duda, A.; Penczek, S. Polymerization of L,L-dilactide initiated by Tin(II) butoxide. Macromolecules 2000, 33, 1964–1971. [Google Scholar] [CrossRef]
- Bourissou, D.; Moebs-Sanchez, S.; Martín-Vaca, B. Recent advances in the controlled preparation of poly(α-hydroxy acids): Metal-free catalysts and new monomers. C. R. Chimie 2007, 10, 775–794. [Google Scholar] [CrossRef]
- Kamber, N.E.; Jeong, W.; Waymouth, R.M.; Pratt, R.C.; Lohmeijer, B.G.G.; Hedrick, J.L. Organocatalytic ring-opening polymerization. Chem. Rev. 2007, 107, 5813–5840. [Google Scholar] [CrossRef]
- Nederberg, F.; Connor, E.F.; Möller, M.; Glauser, T.; Hedrick, J.L. New paradigms for organic catalysts: The first organocatalytic living polymerization. Angew. Chem. Int. Ed. 2001, 40, 2712–2715. [Google Scholar] [CrossRef]
- Zhang, X.; Waymouth, R.M. Zwitterionic Ring opening polymerization with isothioureas. ACS Macro Lett. 2014, 3, 1024–1028. [Google Scholar] [CrossRef]
- Alba, A.; Thillaye du Boullay, O.; Martin-Vaca, B.; Bourissou, D. Direct ring-opening of lactide with amines: Application to the organo-catalyzed preparation of amide end-capped PLA and to the removal of residual lactide from PLA samples. Polym. Chem. 2015, 6, 989–997. [Google Scholar] [CrossRef]
- Lee, G.S.; Moon, B.R.; Jeong, H.; Shin, J.; Kim, J.G. Mechanochemical synthesis of poly(lactic acid) block copolymers: Overcoming the miscibility of the macroinitiator, monomer and catalyst undersolvent-free conditions. Polym. Chem. 2019, 10, 539–545. [Google Scholar] [CrossRef]
- Lohmeijer, R.G.G.; Pratt, R.C.; Leibfarth, F.; Logan, J.W.; Long, D.A.; Dove, A.P.; Nederberg, F.; Choi, J.; Wade, C.; Waymouth, R.M.; et al. Guanidine and amidine organocatalysts for ring-opening polymerization of cyclic esters. Macromolecules 2006, 39, 8574–8583. [Google Scholar] [CrossRef]
- Myers, M.; Connor, E.F.; Glausser, T.; Moeck, A.; Nyce, G.W.; Hedrick, J.L.J. Phosphines: Nucleophilic organic catalysts for the controlled ring-opening polymerization of lactides. J. Polym. Sci. Part A Polym. Chem. 2002, 40, 844. [Google Scholar] [CrossRef]
- Connor, E.F.; Nyce, G.W.; Myers, M.; Moeck, A.; Hedrick, J.L. First example of N-heterocyclic carbenes as catalysts for living polymerization:: Organocatalytic ring-opening polymerization of cyclic esters. J. Am. Chem. Soc. 2002, 124, 914–915. [Google Scholar] [CrossRef]
- Coulembier, O.; Dove, A.P.; Pratt, R.C.; Sentman, A.C.; Culkin, D.A.; Mespouille, L.; Dubois, P.; Waymouth, R.M.; Hedrick, J.L. Latent, thermally activated organic catalysts for the on-demand living polymerization of lactide. Angew. Chem. Int. Ed. 2005, 44, 4964–4968. [Google Scholar] [CrossRef]
- Dove, A.P.; Pratt, R.C.; Lohmeijer, B.G.G.; Waymouth, R.M.; Hedrick, J.L. Thiourea-based bifunctional organocatalysis: Supramolecular recognition for living polymerization. J. Am. Chem. Soc. 2005, 127, 13798–13799. [Google Scholar] [CrossRef] [PubMed]
- Basko, M.; Kubisa, P. Cationic copolymerization of ε-caprolactone and L,L-lactide by an activated monomer mechanism. J. Polym. Sci. A Polym. Chem. 2006, 44, 7071–7081. [Google Scholar] [CrossRef]
- Basko, M.; Kubisa, P. Mechanism of propagation in the cationic polymerization of L,L-lactide. J. Polym. Sci. A Polym. Chem. 2008, 46, 7919–7923. [Google Scholar] [CrossRef]
- Basko, M.; Kubisa, P. Cationic polymerization of L,L-lactide. J. Polym. Sci. A Polym. Chem. 2010, 48, 2650–2658. [Google Scholar] [CrossRef]
- Basko, M. Activated monomer mechanism in the cationic polymerization of L,L-lactide. Pure Appl. Chem. 2012, 84, 2081–2088. [Google Scholar] [CrossRef]
- Lewinski, P.; Kaluzynski, K.; Pretula, J.; Mielniczak, G.; Penczek, S. Catalysis in polymerization of cyclic esters. Catalyst and initiator in one molecule. Polymerization of lactide. J. Catal. 2022, 405, 249–264. [Google Scholar] [CrossRef]
- Kaluzynski, K.; Pretula, J.; Lewinski, P.; Kazmierski, S.; Penczek, S. Catalysis in polymerization of cyclic esters. Catalyst and initiator in one molecule. Polymerization of ε-caprolactone, J. Catal. 2020, 392, 97–107. [Google Scholar] [CrossRef]
- Spassky, N.; Wisniewski, M.; Pluta, C.; LeBorgne, A. Highly stereoelective polymerization of rac-(D,L)-lactide with a chiral Schiff’s base/aluminium alkoxide initiator. Macromol. Chem. Phys. 1996, 197, 2627–2637. [Google Scholar] [CrossRef]
- Nomura, N.; Ishii, R.; Akakura, M.; Aoi, K. Stereoselective ring-opening polymerization of racemic lactide using aluminum-achiral ligand complexes: Exploration of a chain-end control mechanism. J. Am. Chem. Soc. 2002, 124, 5938–5939. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.H.; Yang, Y.K.; Pang, X.; Hu, J.L.; Chen, X.S.; Hu, N.H.; Jing, X.B. Controlled and stereospecific polymerization of rac-lactide with a single-site ethyl aluminum and alcohol initiating system. J. Appl. Polym. Sci. 2005, 98, 102–108. [Google Scholar] [CrossRef]
- Nomura, N.; Ishii, R.; Yamamoto, Y.; Kondo, T. Stereoselective ring-opening polymerization of a racemic lactide by using achiral salen- and homosalen-aluminum complexes. Chem.-A Eur. J. 2007, 13, 4433–4451. [Google Scholar] [CrossRef]
- Radano, C.P.; Baker, G.L.; Smith, M.R. Stereoselective polymerization of a racemic monomer with a racemic catalyst: Direct preparation of the polylactic acid stereocomplex from racemic lactide. J. Am. Chem. Soc. 2000, 122, 1552–1553. [Google Scholar] [CrossRef]
- Ovitt, T.M.; Coates, G.W. Stereoselective ring-opening polymerization of rac-lactide with a single-site, racemic aluminum alkoxide catalyst: Synthesis of stereoblock poly(lactic acid). J. Polym. Sci. Part A Polym. Chem. 2000, 38, 4686–4692. [Google Scholar] [CrossRef]
- Ovitt, T.M.; Coates, G.W. Stereochemistry of lactide polymerization with chiral catalysts: New opportunities for stereocontrol using polymer exchange mechanisms. J. Am. Chem. Soc. 2002, 124, 1316–1326. [Google Scholar] [CrossRef]
- Zhong, Z.Y.; Dijkstra, P.J.; Feijen, J. [(salen)Al]-mediated, controlled and stereoselective ring-opening polymerization of lactide in solution and without solvent: Synthesis of highly isotactic polylactide stereocopolymers from racemic D,L-lactide. Angew. Chem. Int. Ed. 2002, 41, 4510–4513. [Google Scholar] [CrossRef]
- Dijkstra, P.J.; Du, H. Feijen, Single site catalysts for stereoselective ring-opening polymerization of lactides. J. Polym. Chem. 2011, 2, 520–527. [Google Scholar] [CrossRef]
- Tsuji, H. Poly(lactide) stereocomplexes: Formation, structure, properties, degradation, and applications. Macromol. Biosci. 2005, 5, 569–597. [Google Scholar] [CrossRef]
- Hador, R.; Botta, A.; Venditto, V.; Lipstman, S.; Goldberg, I.; Kol, M. The dual-stereocontrol mechanism: Heteroselective polymerization of rac-lactide and syndioselective polymerization of meso-lactide by chiral aluminum salan catalysts. Angew. Chem. Int. Ed. 2019, 58, 14679–14685. [Google Scholar] [CrossRef]
- Rosen, T.; Rajpurohit, J.; Lipstman, S.; Venditto, V.; Kol, M. Isoselective polymerization of rac-lactide by highly active sequential {ONNN} magnesium complexes. Chem. Eur. J. 2020, 26, 17183–17189. [Google Scholar] [CrossRef] [PubMed]
- Roymuhury, S.K.; Mandal, M.; Chakraborty, D.; Ramkumar, V. Homoleptic titanium and zirconium complexes exhibiting unusual Oiminol–metal coordination: Application in stereoselective ring-opening polymerization of lactide. Polym. Chem. 2021, 12, 3953–3967. [Google Scholar] [CrossRef]
- Majerska, K.; Duda, A. Stereocontrolled polymerization of racemic lactide with chiral initiator: Combining stereoelection and chiral ligand-exchange mechanism. J. Am. Chem. Soc. 2004, 126, 1026–1027. [Google Scholar] [CrossRef]
- Sanko, V.; Sahin, I.; Sezer, U.A.; Sezer, S. A versatile method for the synthesis of poly(glycolic acid): High solubility and tunable molecular weights. Polym. J. 2019, 51, 637–647. [Google Scholar] [CrossRef]
- Hurrell, S.; Cameron, R.E. Polyglycolide: Degradation and drug release. Part I: Changes in morphology during degradation. J. Mater. Sci. Mater. Med. 2001, 12, 811–816. [Google Scholar] [CrossRef]
- Hurrell, S.; Cameron, R.E. Polyglycolide: Degradation and drug release. Part II: Drug release. Mater. Sci. Mater. Med. 2001, 12, 817–820. [Google Scholar] [CrossRef]
- Goh, Y.-F.; Shakir, I.; Hussain, R. Electrospun fibers for tissue engineering, drug delivery, and wound dressing. J. Mater. Sci. 2013, 48, 3027–3054. [Google Scholar] [CrossRef]
- Contreras-Cáceres, R.; Cabeza, L.; Perazzoli, G.; Díaz, A.; López-Romero, J.M.; Melguizo, C.; Prados, P. Electrospun nanofibers: Recent applications in drug delivery and cancer therapy. Nanomaterials 2019, 9, 656. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, Y.; Hend, E.; Abdelhakim, H. Drug delivery applications of coaxial electrospun nanofibres in cancer therapy. Molecules 2022, 27, 1803. [Google Scholar] [CrossRef] [PubMed]
- Snoddy, B.; Jayasuriya, A.C. The Use of nanomaterials to treat bone infections. Mater. Sci. Eng. C-Mater. Biol. Appl. 2016, 67, 822–833. [Google Scholar] [CrossRef]
- Reddy, P.G.; Domb, A.J. Formation of micro/nanoparticles and microspheres from polyesters by dispersion ring-opening polymerization. Polym. Adv. Technol. 2021, 32, 3835–3856. [Google Scholar] [CrossRef]
- Procopio, A.; Lagreca, E.; Jamaledin, R.; La Manna, S.; Corrado, B.; Di Natale, C.; Onesto, V. Recent fabrication methods to produce polymer-based drug delivery matrices (experimental and in silico approaches). Pharmaceutics 2022, 14, 872. [Google Scholar] [CrossRef]
- Rusa, C.C.; Tonelli, A.E. Polymer/polymer inclusion compounds as a novel approach to obtaining a PLLA/PCL intimately compatible blend. Macromolecules 2000, 33, 5321–5324. [Google Scholar] [CrossRef]
- Douglas, P.; Andrews, G.; Jones, D.; Walker, G. Analysis of in vitro drug dissolution from PCL melt extrusion. Chem. Eng. J. 2010, 164, 359–370. [Google Scholar] [CrossRef]
- Douglas, P.; Albadarin, A.B.; Al-Muhtaseb, A.H.; Mangwandi, C.; Walker, G.M. Thermo-mechanical propertiesofpolyε-caprolactone/poly L-lactic acid blends: Additionofnalidixicacid and polyethyleneglycoladditives. J. Mechan. Behav. Biomed. Maater. 2015, 45, 154–165. [Google Scholar] [CrossRef]
- Woodruff, M.A.; Hutmacher, D.W. The return of a forgott in the 21st century. Prog. Polym. Sci. 2010, 35, 1217–1256. [Google Scholar] [CrossRef]
- Lewinski, P.; Pretula, P.; Kaluzynski, K.; Kazmierski, S.; Penczek, S. ε-Caprolactone: Activated monomer polymerization; controversy over the mechanism of polymerization catalyzed by phosphorus acids (diarylhydrogen phosphates). Do acids also act as initiators? J. Catal. 2019, 371, 305–312. [Google Scholar] [CrossRef]
- Ito, K.; Hashizuka, Y.; Yamashita, Y. Equilibrium cyclic oligomer formation in the anionic polymerization of ε-caprolactone. Macromolecules 1977, 10, 821–824. [Google Scholar] [CrossRef]
- Ito, K.; Yamashita, Y. Propagation and depropagation rates in the anionic polymerization of ε-caprolactone cyclic oligomers. Macromolecules 1978, 11, 68–72. [Google Scholar] [CrossRef]
- Sosnowski, S.; Slomkowski, S.; Penczek, S.; Reibel, L. Kinetic-caprolactone polymerization and formation of cyclic oligomers. Makromol. Chem. 1983, 1984, 2159–2179. [Google Scholar] [CrossRef]
- Hofman, A.; Slomkowski, S.; Penczek, S. Polymerization of ε-caprolactones with kinetic suppression of macrocycles. Makromol. Chem. Rapid Commun. 1987, 8, 387–391. [Google Scholar] [CrossRef]
- Penczek, S.; Duda, A.; Slomkowski, S. The reactivity-selectivity principle in polymerization. The case of polymerization of ε-caprolactone. Makromol. Chem. Macromol. Symp. 1992, 54–55, 31–40. [Google Scholar] [CrossRef]
- Biela, T.; Kowalski, A.; Libiszowski, J.; Duda, A.; Penczek, S. Progress in polymerization of cyclic esters: Mechanisms and synthetic applications. Macromol. Symp. 2006, 240, 47–55. [Google Scholar] [CrossRef]
- van der Meulen, I.; Li, Y.; Deumens, R.; Joosten, E.A.J.; Koning, C.E.; Heise, A. Copolymers from unsaturated macrolactones: Toward the design of cross-linked biodegradable polyesters. Biomacromolecules 2011, 12, 837–843. [Google Scholar] [CrossRef]
- Claudino, M.; van der Meulen, I.; Trey, S.; Jonsson, M.; Heise, A.; Johansson, M. Photoinduced thiol-ene crosslinking of globalide/ε-caprolactone copolymers: Curing performance and resulting thermoset properties. J. Polym. Sci. A Polym. Chem. 2012, 50, 16–24. [Google Scholar] [CrossRef]
- Wilson, J.A.; Ates, Z.; Pflughaupt, R.L.; Dove, A.P.; Heise, A. Polymers from macrolactones: From pheromones to functional materials. Prog. Polym. Sci. 2019, 91, 29–50. [Google Scholar] [CrossRef]
- Lee, J.; Jung, S.G.; Cheon-Seok Park, C.S.; Kim, H.-Y.; Batt, C.A.; Kim, Y.-R. Tumor-specific hybrid polyhydroxybutyrate nanoparticle: Surface modification of nanoparticle by enzymatically synthesized functional block copolymer. Bioorg. Med. Chem. Lett. 2011, 21, 2941–2944. [Google Scholar] [CrossRef]
- Kajjari, P.B.; Manjeshwar, L.S.; Aminabhavi, T.M. Novel blend microspheres of poly(3-hydroxybutyrate) and pluronic f68/127 for controlled release of 6-mercaptopurine. J. Appl. Polym. Sci. 2014, 131, 40196. [Google Scholar] [CrossRef]
- Lee, J.; Saparbayeva, A.; Hlaing, S.P.; Kwak, D.; Kim, H.; Kim, J.; Lee, E.H.; Yoo, J.-W. Cupriavidus necator-produced polyhydroxybutyrate/EudragitFS hybrid nanoparticles mitigates ulcerative colitis via colon-targeted delivery of cyclosporine A. Pharmaceutics 2022, 14, 2811. [Google Scholar] [CrossRef]
- Pan, C.-T.; Hwang, Y.-M.; Lin, Y.-M.; Zeng, S.-W.; Wang, S.-Y.; Kuo, S.-W.; Ju, S.-P.; Liang, S.-S.; Liu, Z.-H.; Yen, C.-K. Development of polycaprolactone microspheres with controllable and uniform particle size by uniform design experiment in emulsion progress. Sens. Mater. 2019, 31, 311–318. [Google Scholar] [CrossRef]
- Ponjavić, M.; Nikolić, M.S.; Jevtić, S.; Jeremić, S.; Dokić, L.; Donlagić, J. Star-shaped poly(ε-caprolactones) with well-defined architecture as potential drug carriers. J. Serb. Chem. Soc. 2022, 87, 1075–1090. [Google Scholar] [CrossRef]
- Mozafari, M. Synthesis and characterisation of poly(lactide-co-glycolide) nanospheres using vitamin E emulsifier prepared through one-step oil-in-water emulsion and solvent evaporation techniques. IET Nanobiotechnol. 2014, 8, 257–262. [Google Scholar] [CrossRef]
- Harguindey, A.; Domaille, D.W.; Fairbanks, B.D.; Wagner, J.; Bowman, C.N.; Cha, J.N. Synthesis and assembly of click-nucleic-acid-containin PEG–PLGA nanoparticles for DNA delivery. Adv. Mater. 2017, 29, 1700743. [Google Scholar] [CrossRef]
- Takeuchi, I.; Kimura, Y.; Makino, K. Effect of the conformation of poly(L-lactide-co-glycolide) molecules in organic solvents onnanoparticle. size. J. Oleo Sci. 2020, 69, 1125–1132. [Google Scholar] [CrossRef]
- Han, C.-S.; Kang, J.-H.; Kim, Y.-J.; Kim, D.-W.; Park, C.W. Inhalable nano-dimpled microspheres containing Budesonide-PLGA for improved aerodynamic performance. Int. J. Nanomed. 2022, 17, 3405–3419. [Google Scholar] [CrossRef]
- Slomkowski, S. Polyester nano- and microparticles by polymerization and self-assembly of macromolecules. In Nanoparticles for Pharmaceutical; Domb, A.J., Tabata, Y., Kumar, M.N.V.R., Farber, S., Eds.; American Scientific Publishers: California, CA, USA, 2007. [Google Scholar]
- Heslinga, M.J.; Mastria, E.M.; Eniola-Adefeso, O. Fabrication of biodegradable spheroidal microparticles for drug delivery applications. J. Control. Release 2009, 138, 235–242. [Google Scholar] [CrossRef]
- Cohen, H.; Levy, R.J.; Gao, J.; Fishbein, I.; Kousaev, V.; Sosnowski, S.; Slomkowski, S.; Golomb, G. Sustained delivery and expression of DNA encapsulated in polymeric nanoparticles. Gene Ther. 2000, 7, 1896–1905. [Google Scholar] [CrossRef]
- You, G.; Kim, Y.; Lee, J.H.; Song, J.; Mok, H. Exosome-modified PLGA microspheres for improve internalization into dendritic cells and macrophages. Biotechnol. Bioproc. Eng. 2020, 25, 521–527. [Google Scholar] [CrossRef]
- Duan, J.; Liu, C.; Liang, X.; Li, X.; Chen, Y.; Chen, Z.; Wang, X.; Kong, D.; Li, Y.; Yang, J. Protein delivery nanosystem of six-arm copolymer poly(ε-caprolactone)–poly(ethylene glycol) for long-term sustained release. Int. J. Nanomed. 2018, 13, 2743–2754. [Google Scholar] [CrossRef]
- Gökberk, B.D.; Erdinç, N. Design, Optimization, and characterization of lysozyme-loaded poly(ε-caprolactone) microparticles for pulmonary delivery. J. Pharm. Innov. 2023, 18, 325–338. [Google Scholar] [CrossRef]
- Chen, L.; Mei, L.; Feng, D.; Huang, D.; Tong, X.; Pan, X.; Zhu, C.; Wu, C. Anhydrous reverse micelle lecithin nanoparticles/PLGA compositemicrospheres for long-term protein delivery with reduced initial burst. Colloids Surf. B Biointerfaces 2018, 163, 146–154. [Google Scholar] [CrossRef]
- Fessi, H.; Puisieux, F.; Devissaguet, J.P.; Ammoury, N.; Benita, S. Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int. J. Pharm. 1989, 55, R1–R4. [Google Scholar] [CrossRef]
- Rivas, C.J.M.; Tarhini, M.; Badri, W.; Miladi, K.; Greige-Gerges, H.; Nazari, Q.A.; Rodríguez, S.A.G.; Román, R.A.; Fessi, H.; Elaissari, A. Nanoprecipitation process: From encapsulation to drug delivery. Int. J. Pharm. 2017, 532, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Prabhuraj, R.S.; Bomb, K.; Srivastava, R.; Bandyopadhyaya, R. Dual drug delivery of curcumin and niclosamide using PLGA nanoparticles for improved therapeutic effect on breast cancer cells. J. Polym. Res. 2020, 27, 133. [Google Scholar] [CrossRef]
- Alsulays, B.B.; Anwer, M.K.; Aldawsari, M.F.; Aodah, A.; Adam, E.; Alshehri, S.; Abdel-Kader, M.S. Preparation and evaluation of a stable and sustained releaseof lansoprazole-loaded poly(d,l-lactide-co-glycolide) polymeric nanoparticles. J. Polym. Eng. 2019, 39, 822–829. [Google Scholar] [CrossRef]
- Xu, J.; Chen, Y.; Jiang, X.; Gui, Z.; Zhang, L. Development of hydrophilic drug encapsulation and controlled release using a modified nanoprecipitation method. Processes 2019, 7, 331. [Google Scholar] [CrossRef]
- Marante, T.; Viegas, C.; Duarte, I.; Macedo, A.S.; Fonte, P. An overview on spray-drying of protein-loaded polymeric nanoparticles for dry powder inhalation. Pharmaceutics 2020, 12, 1032. [Google Scholar] [CrossRef]
- Heidari, M.; Golenser, J.; Greiner, A. Meeting the needs of a potent carrier for malaria treatment: Encapsulation of Artemisone in poly(lactide-coglycolide). Part. Part. Syst. Charact. 2022, 39, 2100152. [Google Scholar] [CrossRef]
- Nosrati, Z.; Li, N.; Michaud, F.; Ranamukhaarachchi, S.; Karagiozov, S.; Soulez, G.; Martel, S.; Saatchi, K.; Häfeli, U.O. Development of a coflowing device for the size-controlled preparation of magnetic-polymeric microspheres as embolization agents in magnetic resonance navigation technology. ACS Biomater. Sci. Eng. 2018, 4, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.; Tian, M.; Chen, Y.; You, P.; Song, X.; Xu, B.; Duan, C.; Jin, D. Preparation of PLGA microspheres loaded with niclosamide via microfluidic technology and their inhibition of Caco-2 cell activity in vitro. Front. Chem. 2023, 11, 1249293. [Google Scholar] [CrossRef] [PubMed]
- Lababidi, N.; Montefusco-Pereira, C.V.; Carvalho-Wodarz, C.S.; Lehr, C.-M.; Schneider, M. Spray-dried multidrug particles for pulmonary co-delivery of antibiotics with N-acetylcysteine and curcumin-loaded PLGA-nanoparticles. Eur. J. Pharm. Biopharm. 2020, 157, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Sosnowski, S.; Gadzinowski, M.; Slomkowski, S.; Penczek, S. Synthesis of bioerodible poly(ε-caprolactone) latexes and poly(D,L-lactide) microspheres by ring-opening polymerization. J. Bioact. Compat. Polym. 1994, 9, 345–366. [Google Scholar] [CrossRef]
- Sosnowski, S.; Gadzinowski, M.; Slomkowski, S. Poly(L,L-lactide) microspheres by ring-opening polymerization. Macromolecules 1996, 29, 4556–4564. [Google Scholar] [CrossRef]
- Slomkowski, S.; Sosnowski, S.; Gadzinowski, M. Polyesters from lactides and caprolactone. Dispersion polymerization versus polymerization in solution. Polym. Degrad. Stab. 1998, 59, 153–160. [Google Scholar] [CrossRef]
- Gadzinowski, M.; Sosnowski, S.; Slomkowski, S. Kinetics of the dispersion ring-opening polymerization of ε-caprolactone initiated with diethylaluminum ethoxide. Macromolecules 1996, 29, 6404–6407. [Google Scholar] [CrossRef]
- Slomkowski, S.; Gadzinowski, M.; Sosnowski, S. Mechanism of particle formation and kinetics of the dispersion polymerization of cyclic esters. Macromol. Symp. 1998, 132, 451–462. [Google Scholar] [CrossRef]
- Slomkowski, S.; Sosnowski, S.; Gadzinowski, M.; Pichot, C.; Eaissari, A. Tailored synthesis of polyesters by dispersion ring opening polymerization of ε-caprolactone and lactides. Macromol. Symp. 2000, 150, 259–268. [Google Scholar] [CrossRef]
- Muranaka, M.; Kitamura, Y.; Yoshizawa, H. Preparation of biodegradablemicrospheres by anionic dispersion polymerization with PLA copolymeric dispersion stabilizer. Colloid Polym. Sci. 2007, 285, 1441–1448. [Google Scholar] [CrossRef]
- Muranaka, M.; Ono, T. Preparation of monodisperse polylactide microspheresby dispersion polymerization using a polymeric stabilizer with hydroxy groups. Macromol. Rapid Commun. 2009, 30, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Muranaka, M.; Yoshizawa, H.; Ono, T. Design of polylactide-grafted copolymeric stabilizer for dispersion polymerization of D,L-lactide. Colloid Polym. Sci. 2009, 287, 525–532. [Google Scholar] [CrossRef]
- Gadzinowski, M.; Slomkowski, S.; Elaïssari, A.; Pichot, C. Phase transfer and characterization of poly(ε-caprolactone) and poly(L-lactide) microspheres. J. Biomater. Sci. Polym. Ed. 2000, 11, 459–480. [Google Scholar] [CrossRef] [PubMed]
- Slomkowski, S. Preparation of biodegradable particles by polymerization processes. In Colloidal Biomolecules, Biomaterials and Biomedical Applications; Elaissari, A., Ed.; Surfactant Science Series; Marcel Deker: New York, NY, USA, 2003; Volume 16. [Google Scholar]
- Slomkowski, S.; Sosnowski, S.; Gadzinowski, M.; Pichot, C.; Elaissari, A. Direct synthesis of polyester microspheres, potential carriers of bioactive compounds. ACS Symp. Ser. 1998, 709, 143–153. [Google Scholar] [CrossRef]
- Gizurarson, S. Anatomical and histological factors affecting intranasal drug and vaccine delivery. Curr. Drug Deliv. 2012, 9, 566–582. [Google Scholar] [CrossRef]
- Taylor, J.P.; Hardy, J.; Fischbeck, K.H. Toxic proteins in neurodegenerative disease. Science 2002, 296, 1991–1995. [Google Scholar] [CrossRef]
- Ahmad, N. Rasagiline-encapsulated chitosan-coated PLGA nanoparticles targeted to the brain in the treatment of Parkinson’s disease. J. Liquid Chrom. Related Technol. 2017, 40, 677–690. [Google Scholar] [CrossRef]
- Katila, N.; Duwa, R.; Bhurtel, S.; Khanal, S.; Maharjan, S.; Jee-Heon Jeong, J.-H.; Lee, S.; Choi, D.-Y.; Yook, S. Enhancement of blood–brain barrier penetration and the neuroprotective effect of resveratrol. J. Control. Release 2022, 346, 1–19. [Google Scholar] [CrossRef]
- Pahuja, R.; Seth, K.; Shukla, A.; Shukla, R.K.; Bhatnagar, P.; Chauhan, L.K.S.; Saxena, P.N.; Arun, J.; Chaudhari, B.P.; Patel, D.K.; et al. Trans-blood brain barrier delivery of dopamine-loaded nanoparticles reverses functional deficits in Parkinsonian rats. ACS Nano 2015, 9, 4850–4871. [Google Scholar] [CrossRef]
- Tang, S.; Wang, A.; Yan, X.; Chu, L.; Yang, X.; Song, Y.; Sun, K.; Yu, X.; Liu, R.; Wu, Z.; et al. Brain-targeted intranasal delivery of dopamine with borneol and lactoferrin co-modified nanoparticles for treating Parkinson’s disease. Drug Deliv. 2019, 26, 700–707. [Google Scholar] [CrossRef]
- Monge-Fuentes, V.; Mayer, A.B.; Lima, M.R.; Geraldes, L.R.; Zanotto, L.N.; Moreir, K.G.; Martins, O.P.; Piva, H.L.; Felipe, M.S.S.; Amaral, A.C.; et al. Dopamine-loaded nanoparticle systems circumvent the blood-brain barrier restoring motor function in mouse model. Sci. Rep. 2021, 11, 15185. [Google Scholar] [CrossRef]
- Sánchez-López, E.; Ettcheto, M.; Egea, M.A.; Espina, M.; Cano, A.; Calpena, A.C.; Camins, A.; Carmona, N.; Silva, A.M.; Souto, E.B.; et al. Memantine loaded PLGA PEGylated nanoparticles for Alzheimer’s disease: In vitro and in vivo characterization. J. Nanobiotechnol. 2018, 16, 32. [Google Scholar] [CrossRef]
- Wu, Q.; Karthivashan, G.; Nakhaei-Nejad, M.; Anand, B.G.; Giuliani, F.; Kar, S. Native PLGA nanoparticles regulate APP metabolism and protect neurons against β-amyloid toxicity: Potential significance in Alzheimer’sdisease pathology. Int. J. Biol. Macromol. 2022, 219, 180–1196. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Pan, B.; Xu, H.; Zhao, Z.; Shen, J.; Lu, J.; Yu, R.; Liu, H. Co-delivery of GOLPH3 siRNA and gefitinib by cationic lipid-PLGA nanoparticles improves EGFR-targeted therapy for glioma. J. Mol. Med. 2019, 97, 1575–1588. [Google Scholar] [CrossRef]
- Cui, Y.; Sun, J.; Hao, W.; Chen, M.; Wang, Y.; Xu, F.; Gao, C. Dual-target peptide-modified erythrocyte membrane-enveloped PLGA nanoparticles for the treatment of glioma. Front. Oncol. 2020, 10, 563938. [Google Scholar] [CrossRef]
- Caban-Toktas, S.; Sahin, A.; Lulei, S.; Esendagli, G.; Vural, I.; Oguz, K.K.; Soylemezoglu, F.; Mut, M.; Dalkara, T.; Mansoor Khan, M.; et al. Combination of paclitaxel and R-flurbiprofen loaded PLGA nanoparticles suppresses glioblastoma growth on systemic administration. Int. J. Pharm. 2020, 578, 119076. [Google Scholar] [CrossRef]
- Acharya, S.; Praveena, P.; Raja Guru, B.R. In vitro studies of prednisolone loaded PLGA nanoparticles-surface functionalized with folic acid on glioma and macrophage cell lines. Pharm. Sci. 2021, 27, 407–417. [Google Scholar] [CrossRef]
- Ma, J.; Dai, L.; Yu, J.; Cao, H.; Bao, Y.; Hu, J.; Zhou, L.; Yang, J.; Sofia, A.; Chen, H.; et al. Tumor microenvironment targeting system for glioma treatment via fusion cell membrane coating nanotechnology. Biomaterials 2023, 295, 122026. [Google Scholar] [CrossRef] [PubMed]
- Alsaab, H.O.; Alharbi, F.D.; Alhibs, A.S.; Alanazi, N.B.; Alshehri, B.Y.; Saleh, M.A.; Alshehri, F.S.; Algarni, M.A.; Almugaiteeb, T.; Uddin, M.N.; et al. PLGA-based nanomedicine: History of advancement and development in clinical applications of multiple diseases. Pharmaceutics 2022, 14, 2728. [Google Scholar] [CrossRef]
- Loureiro, J.A.; Ramalho, M.U.; do Carmo Pereira, M. Immuno-nanocarriers for brain delivery: Limitations from in vitro to preclinical and clinical studies. Nanomedicine 2020, 15, 543–545. [Google Scholar] [CrossRef]
- Available online: https://www.cancer.gov/nano/cancer-nanotechnology/current-treatments (accessed on 4 February 2024).
- Lee, S.-W.; Kim, Y.-M.; Cho, C.H.; Kim, Y.T.; Kim, S.M.; Hur, S.Y.; Kim, J.-H.; Kim, B.-G.; Kim, S.-C.; Ryu, H.-S.; et al. An open-label, randomized, parallel, phase II trial to evaluate the efficacy and safety of a cremophor-free polymeric micelle formulation of paclitaxel as first-line treatment for ovarian cancer: A Korean Gynecologic Oncology Group Study (KGO G-3021). Cancer Res. Treat. 2018, 50, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Wang, X.-Y.; Shimizu, T.; Nawata, K.; Kiwada, H. PEGylated liposomes elicit an anti-PEG IgM response in a T cell-independent manner. J. Control. Release 2007, 122, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Tagami, T.; Nakamura, K.; Shimizu, T.; Ishida, T.; Kiwada, H. Effect of siRNA in PEG-coated siRNA-lipoplex on anti-PEG IgM production. J. Control. Release 2009, 137, 234–240. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Abu Lila, A.S.; Shimizu, T.; Ishida, T.; Kiwada, H.J. B cell-intrinsic toll-like receptor 7 is responsible for the enhanced anti-PEG IgM production following injection of siRNA-containing PEGylated lipoplex in mice. J. Control. Release 2014, 184, 1–8. [Google Scholar] [CrossRef]
- Bordes, C.; Fréville, V.; Ruffin, E.; Marote, P.; Gauvrit, J.Y.; Briançon, S.; Lantéri, P. Determination of poly(ε-caprolactone) solubility parameters: Application to solvent substitution in a microencapsulation process. Int. J. Pharm. 2010, 383, 236–243. [Google Scholar] [CrossRef] [PubMed]









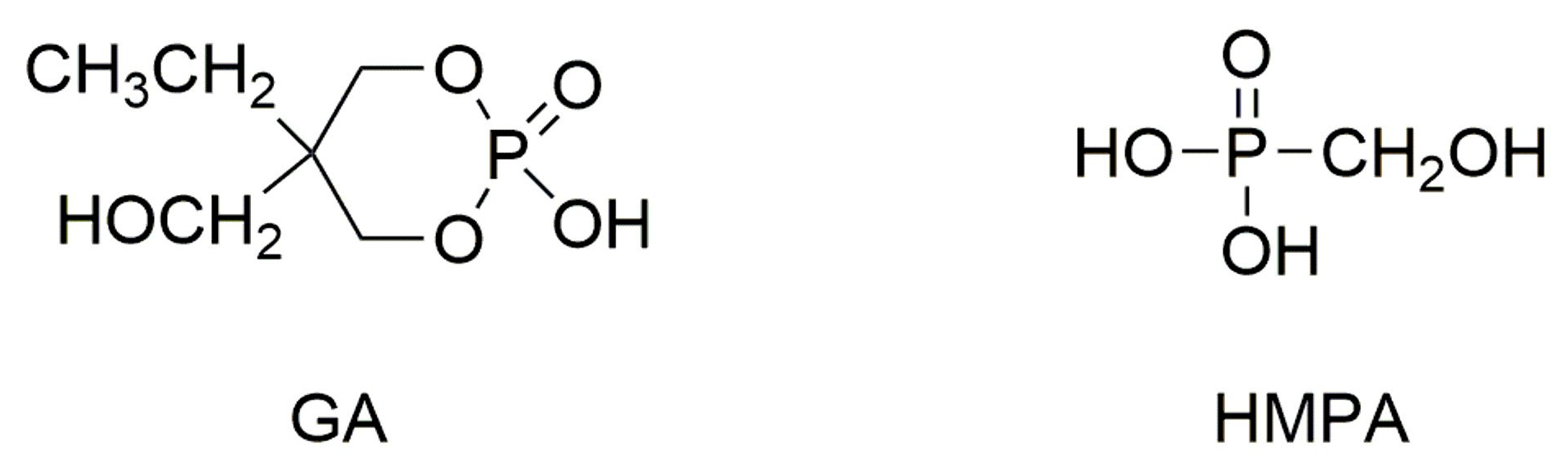





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Slomkowski, S.; Basinska, T.; Gadzinowski, M.; Mickiewicz, D. Polyesters and Polyester Nano- and Microcarriers for Drug Delivery. Polymers 2024, 16, 2503. https://doi.org/10.3390/polym16172503
Slomkowski S, Basinska T, Gadzinowski M, Mickiewicz D. Polyesters and Polyester Nano- and Microcarriers for Drug Delivery. Polymers. 2024; 16(17):2503. https://doi.org/10.3390/polym16172503
Chicago/Turabian StyleSlomkowski, Stanislaw, Teresa Basinska, Mariusz Gadzinowski, and Damian Mickiewicz. 2024. "Polyesters and Polyester Nano- and Microcarriers for Drug Delivery" Polymers 16, no. 17: 2503. https://doi.org/10.3390/polym16172503