

Tailoring Hydrogel Sheet Properties through Co-Monomer Selection in AMPS Copolymer Macromers

 , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
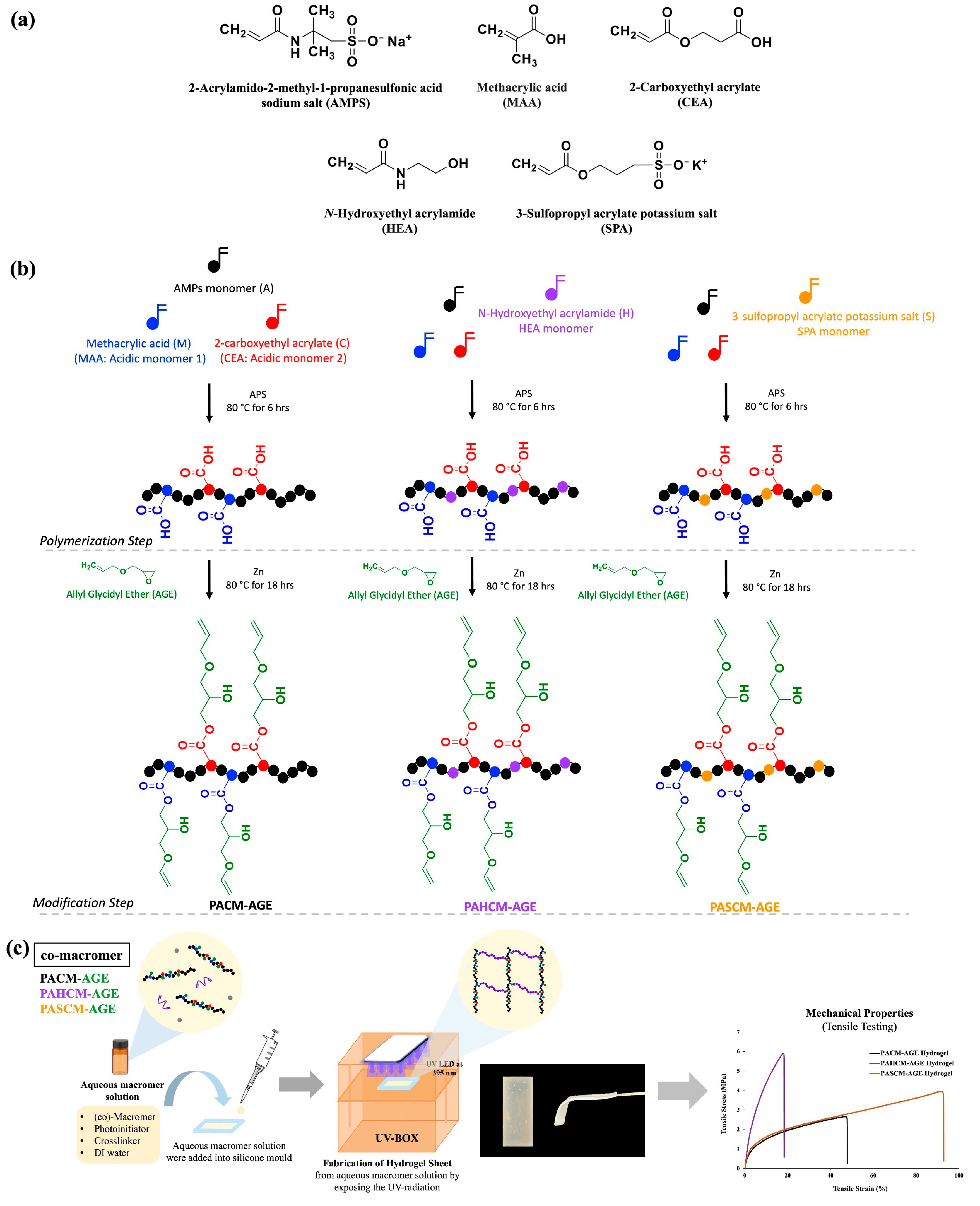
2.2. Synthesis of AMPS-Based Copolymers and AMPS-Based Macromers
2.3. Synthesis of AMPS-Based Macromers
2.4. Fabrication of Hydrogel Sheet
2.5. Characterization of Copolymers and Macromers
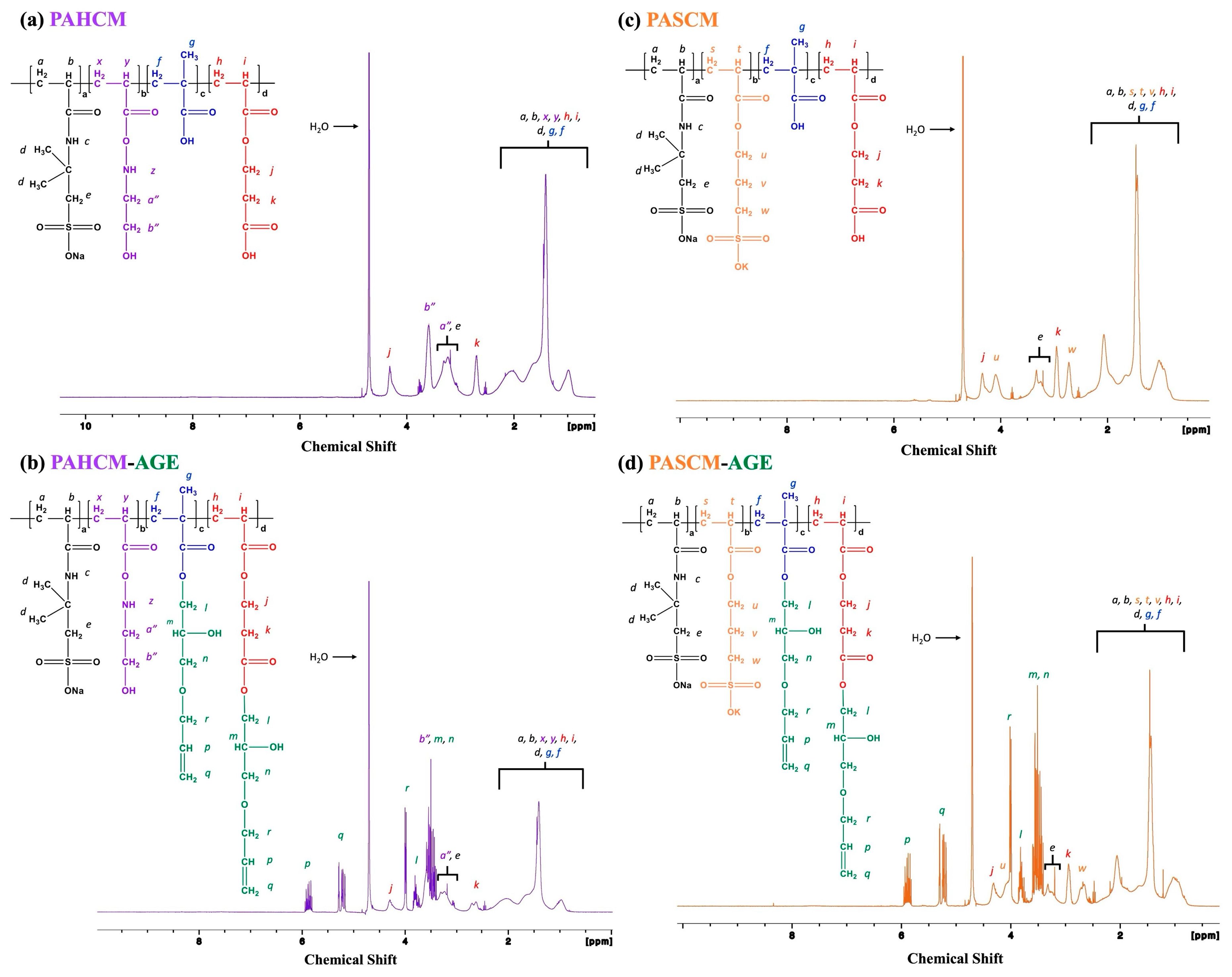
2.5.1. Chemical Structures: 1H NMR Spectroscopy
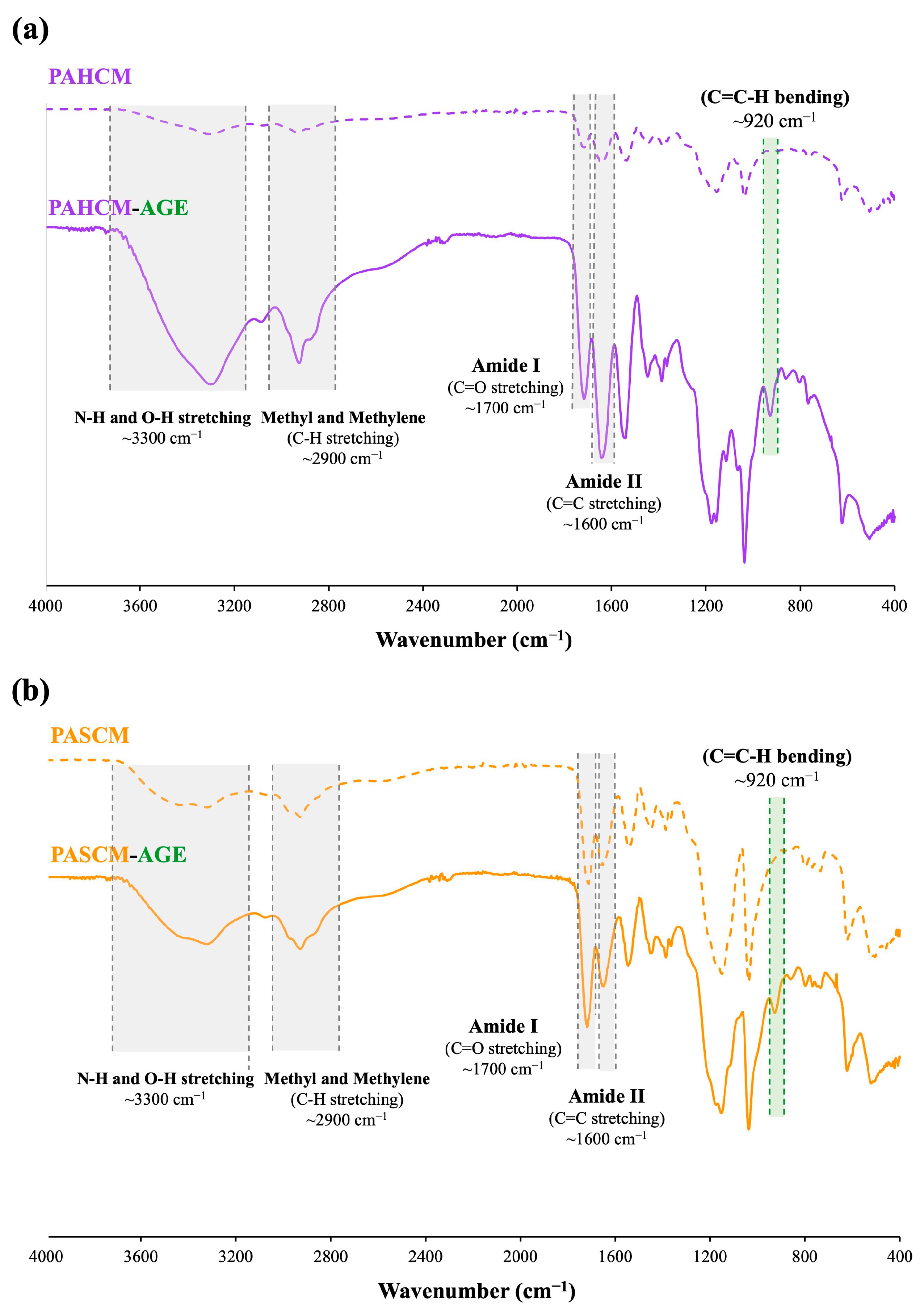
2.5.2. Chemical Functional Groups: FT-IR Spectroscopy
2.5.3. Molecular Weight: GPC
2.6. Characterization of Hydrogel Sheets
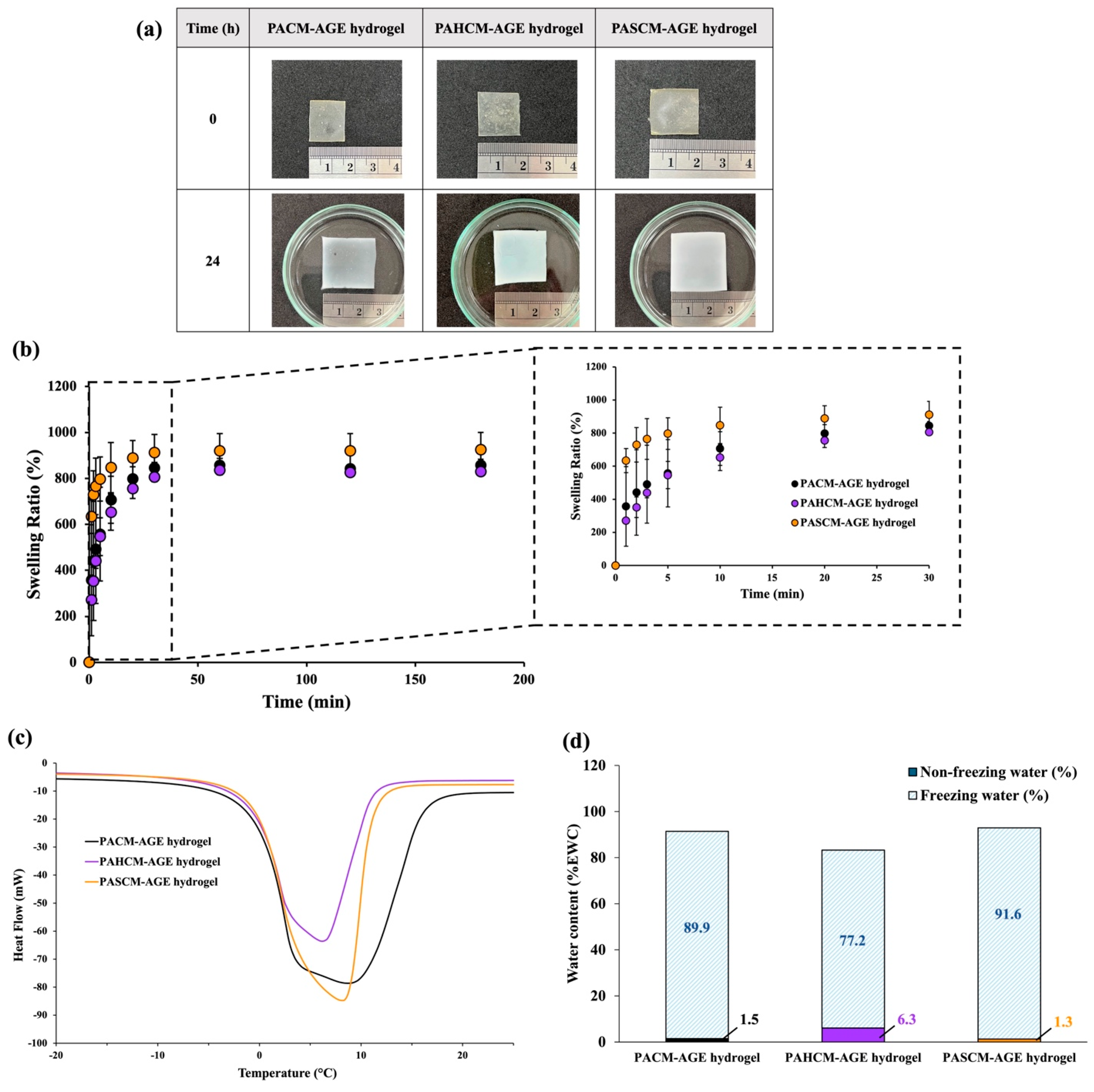
2.6.1. Equilibrium Water Content
2.6.2. Differential Scanning Calorimetry (DSC)
2.6.3. Swelling Test
2.6.4. Tensile Testing
2.6.5. Dye Uptake and Release
2.6.6. In Vitro Anti-Bacterial Experiments
3. Results and Discussion
3.1. Copolymers and Macromers
3.1.1. Chemical Structure Verification by 1H NMR Spectroscopy
3.1.2. Chemical Structure Verification by FT-IR Spectroscopy
3.1.3. Molar Mass Analysis by GPC
3.2. Hydrogel Sheets
3.2.1. Hydrogel Swelling Behavior and Morphology
3.2.2. Mechanical Performance of Hydrogels
3.2.3. Dye Release Profiles
3.2.4. Anti-Bacterial Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, K.Y.; Mooney, D.J. Hydrogels for Tissue Engineering. Chem. Rev. 2001, 101, 1869–1880. [Google Scholar] [CrossRef]
- Wang, W.; Ummartyotin, S.; Narain, R. Advances and Challenges on Hydrogels for Wound Dressing. Curr. Opin. Biomed. Eng. 2023, 26, 100443. [Google Scholar] [CrossRef]
- Yang, X.; Li, J.; Chen, X.; Wang, T.; Li, G.; Zhang, K.; Yin, J.; Cui, H. Multifunctional Hydrogels for Wound Healing. J. Polym. Eng. 2024, 44, 173–194. [Google Scholar] [CrossRef]
- Tavakoli, S.; Klar, A.S. Advanced Hydrogels as Wound Dressings. Biomolecules 2020, 10, 1169. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.; Simões, M.; Vitorino, C.; Mascarenhas-Melo, F. Hydrogels in Cutaneous Wound Healing: Insights into Characterization, Properties, Formulation and Therapeutic Potential. Gels 2024, 10, 188. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Newton, M.A.A.; Cheng, H.; Zhang, Q.; Gao, W.; Zheng, Y.; Lu, Z.; Dai, Z.; Zhu, J. Progress of Hydrogel Dressings with Wound Monitoring and Treatment Functions. Gels 2023, 9, 694. [Google Scholar] [CrossRef]
- Gupta, A.; Kowalczuk, M.; Heaselgrave, W.; Britland, S.T.; Martin, C.; Radecka, I. The Production and Application of Hydrogels for Wound Management: A Review. Eur. Polym. J. 2019, 111, 134–151. [Google Scholar] [CrossRef]
- Bashir, S.; Hina, M.; Iqbal, J.; Rajpar, A.H.; Mujtaba, M.A.; Alghamdi, N.A.; Wageh, S.; Ramesh, K.; Ramesh, S. Fundamental Concepts of Hydrogels: Synthesis, Properties, and Their Applications. Polymers 2020, 12, 2702. [Google Scholar] [CrossRef]
- Naseri, E.; Ahmadi, A. A Review on Wound Dressings: Antimicrobial Agents, Biomaterials, Fabrication Techniques, and Stimuli-Responsive Drug Release. Eur. Polym. J. 2022, 173, 111293. [Google Scholar] [CrossRef]
- Prete, S.; Dattilo, M.; Patitucci, F.; Pezzi, G.; Parisi, O.I.; Puoci, F. Natural and Synthetic Polymeric Biomaterials for Application in Wound Management. J. Funct. Biomater. 2023, 14, 455. [Google Scholar] [CrossRef]
- Pinthong, T.; Yooyod, M.; Daengmankhong, J.; Tuancharoensri, N.; Mahasaranon, S.; Viyoch, J.; Jongjitwimol, J.; Ross, S.; Ross, G.M. Development of Natural Active Agent-Containing Porous Hydrogel Sheets with High Water Content for Wound Dressings. Gels 2023, 9, 459. [Google Scholar] [CrossRef]
- Seo, H.S.; Bae, J.Y.; Kwon, K.; Shin, S. Synthesis and Assessment of AMPS-Based Copolymers Prepared via Electron-Beam Irradiation for Ionic Conductive Hydrogels. Polymers 2022, 14, 2547. [Google Scholar] [CrossRef] [PubMed]
- Üzüm, Ö.B.; Kundakci, S.; Karadaǧ, E. Polymeric Absorbent for Water Sorption Based on Chemically Crosslinked Poly (Acrylamide/2-Acrylamido-2-Methyl-1-Propanesulfonic Acid Sodium Salt) Hydrogels. Polym. Bull. 2006, 57, 703–712. [Google Scholar] [CrossRef]
- Amiri, F.; Kabiri, K.; Bouhendi, H.; Abdollahi, H.; Najafi, V.; Karami, Z. High Gel-Strength Hybrid Hydrogels Based on Modified Starch through Surface Cross-Linking Technique. Polym. Bull. 2019, 76, 4047–4068. [Google Scholar] [CrossRef]
- Su, E.; Yurtsever, M.; Okay, O. A Self-Healing and Highly Stretchable Polyelectrolyte Hydrogel via Cooperative Hydrogen Bonding as a Superabsorbent Polymer. Macromolecules 2019, 52, 3257–3267. [Google Scholar] [CrossRef]
- Martínez-Cornejo, V.; Velázquez-Roblero, J.; Rosiles-González, V.; Correa-Duran, M.; Avila-Ortega, A.; Hernández-Núñez, E.; Le Lagadec, R.; González-Díaz, M.O. Synthesis of Poly(2-Acrylamido-2-Methylpropane Sulfonic Acid) and Its Block Copolymers with Methyl Methacrylate and 2-Hydroxyethyl Methacrylate by Quasiliving Radical Polymerization Catalyzed by a Cyclometalated Ruthenium(Ii) Complex. Polymers 2020, 12, 1663. [Google Scholar] [CrossRef] [PubMed]
- Sekizkardes, B.; Su, E.; Okay, O. Mechanically Strong Superabsorbent Terpolymer Hydrogels Based on AMPS via Hydrogen-Bonding Interactions. ACS Appl. Polym. Mater. 2023, 5, 2043–2050. [Google Scholar] [CrossRef]
- Ning, X.; Huang, J.; A, Y.; Yuan, N.; Chen, C.; Lin, D. Research Advances in Mechanical Properties and Applications of Dual Network Hydrogels. Int. J. Mol. Sci. 2022, 23, 15757. [Google Scholar] [CrossRef]
- Daengmankhong, J.; Ross, S.; Pinthong, T.; Mahasaranon, S.; Viyoch, J.; Tighe, B.J.; Derry, M.J.; Topham, P.D.; Ross, G. Water-Soluble Macromers Based on 2-Acrylamido-2-Methyl-1-Propanesulfonic Acid Sodium Salt (Na-AMPS) for Rapid in Situ Hydrogel Film Formation. Polym. Chem. 2024, 15, 1620–1634. [Google Scholar] [CrossRef]
- Bhattacharyya, R.; Chowdhury, P. Hydrogels of Acryloyl Guar Gum-g-(Acrylic Acid-Co-3sulfopropylacrylate) for High-Performance Adsorption and Release of Gentamicin Sulphate. J. Polym. Res. 2021, 28, 286. [Google Scholar] [CrossRef]
- Narimani, F.; Zohuriaan-Mehr, M.J.; Kabiri, K.; Bouhendi, H.; Omidian, H.; Najafi, V. Overentrant Swelling Behaviour of Poly(Potassium, 3-Sulfopropyl Acrylate-Acrylic Acid) Gels. J. Polym. Res. 2012, 19, 7. [Google Scholar] [CrossRef]
- Lu, J.; Fan, X.; Hu, J.; Li, J.; Rong, J.; Wang, W.; Chen, Y.; Liu, W.; Chen, J.; Chen, Y. Construction and Function of Robust and Moist Bilayer Chitosan-Based Hydrogel Wound Dressing. Mater. Des 2023, 226, 111604. [Google Scholar] [CrossRef]
- Su, J.; Li, J.; Liang, J.; Zhang, K.; Li, J. Hydrogel Preparation Methods and Biomaterials for Wound Dressing. Life 2021, 11, 1016. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, Y.; Wu, N.; Chen, Q.; Wu, K. Removal of Heavy Metal Ions from Wastewater by a Novel HEA/AMPS Copolymer Hydrogel: Preparation, Characterization, and Mechanism. Environ. Sci. Pollut. Res. 2013, 20, 1511–1525. [Google Scholar] [CrossRef] [PubMed]
- Sanna, R.; Alzari, V.; Nuvoli, D.; Scognamillo, S.; Marceddu, S.; Mariani, A. Polymer Hydrogels of 2-Hydroxyethyl Acrylate and Acrylic Acid Obtained by Frontal Polymerization. J. Polym. Sci. A Polym. Chem. 2012, 50, 1515–1520. [Google Scholar] [CrossRef]
- Copling, A.; Akantibila, M.; Kumaresan, R.; Fleischer, G.; Cortes, D.; Tripathi, R.S.; Carabetta, V.J.; Vega, S.L. Recent Advances in Antimicrobial Peptide Hydrogels. Int. J. Mol. Sci. 2023, 24, 7563. [Google Scholar] [CrossRef]
- Vigata, M.; Meinert, C.; Bock, N.; Dargaville, B.L.; Hutmacher, D.W. Deciphering the Molecular Mechanism of Water Interaction with Gelatin Methacryloyl Hydrogels: Role of Ionic Strength, Ph, Drug Loading and Hydrogel Network Characteristics. Biomedicines 2021, 9, 574. [Google Scholar] [CrossRef]
- ASTM D638; Standard Test Method for Tensile Properties of Plastics. ASTM International: West Conshohocken, PA, USA, 2014.
- Nakonechny, F.; Barel, M.; David, A.; Koretz, S.; Litvak, B.; Ragozin, E.; Etinger, A.; Livne, O.; Pinhasi, Y.; Gellerman, G.; et al. Dark Antibacterial Activity of Rose Bengal. Int. J. Mol. Sci. 2019, 20, 3196. [Google Scholar] [CrossRef]
- Tian, R.B.D.; Asmar, S.; Napez, C.; Lépidi, H.; Drancourt, M. Effectiveness of Purified Methylene Blue in an Experimental Model of Mycobacterium Ulcerans Infection. Int. J. Antimicrob. Agents 2017, 49, 290–295. [Google Scholar] [CrossRef]
- Cushnie, T.P.T.; Lamb, A.J. Antimicrobial Activity of Flavonoids. Int. J. Antimicrob. Agents 2005, 26, 343–356. [Google Scholar] [CrossRef]
- Rattanachak, N.; Weawsiangsang, S.; Jongjitvimol, T.; Baldock, R.A.; Jongjitwimol, J. Hydroquinine Possesses Antibacterial Activity, and at Half the MIC, Induces the Overexpression of RND-Type Efflux Pumps Using Multiplex Digital PCR in Pseudomonas Aeruginosa. Trop Med. Infect Dis. 2022, 7, 156. [Google Scholar] [CrossRef] [PubMed]
- Kongprayoon, A.; Ross, G.; Limpeanchob, N.; Mahasaranon, S.; Punyodom, W.; Topham, P.D.; Ross, S. Bio-Derived and Biocompatible Poly(Lactic Acid)/Silk Sericin Nanogels and Their Incorporation within Poly(Lactide-Co-Glycolide) Electrospun Nanofibers. Polym. Chem. 2022, 13, 3343–3357. [Google Scholar] [CrossRef]
- Yooyod, M.; Pinthong, T.; Mahasaranon, S.; Viyoch, J.; Ross, S.; Ross, G.M. Characterization and Performance Analysis of Hydrolyzed versus Non-Hydrolyzed Poly(NVF-Co-HEA) Hydrogels for Cosmetic Applications. Gels 2024, 10, 311. [Google Scholar] [CrossRef]
- Patel, J.B. Performance Standards for Antimicrobial Susceptibility Testing; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2017; ISBN 1562388045. [Google Scholar]
- Ser, J.; Lee, J.Y.; Kim, Y.H.; Cho, H. Enhanced Efficacy of Photodynamic Therapy by Coupling a Cell-Penetrating Peptide with Methylene Blue. Int. J. Nanomed. 2020, 15, 5803–5811. [Google Scholar] [CrossRef]
- Engel Naves Freire, A.; Macedo Iunes Carrera, T.; de Oliveira, G.J.P.L.; Pigossi, S.C.; Vital Ribeiro Júnior, N. Comparison between Antimicrobial Photodynamic Therapy and Low-Level Laser Therapy on Non-Surgical Periodontal Treatment: A Clinical Study. Photodiagn. Photodyn. Ther. 2020, 31, 101756. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Synthesized Samples | Macromers (mol eq.) | Reactants (mol eq.) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| AMPS (A) | HEA (H) | SPA (S) | CEA (C) | MAA (M) | APS | AGE | Zn | ||
| Copolymers | |||||||||
| PACM | Poly(AMPS-stat-CEA-stat-MAA) | 1 | - | - | 0.4 | 0.4 | 0.02 | - | - |
| PAHCM | Poly(AMPS-stat-HEA-stat-CEA-stat-MAA) | 0.75 | 0.25 | - | 0.4 | 0.4 | 0.02 | - | - |
| PASCM | Poly(AMPS-stat-SPA-stat-CEA-stat-MAA) | 0.75 | - | 0.25 | 0.4 | 0.4 | 0.02 | - | - |
| Macromers | |||||||||
| PACM-AGE | Poly(AMPS-stat-CEA-stat-MAA)-graft-AGE | 1 | - | - | 0.4 | 0.4 | 0.02 | 0.8 | 0.06 |
| PAHCM-AGE | Poly(AMPS-stat-HEA-stat-CEA-stat-MAA)-graft-AGE | 0.75 | 0.25 | - | 0.4 | 0.4 | 0.02 | 0.8 | 0.06 |
| PASCM-AGE | Poly(AMPS-stat-SPA-stat-CEA-stat-MAA)-graft-AGE | 0.75 | - | 0.25 | 0.4 | 0.4 | 0.02 | 0.8 | 0.06 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daengmankhong, J.; Pinthong, T.; Promkrainit, S.; Yooyod, M.; Mahasaranon, S.; Punyodom, W.; Ross, S.; Jongjitwimol, J.; Tighe, B.J.; Derry, M.J.; et al. Tailoring Hydrogel Sheet Properties through Co-Monomer Selection in AMPS Copolymer Macromers. Polymers 2024, 16, 2522. https://doi.org/10.3390/polym16172522
Daengmankhong J, Pinthong T, Promkrainit S, Yooyod M, Mahasaranon S, Punyodom W, Ross S, Jongjitwimol J, Tighe BJ, Derry MJ, et al. Tailoring Hydrogel Sheet Properties through Co-Monomer Selection in AMPS Copolymer Macromers. Polymers. 2024; 16(17):2522. https://doi.org/10.3390/polym16172522
Chicago/Turabian StyleDaengmankhong, Jinjutha, Thanyaporn Pinthong, Sudarat Promkrainit, Maytinee Yooyod, Sararat Mahasaranon, Winita Punyodom, Sukunya Ross, Jirapas Jongjitwimol, Brian J. Tighe, Matthew J. Derry, and et al. 2024. "Tailoring Hydrogel Sheet Properties through Co-Monomer Selection in AMPS Copolymer Macromers" Polymers 16, no. 17: 2522. https://doi.org/10.3390/polym16172522
APA StyleDaengmankhong, J., Pinthong, T., Promkrainit, S., Yooyod, M., Mahasaranon, S., Punyodom, W., Ross, S., Jongjitwimol, J., Tighe, B. J., Derry, M. J., Topham, P. D., & Ross, G. M. (2024). Tailoring Hydrogel Sheet Properties through Co-Monomer Selection in AMPS Copolymer Macromers. Polymers, 16(17), 2522. https://doi.org/10.3390/polym16172522