
Thermokinetic and Chemorheology of the Geopolymerization of an Alumina-Rich Alkaline-Activated Metakaolin in Isothermal and Dynamic Thermal Scans


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials and Sample Preparation Procedures
2.2. Thermoanalysis Methods and Testing Procedures
2.2.1. Differential Scanning Calorimetry
2.2.2. DSC Thermograms Deconvolution
- , is the peak amplitude (in mW);
- , is the peak temperature or time (in °C for dynamic and hours for isotherm tests);
- , is the peak with (in °C for dynamic and hr for isotherm tests);
- , is the peak ordinate baseline (in mW);
- , is the thermogram abscissa (in °C for dynamic or hr, for isothermal tests);
- , is the thermogram ordinate (in mW).
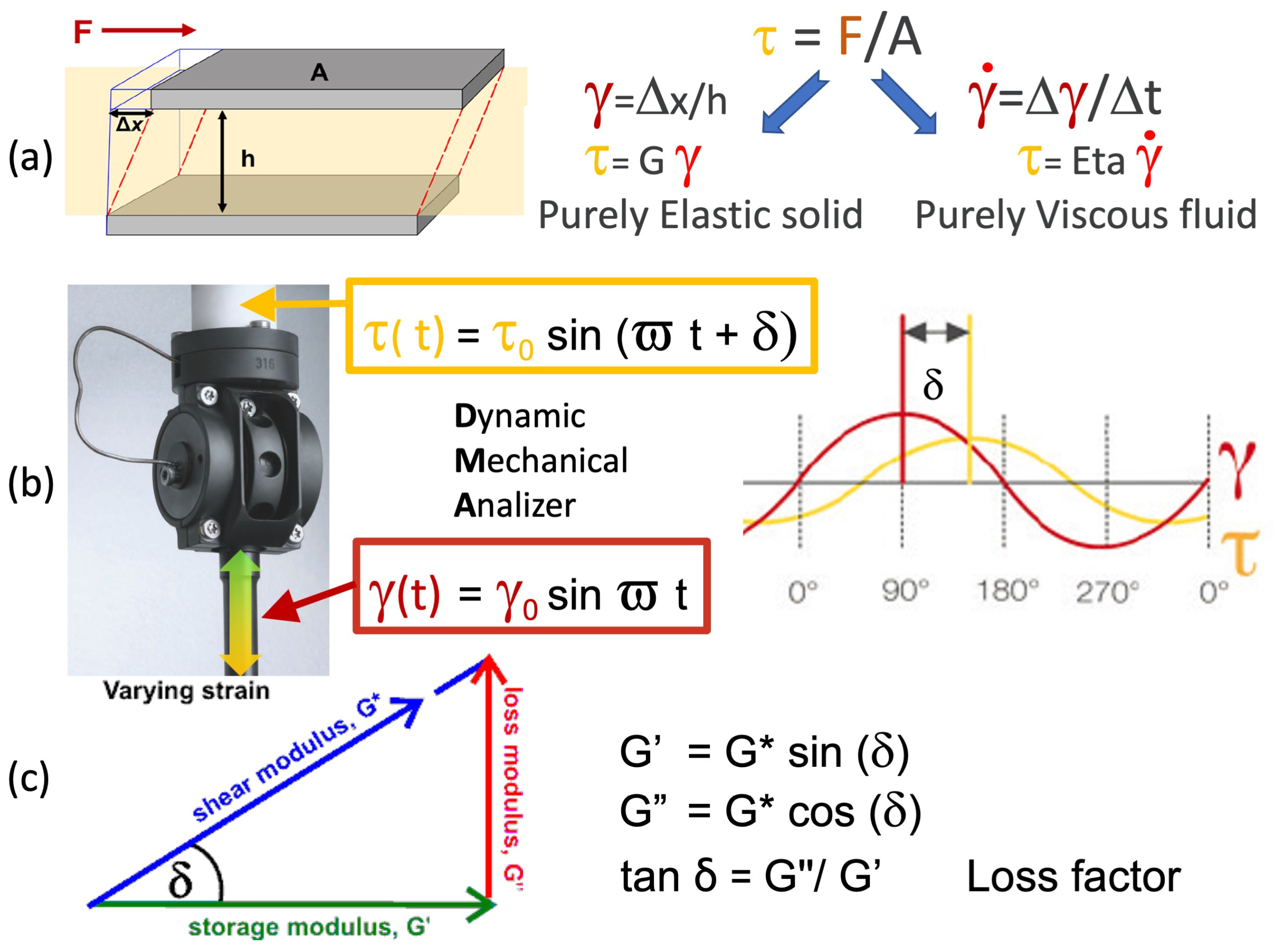
2.2.3. Dynamic Mechanical Analysis Test Procedures
Eta″ = G′/ω
3. Results
3.1. Differential Scanning Calorimetry
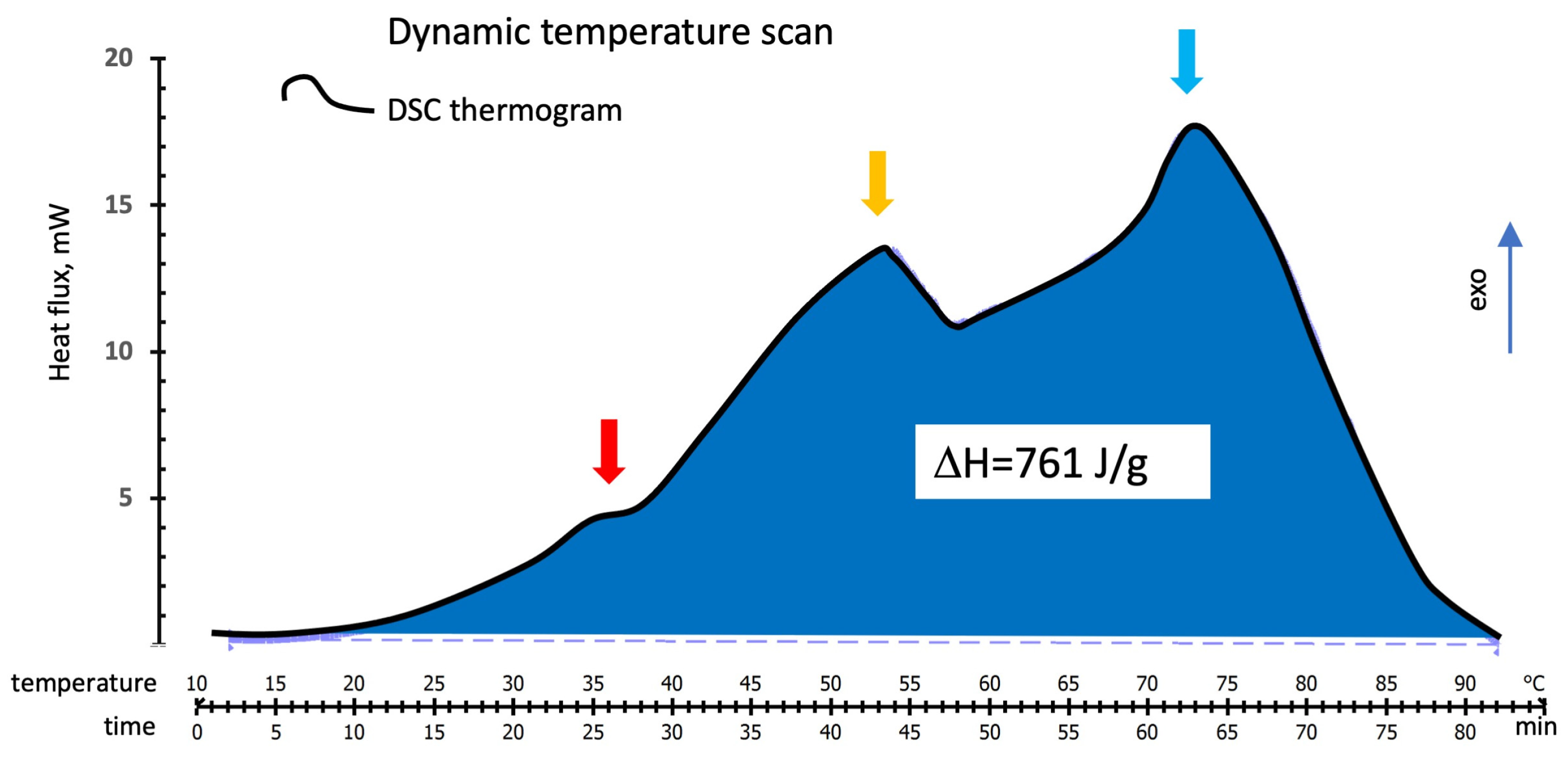
3.1.1. DSC Temperature Scan Test
3.1.2. DSC Isothermal Cure Tests
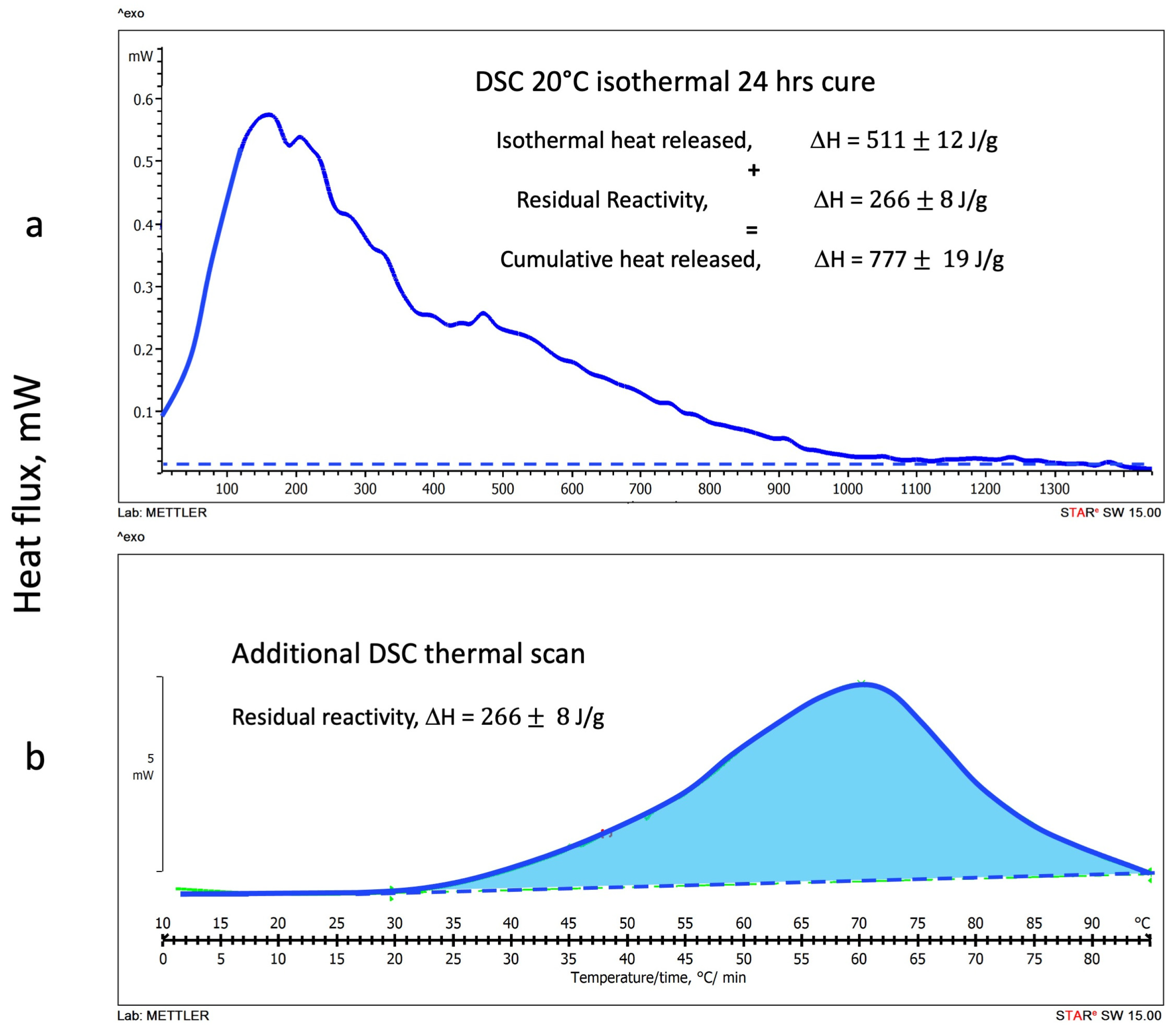

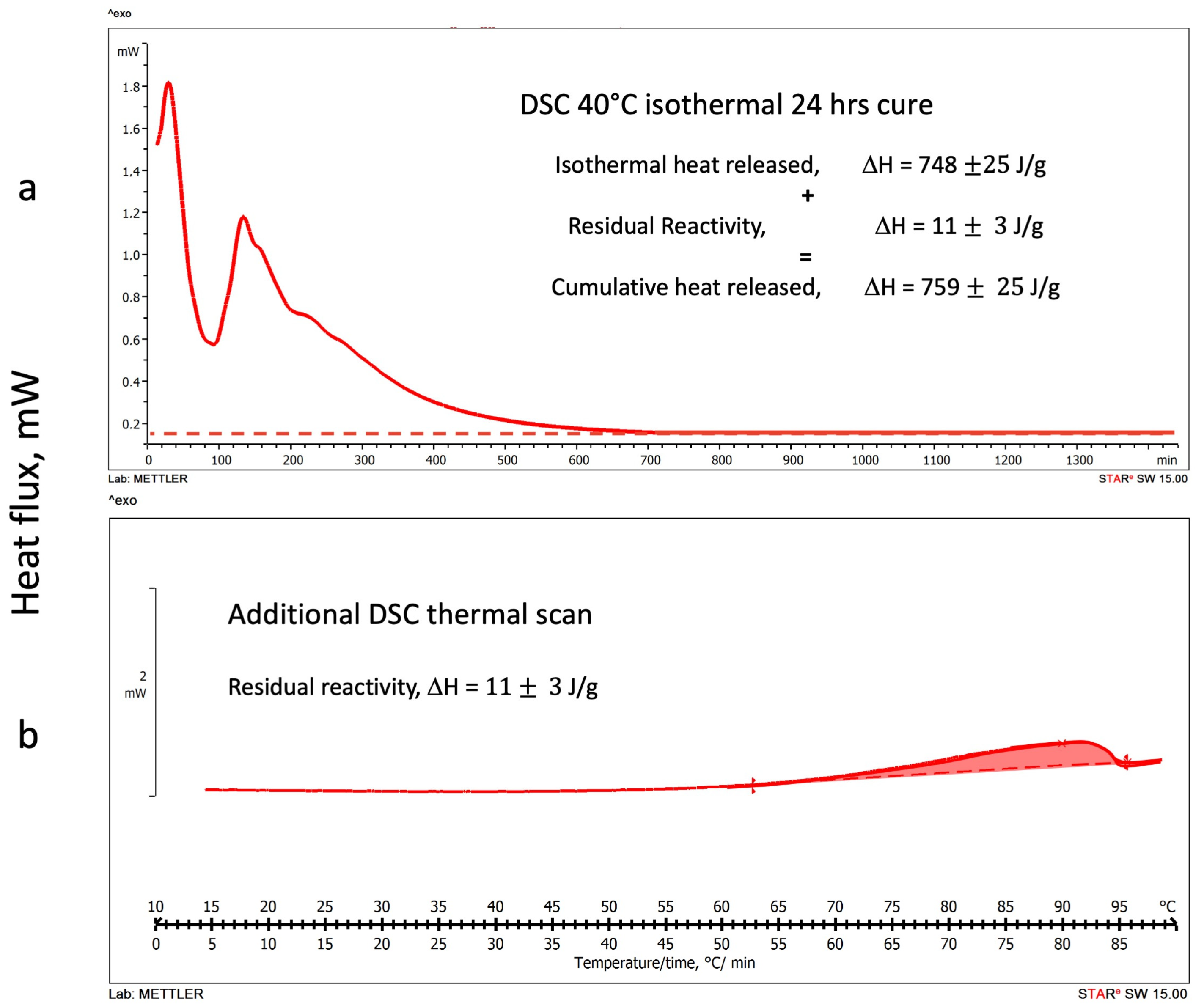
3.2. DSC Thermograms Deconvolution
3.2.1. DSC Temperature Scan Test
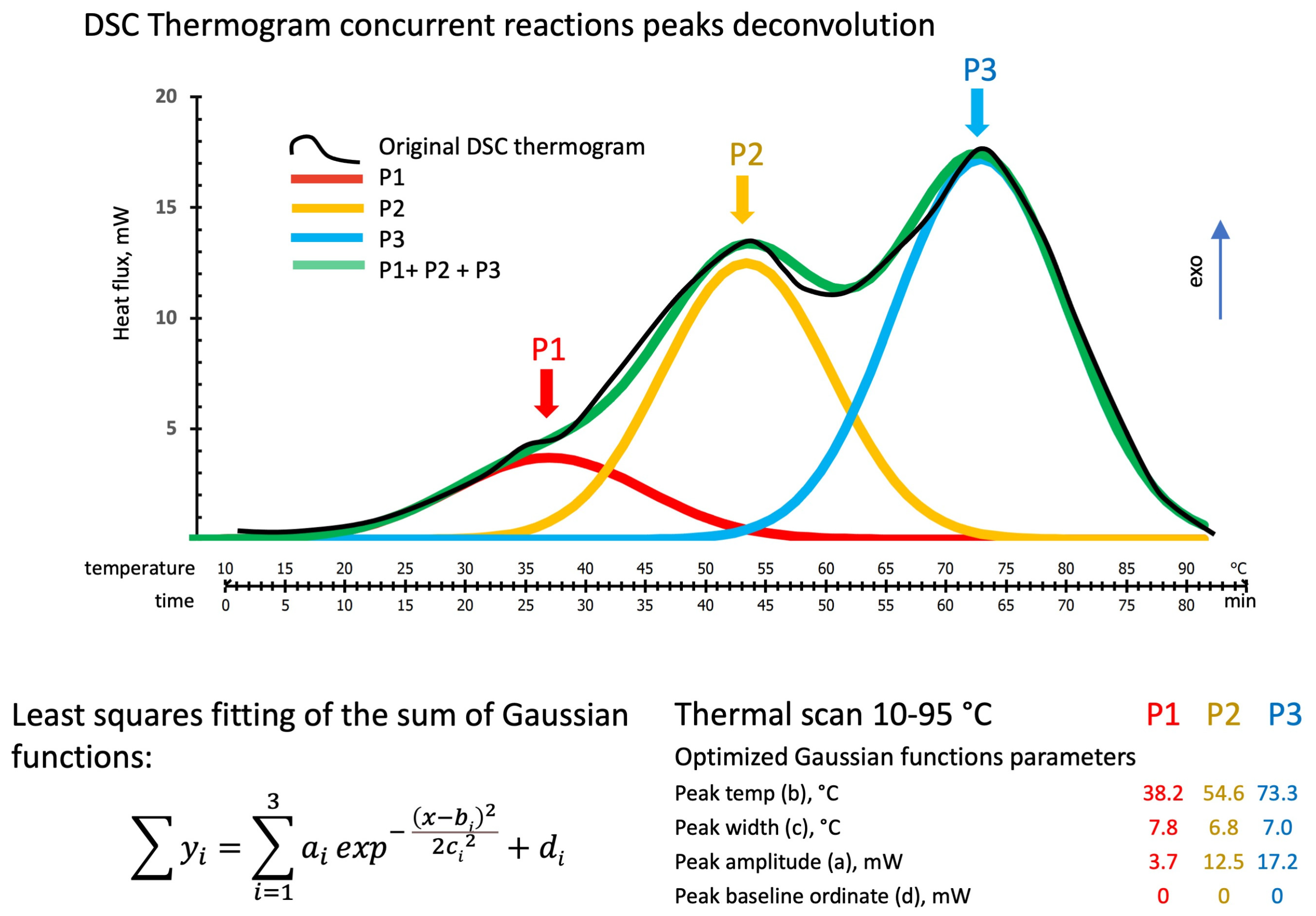
3.2.2. DSC Isothermal Scan Test
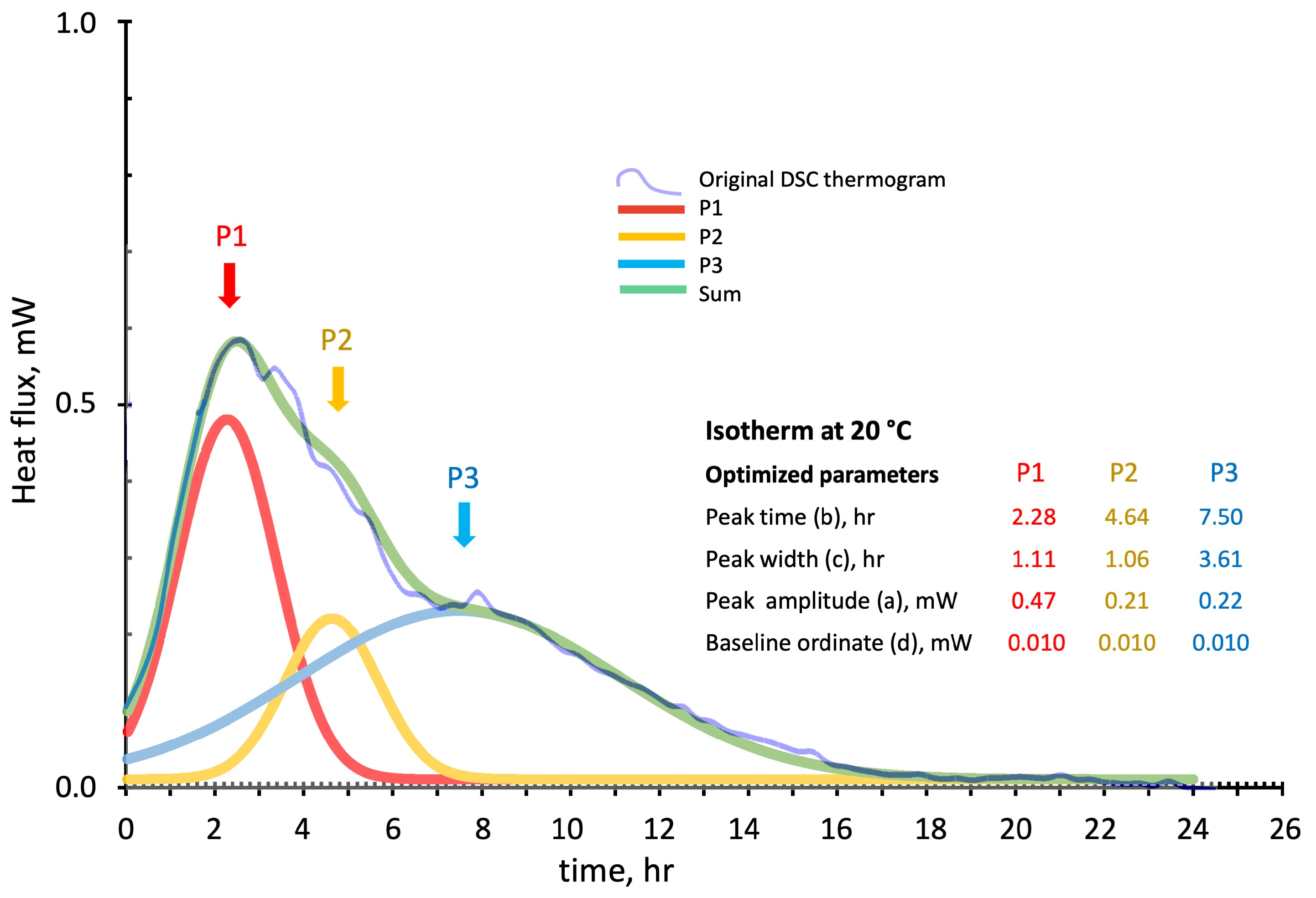


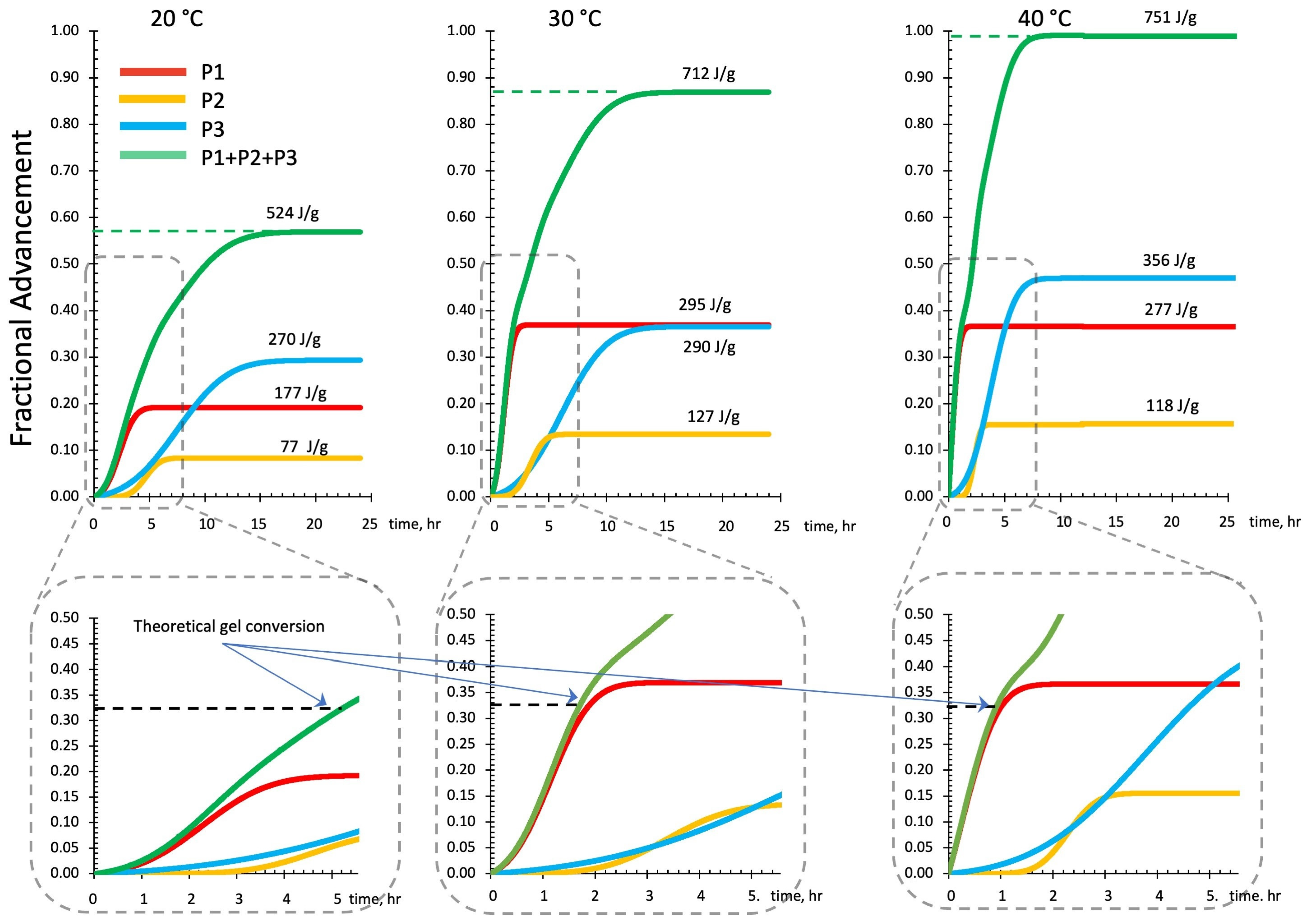
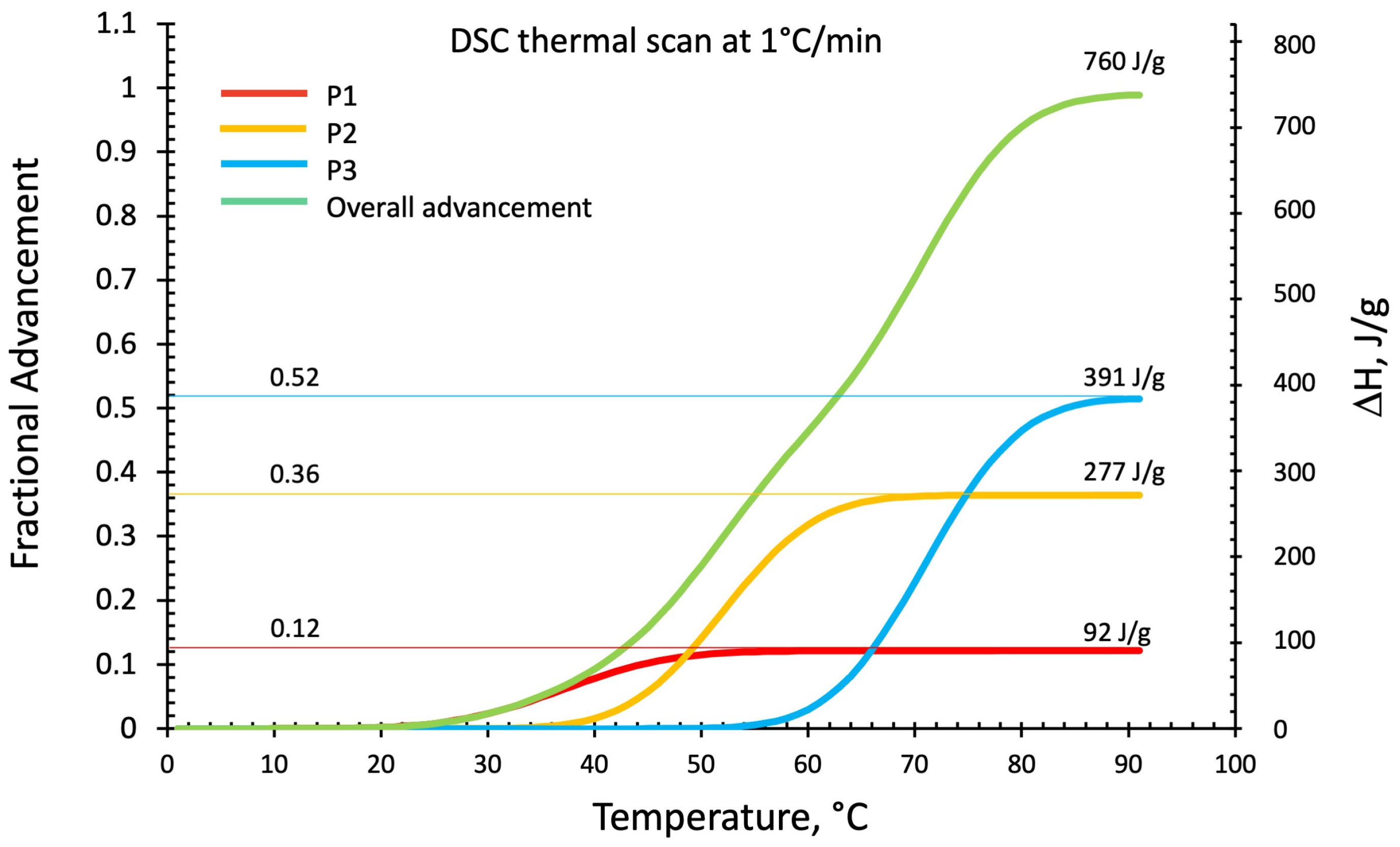
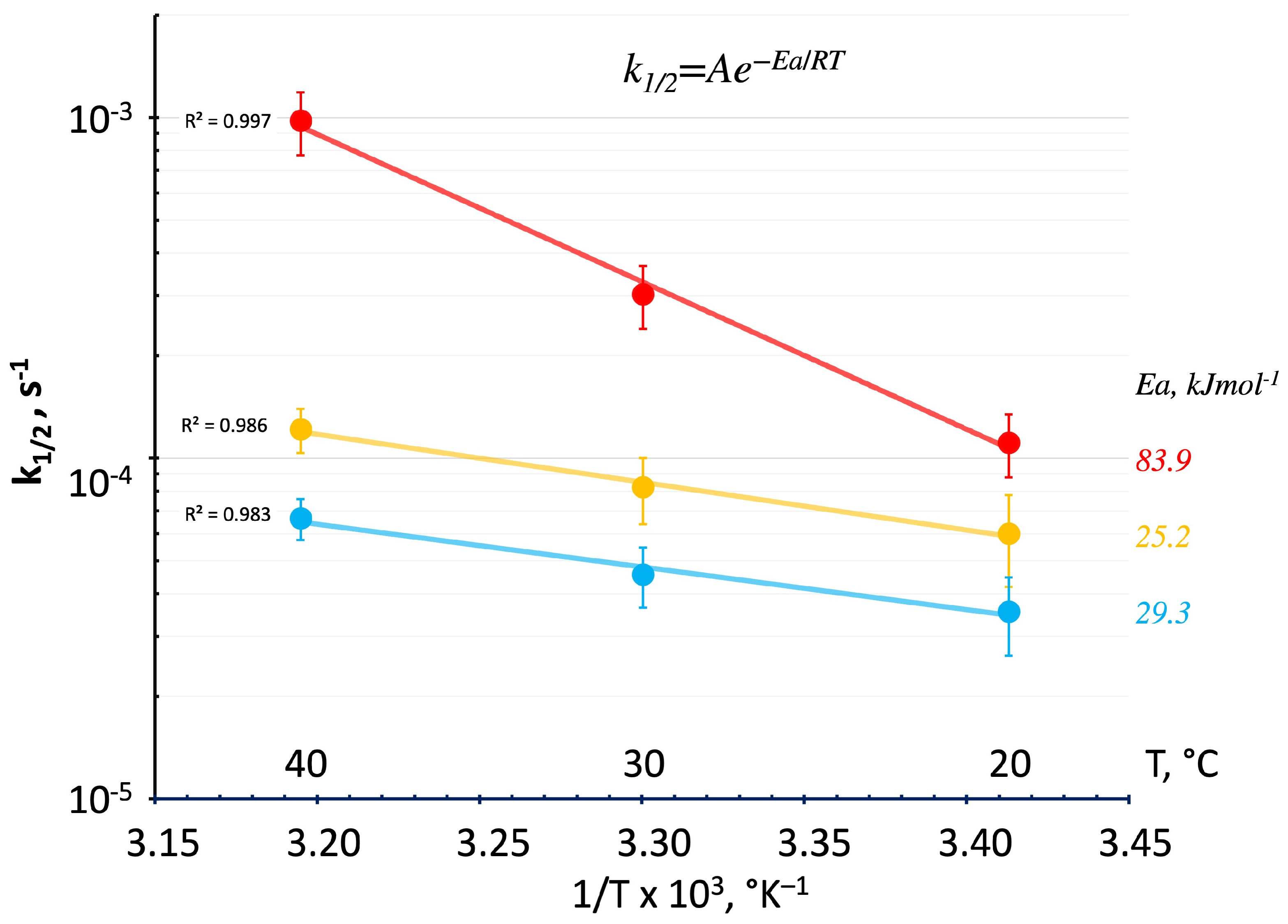
3.2.3. DSC Kinetics from Deconvoluted Peaks
- k1/2 is the rate constant;
- T is the absolute temperature (in °K);
- A is a constant reaction frequency factor;
- Ea is the activation energy for the reaction (in J mol−1);
- R is the universal gas constant, 8.314 J mol−1 °K−1.
3.2.4. Residual Reactivity from DSC Temperature Scan Test of Isothermally Cured Samples
4. Discussion
4.1. Thermosets Polymerisation and Theoretical Evaluation of the Gelation Critical Conversion
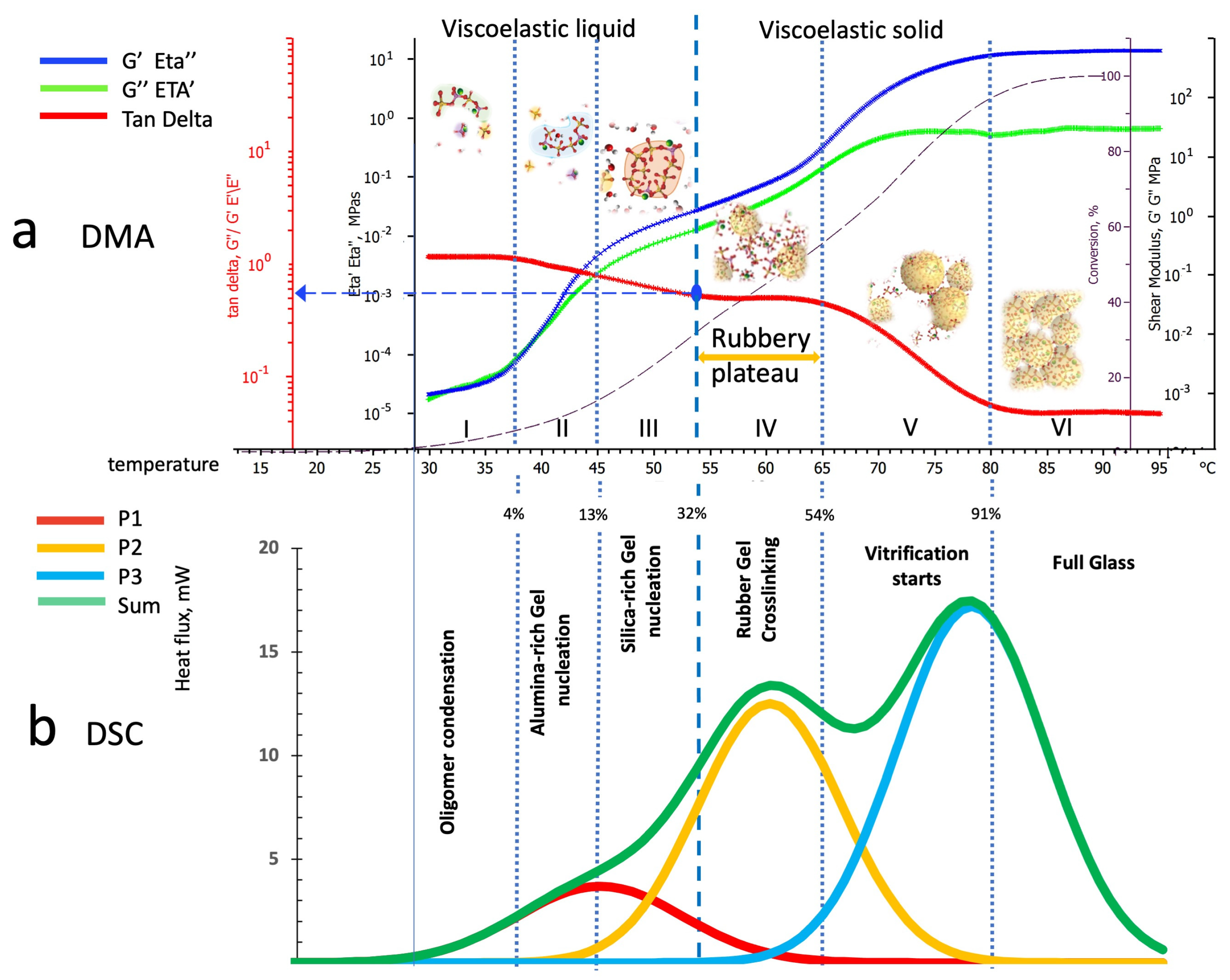
4.2. Parallel Interpretation of Dynamic Mechanical Analysis and Deconvoluted Differential Scanning Calorimetry Thermograms
- Zone I: Kaolin deconstruction and silico-aluminate oligomers formation—viscoelastic liquid;
- Zone II: Nucleation of alumina-rich gel particles—a viscoelastic liquid solution containing alumina-rich gel particles;
- Zone III: Nucleation of silica-rich gel particles—a viscoelastic liquid solution containing alumina-rich and silica-rich gel particles;
- Zone IV: Silico-aluminate rubber gel—amorphous viscoelastic rubber;
- Zone V: Silico-aluminate vitrification starting (still crosslinking can occur);
- Zone VI: Fully polymerized silico-aluminate glassy geopolymer.
4.2.1. Dynamic Mechanical Analysis and Differential Scanning Calorimetry Peaks Deconvolution in Temperature Scans at 1 °C/min Heating Rate
4.2.2. Dynamic Mechanical Analysis and Differential Scanning Calorimetry Peaks Deconvolution in Isothermal Cure Cycles at 20 °C, 30 °C, and 40 °C
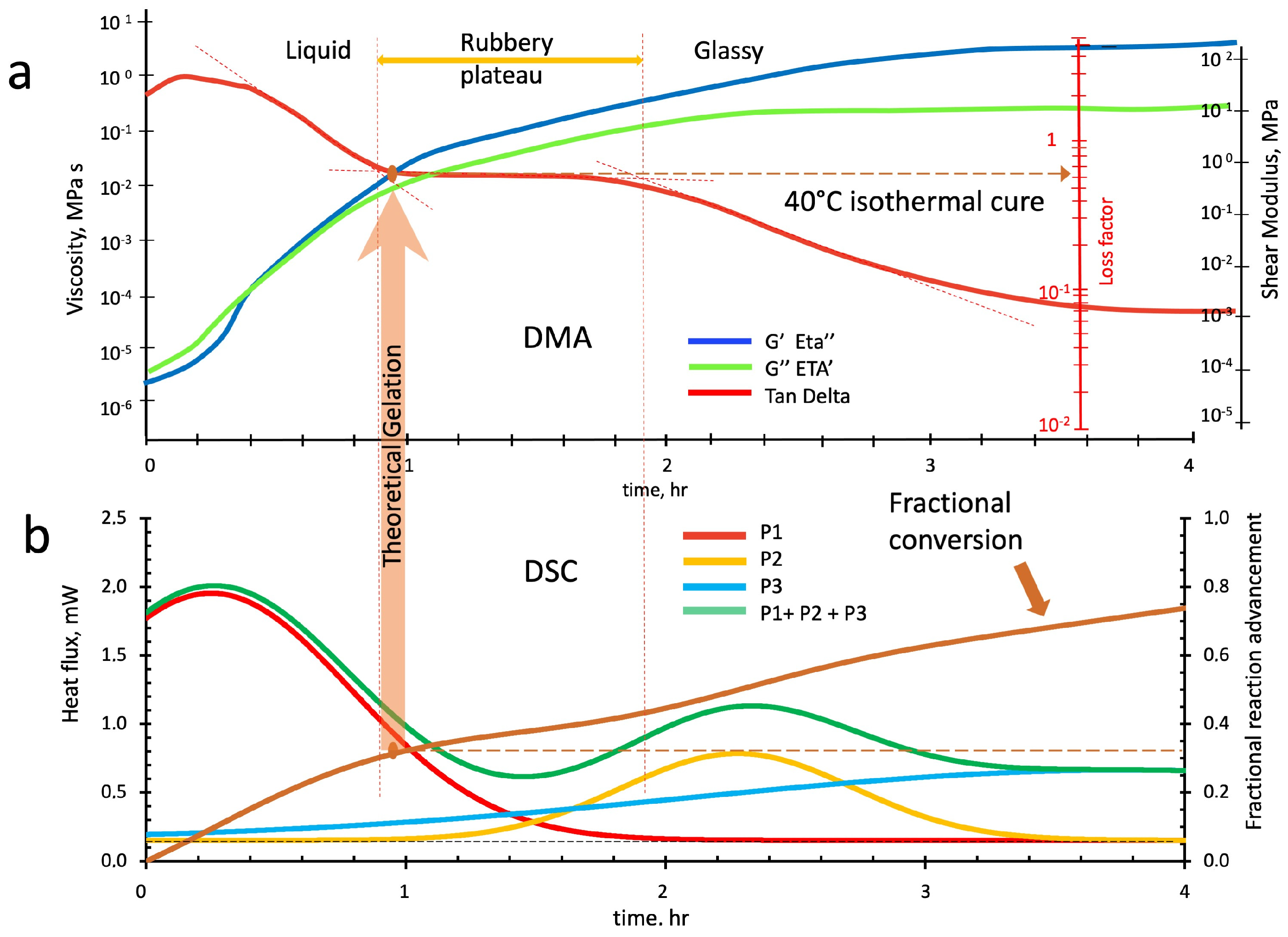


5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Davidovits, J. Geopolymers. J. Therm. Anal. 1991, 37, 1633–1656. [Google Scholar] [CrossRef]
- North, M.R.; Swaddle, T.W. Kinetics of silicate exchange in alkaline aluminosilicate solutions. Inorg. Chem. 2000, 39, 2661–2665. [Google Scholar] [CrossRef]
- Davidovits, J. Geopolymers: Ceramic-like inorganic polymers. J. Ceram. Sci. Technol. 2017, 8, 335–350. [Google Scholar] [CrossRef]
- Ricciotti, L.; Apicella, A.; Perrotta, V.; Aversa, R. Geopolymer Materials for Extrusion-Based 3D-Printing: A Review. Polymers 2023, 15, 4688. [Google Scholar] [CrossRef] [PubMed]
- Long, W.; Wu, Z.; Khayat, K.H.; Wei, J.; Dong, B.; Xing, F.; Zhang, J. Design, dynamic performance and ecological efficiency of fiber-reinforced mortars with different binder systems: Ordinary Portland cement, limestone calcined clay cement and alkali-activated slag. J. Clean. Prod. 2022, 337, 130478. [Google Scholar] [CrossRef]
- Liew, K.; Sojobi, A.; Zhang, L. Green concrete: Prospects and challenges. Construct. Build. Mater. 2017, 156, 1063–1095. [Google Scholar] [CrossRef]
- Aversa, R.; Petrescu, R.V.V.; Sorrentino, R.; Petrescu, F.I.T.; Apicella, A. Hybrid ceramo-polymeric nanocomposite for biomimetic scaffolds design and preparation. Am. J. Eng. Appl. Sci. 2016, 9, 1096–1105. [Google Scholar] [CrossRef]
- Swagato Das, Purnachandra Saha, Swatee Prajna Jena, Pratyush Panda, Geopolymer concrete: Sustainable green concrete for reduced greenhouse gas emission—A review. Mater. Today Proc. 2022, 60, 62–71. [CrossRef]
- Mikulčić, H.; Klemeš, J.J.; Vujanović, M.; Urbaniec, K.; Duić, N. Reducing greenhouse gasses emissions by fostering the deployment of alternative raw materials and energy sources in the cleaner cement manufacturing process. J. Clean. Prod. 2016, 136, 119–132. [Google Scholar] [CrossRef]
- The European Cement Industry Association (CEMBUERAU). The Role of Cement in the 2050 Low Carbon Economy—Full Report; CEMBUERAU: Brussels, Belgium, 2014. [Google Scholar]
- Zhuang, X.Y.; Chen, L.; Komarneni, S.; Zhou, C.H.; Tong, D.S.; Yang, H.M.; Yu, W.H.; Wang, H. Fly ash-based geopolymer: Clean production, properties and applications. J. Clean. Prod. 2016, 125, 253–267. [Google Scholar] [CrossRef]
- Zhong, H.; Zhang, M. 3D printing geopolymers: A review. Cem. Concr. Compos. 2022, 128, 104455. [Google Scholar] [CrossRef]
- Aversa, R.; Petrescu, R.V.V.; Petrescu, F.I.T.; Apicella, A. Biomimetic and evolutionary design driven innovation in sustainable products development. Am. J. Eng. Appl. Sci. 2016, 9, 1027–1036. [Google Scholar] [CrossRef]
- Ricciotti, L.; Apicella, A.; Perrotta, V.; Aversa, R. Geopolymer Materials for Bone Tissue Applications: Recent Advances and Future Perspectives. Polymers 2023, 15, 1087. [Google Scholar] [CrossRef]
- Apicella, A. Effect of Chemorheology on Epoxy Resin Properties. In Developments in Reinforced Plastics—5; Pritchard, G., Ed.; Springer: Dordrecht, The Netherlands, 1986. [Google Scholar] [CrossRef]
- de Jong, B.H.W.S.; Brown, G.E. Polymerization of silicate and aluminate tetrahedra in glasses, melts, and aqueous solutions: Electronic structure of H6Si2O7, H6AlSiO7, and H6Al2O7. Geochim. Cosmochim. Acta 1980, 44, 491–511. [Google Scholar] [CrossRef]
- Koleżyński, A.; Król, M.; Żychowicz, M. The structure of geopolymers. Theoretical studies. J. Mol. Struct. 2018, 1163, 465–471. [Google Scholar] [CrossRef]
- Scherb, S.; Köberl, M.; Beuntner, N.; Thienel, K.-C.; Neubauer, J. Reactivity of Metakaolin in Alkaline Environment: Correlation of Results from Dissolution Experiments with XRD Quantifications. Materials 2020, 13, 2214. [Google Scholar] [CrossRef]
- Le, V.Q.; Do, Q.M.; Hoang, M.D.; Nguyen, H.T. The role of active silica and alumina in geopolymerization. Vietnam. J. Sci. Technol. Eng. 2018, 60, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Aversa, R.; Petrescu, R.V.V.; Petrescu, F.I.T.; Apicella, A. Smart-factory: Optimization and process control of composite centrifuged pipes. Am. J. Appl. Sci. 2016, 13, 1330–1341. [Google Scholar] [CrossRef]
- Villaquirán-Caicedo, M.A.; Fernández-González, A.; Fernández-García, D.A.; Mejía de Gutiérrez, R. Valorization of a low-quality coal ash, in the preparation of alkali activated inks for applications in 3D additive manufacturing. Constr. Build. Mater. 2023, 399, 132598. [Google Scholar] [CrossRef]
- Aversa, R.; Ricciotti, L.; Perrotta, V.; Apicella, A. Chemorheology of a Si/Al > 3 Alkali Activated Metakaolin Paste through Parallel Differential Scanning Calorimetry (DSC) and Dynamic Mechanical Analysis (DMA). Polymers 2023, 15, 3922. [Google Scholar] [CrossRef]
- Hu, Z.; Wyrzykowski, M.; Lura, P. Estimation of reaction kinetics of geopolymers at early ages. Cem. Concr. Res. 2020, 129, 105971. [Google Scholar] [CrossRef]
- Flory, P.J. Thermodynamics of high polymer solutions. J. Chem. Phys. 1942, 10, 51–61. [Google Scholar] [CrossRef]
- Flory, P.J. Molecular Size Distribution in Three Dimensional Polymers II. Trifunctional Branching Units. J. Am. Chem. Soc. 1941, 63, 3091. [Google Scholar] [CrossRef]
- Flory, P.J. Molecular Size Distribution in Three Dimensional Polymers III. Tetrafunctional Branching Units. J. Am. Chem. Soc. 1941, 63, 3096. [Google Scholar] [CrossRef]
- Barbosa, V.F.; MacKenzie, K.J.; Thaumaturgo, C. Synthesis and characterisation of materials based on inorganic polymers of alumina and silica: Sodium polysialate polymers. Int. J. Inorg. Mater. 2000, 2, 309–317. [Google Scholar] [CrossRef]
- Provis, J.; Duxon, P.; Van Deventer, J.; Luckey, J.C. The Role of Mathematical Modelling and Gel Chemistry in Advancing Geopolymer Technology. Chem. Eng. Res. Des. 2005, 83, 853–860. [Google Scholar] [CrossRef]
- Ng, C.; Alengaram, U.J.; Wong, L.S.; Mo, K.H.; Jumaat, M.Z.; Ramesh, S. A review on microstructural study and compressive strength of geopolymer mortar, paste and concrete. Constr. Build. Mater. 2018, 186, 550–576. [Google Scholar] [CrossRef]
- Liew, Y.-M.; Heah, C.-Y.; Mustafa, A.B.M.; Kamarudin, H. Structure and properties of clay-based geopolymer cements: A review. Prog. Mater. Sci. 2016, 83, 595–629. [Google Scholar] [CrossRef]
- Chen, X.; Kim, E.; Suraneni, P.; Struble, L. Quantitative Correlation between the Degree of Reaction and Compressive Strength of Metakaolin-Based Geopolymers. Materials 2020, 13, 5784. [Google Scholar] [CrossRef] [PubMed]
- Görhan, G.; Aslaner, R.; Şinik, O. The effect of curing on the properties of metakaolin and fly ash-based geopolymer paste. Compos. Part B Eng. 2016, 97, 329–335. [Google Scholar] [CrossRef]
- Dimas, D.; Giannopoulou, I.; Panias, D. Polymerization in sodium silicate solutions: A fundamental process in geopolymerization technology. J. Mater. Sci. 2009, 44, 3719–3730. [Google Scholar] [CrossRef]
- Buchwald, A.; Zellmann, H.; Kaps, C. Condensation of aluminosilicate gels—Model system for geopolymer binders. J. Non-Cryst. Solids 2011, 357, 1376–1382. [Google Scholar] [CrossRef]
- Dilawar Riaz, R.; Usman, M.; Ali, A.; Majid, U.; Faizan, M.; Jalil Malik, U. Inclusive characterization of 3D printed concrete (3DPC) in additive manufacturing: A detailed review. Constr. Build. Mater. 2023, 394, 132229. [Google Scholar] [CrossRef]
- Reiner, M. The Deborah Number. Phys. Today 1964, 17, 62. [Google Scholar] [CrossRef]
- Poole, R.J. The Deborah and Weissenberg numbers. Rheol. Bull. 2012, 53, 32–39. [Google Scholar]
- ASTM C-618; Standard Specification for Coal Fly Ash and Raw or Calcined Natural Pozzolan for Use in Concrete. ASTM International: West Conshohocken, PA, USA,, 2017.
- Roviello, G.; Ricciotti, L.; Ferone, C.; Colangelo, F.; Cioffi, R.; Tarallo, O. Synthesis and Characterization of Novel Epoxy Geopolymer Hybrid Composites. Materials 2013, 6, 3943–3962. [Google Scholar] [CrossRef]
- Bampi, M.; Sereno, A.M.; Schmidt, F.C.; Laurindo, J.B. Evaluation of different software tools for deconvolving differential scanning calorimetry thermograms of salted beef. Food Sci. Technol. 2016, 36, 694–700. [Google Scholar] [CrossRef]
- Kriven, W.M.; Bell, J.; Gordon, M. Microstructure and micro-chemistry of fully-reacted geopolymers and geopolymer matrix composites. Ceram. Trans. 2003, 153, 227–250. [Google Scholar]
- Zoulgami, M.; Lucas-Girot, A.; Michaud, V.; Briard, P.; Gaudé, J.; Oudadesse, H. Synthesis and physico-chemical characterization of a polysialate-hydroxyapatite composite for potential biomedical application. Eur. Phys. J. Appl. Phys. 2002, 19, 173–179. [Google Scholar] [CrossRef]
- Nath, P.; Sarker, P.K.; Rangan, V.B. Early Age Properties of Low-calcium Fly Ash Geopolymer Concrete Suitable for Ambient Curing. Procedia Eng. 2015, 25, 601–607. [Google Scholar] [CrossRef]
- Rożek, P.; Król, M.; Mozgawa, W. Geopolymer-zeolite composites: A review. J. Clean. Prod. 2019, 230, 557–579. [Google Scholar] [CrossRef]
- Khale, D.; Chaudhary, R. Mechanism of geopolymerization and factors influencing its development: A review. J. Mater. Sci. 2007, 42, 729–746. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, H.; Provis, J.L.; Bullen, F.; Reid, A.; Zhu, Y. Quantitative kinetic and structural analysis of geopolymers. Part 1. The activation of metakaolin with sodium hydroxide. Thermochim. Acta 2012, 539, 23–33. [Google Scholar] [CrossRef]
- Welle, F. Diffusion Coefficients and Activation Energies of Diffusion of Organic Molecules in Polystyrene below and above Glass Transition Temperature. Polymers 2021, 13, 1317. [Google Scholar] [CrossRef]
- Mierzwiński, D.; Łach, M.; Gądek, S.; Lin, W.-T.; Tran, D.H.; Korniejenko, K. A brief overview of the use of additive manufacturing of con-create materials in construction. Acta Innov. 2023, 48, 22–37. [Google Scholar] [CrossRef]
- Chen, Y.; Jia, L.; Liu, C.; Zhang, Z.; Ma, L.; Chen, C.; Banthia, N.; Zhang, Y. Mechanical anisotropy evolution of 3D-printed alkali-activated materials with different GGBFS/FA combinations. J. Build. Eng. 2022, 501, 104126. [Google Scholar] [CrossRef]
- Khan, S.A.; İlcan, H.; Aminipour, E. Buildability analysis on effect of structural design in 3D concrete printing (3DCP): An experimental and numerical study. Case Stud. Constr. Mater. 2023, 19, e02295. [Google Scholar] [CrossRef]
- Marczyk, J.; Ziejewska, C.; Łach, M. Possibilities of using the 3D printing process in the concrete and geopolymers application. IOP Conf. Ser. Mater. Sci. Eng. 2019, 706, 012019. [Google Scholar] [CrossRef]
- Sahini, M.; Sahimi, M. Applications of Percolation Theory; CRC Press: Boca Raton, FL, USA, 2003; ISBN 978-0-203-22153-2. [Google Scholar]
- Kryven, I. Emergence of the giant weak component in directed random graphs with arbitrary degree distributions. Phys. Rev. E 2016, 94, 012315. [Google Scholar] [CrossRef]
- Flory, P.J. Molecular Size Distribution in Three Dimensional Polymers I. Gelation. J. Am. Chem. Soc. 1941, 63, 3083. [Google Scholar] [CrossRef]
- Stockmayer, W.H. Theory of Molecular Size Distribution and Gel Formation in Branched Polymers II. General Cross Linking. J. Chem. Phys. 1944, 12, 125. [Google Scholar] [CrossRef]
- Duxson, P.; Provis, J.; Lukey, G.; Mallicoat, S.; Kriven, W.; van Deventera, J. Understanding the relationship between geopolymer composition, microstructure and mechanical properties. Colloids Surf. A 2005, 269, 47–58. [Google Scholar] [CrossRef]
- Walkley, B.; Rees, G.J.; San Nicolas, R.; van Deventer, J.S.; Hanna, J.V.; Provis, J.L. New structural model of sodium aluminosilicate gels and the role of charge balancing extra-framework. Al. J. Phys. Chem. C 2018, 122, 5673–5685. [Google Scholar] [CrossRef]
- Rifaai, Y.; Yahia, A.; Mostafa, A.; Aggoun, S.; Kadri, E.H. Rheology of fly ash-based geopolymer: Effect of NaOH concentration. Constr. Build. Mater. 2019, 223, 583–594. [Google Scholar] [CrossRef]
- Castillo, H.; Collado, H.; Droguett, T.; Sánchez, S.; Vesely, M.; Garrido, P.; Palma, S. Factors Affecting the Compressive Strength of Geopolymers: A Review. Minerals 2021, 11, 1317. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oxide 1 | Metakaolin | Sodium Silicate |
|---|---|---|
| SiO2 | 52.30 | 28.36 |
| Al2O3 | 45.20 | - |
| Fe2O3 | 0.42 | - |
| K2O | 0.15 | - |
| Na2O | - | 8.60 |
| MgO | 0.04 | |
| H2O | - | 63.04 |
| DSC, Operating Conditions | Mode | ΔH Partial, J/g | ΔH Total, * J/g |
|---|---|---|---|
| Dynamic 10–95 °C scan, fresh sample | Exp. | ||
| Model | |||
| Iso 20 °C | Exp. | ||
| Model | |||
| Iso 30 °C | Exp. | 726 | 762 |
| Model | 747 | ||
| Iso 40 °C | Exp. | 748 | 759 |
| Model | 763 | ||
| Dynamic 10–95 °C scan After 20 °C cure | Exp. | - | |
| Model | - | ||
| Dynamic 10–95 °C scan After 30 °C cure | Exp. | 36 | - |
| Model | - | ||
| Dynamic 10–95 °C scan After 40 °C cure | Exp. | 11 | - |
| Model | - |
| DSC, Operating Conditions | P1 Fractional Conversions | Fractions Sum | P2 Fractional Conversions | Fractions Sum | P3 Fractional Conversions | Fractions Sum |
|---|---|---|---|---|---|---|
| Dynamic 10–95 °C scan on fresh sample | ||||||
| Iso 20 °C+ Dynamic 10–95 °C | 9 | |||||
| Iso 30 °C+ Dynamic 10–95 °C | ||||||
| Iso 40 °C+ Dynamic 10–95 °C | 6 | |||||
| ZONE | Loss Viscosity, MPa*s | Storage Modulus, MPa | Loss Factor, tanδ |
|---|---|---|---|
| I | 10−5 | - | |
| 10−5 | |||
| II | - | - | - |
| 10−3 | |||
| III | - | - | - |
| 10−2 | |||
| IV | - | - | - |
| 10 | |||
| V | - | - | - |
| 102 | |||
| VI | - | - | - |
| 102 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aversa, R.; Ricciotti, L.; Perrotta, V.; Apicella, A. Thermokinetic and Chemorheology of the Geopolymerization of an Alumina-Rich Alkaline-Activated Metakaolin in Isothermal and Dynamic Thermal Scans. Polymers 2024, 16, 211. https://doi.org/10.3390/polym16020211
Aversa R, Ricciotti L, Perrotta V, Apicella A. Thermokinetic and Chemorheology of the Geopolymerization of an Alumina-Rich Alkaline-Activated Metakaolin in Isothermal and Dynamic Thermal Scans. Polymers. 2024; 16(2):211. https://doi.org/10.3390/polym16020211
Chicago/Turabian StyleAversa, Raffaella, Laura Ricciotti, Valeria Perrotta, and Antonio Apicella. 2024. "Thermokinetic and Chemorheology of the Geopolymerization of an Alumina-Rich Alkaline-Activated Metakaolin in Isothermal and Dynamic Thermal Scans" Polymers 16, no. 2: 211. https://doi.org/10.3390/polym16020211