
Charge Mediated Changes to the Intrinsic Viscosity of Biopolymer Systems


Abstract

1. Introduction
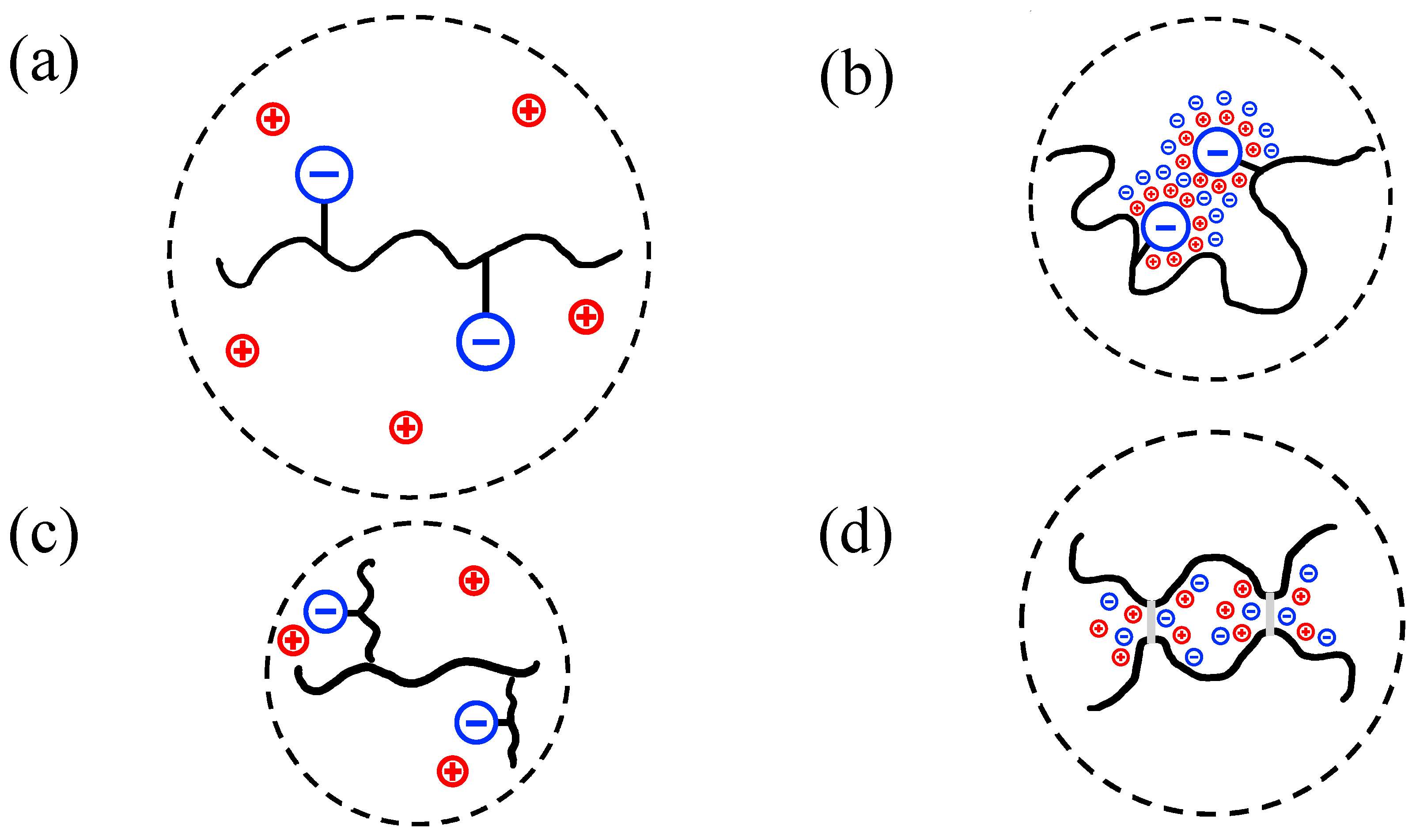
2. Theoretical Review
3. Materials and Methods
3.1. Materials
3.2. Methods
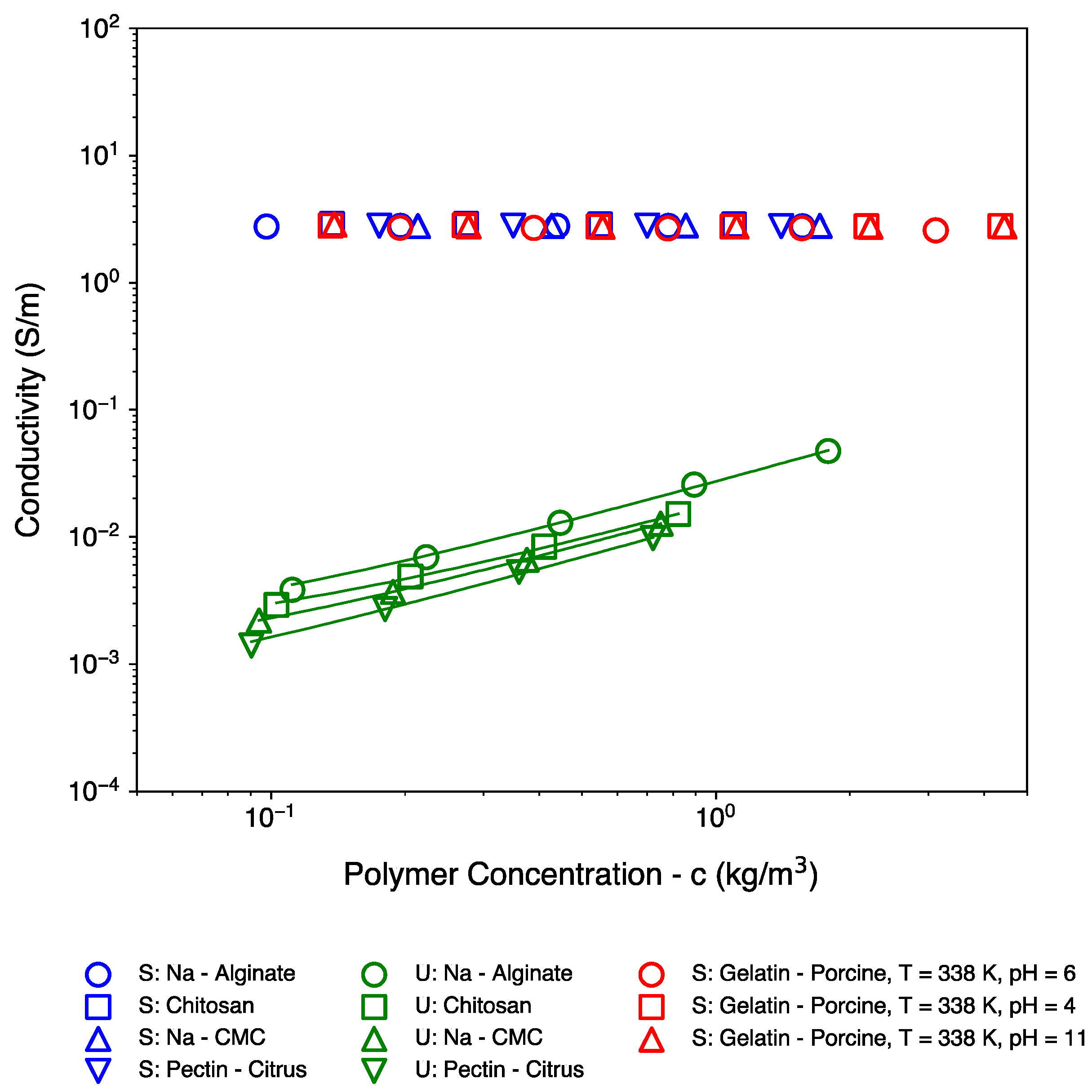
4. Results
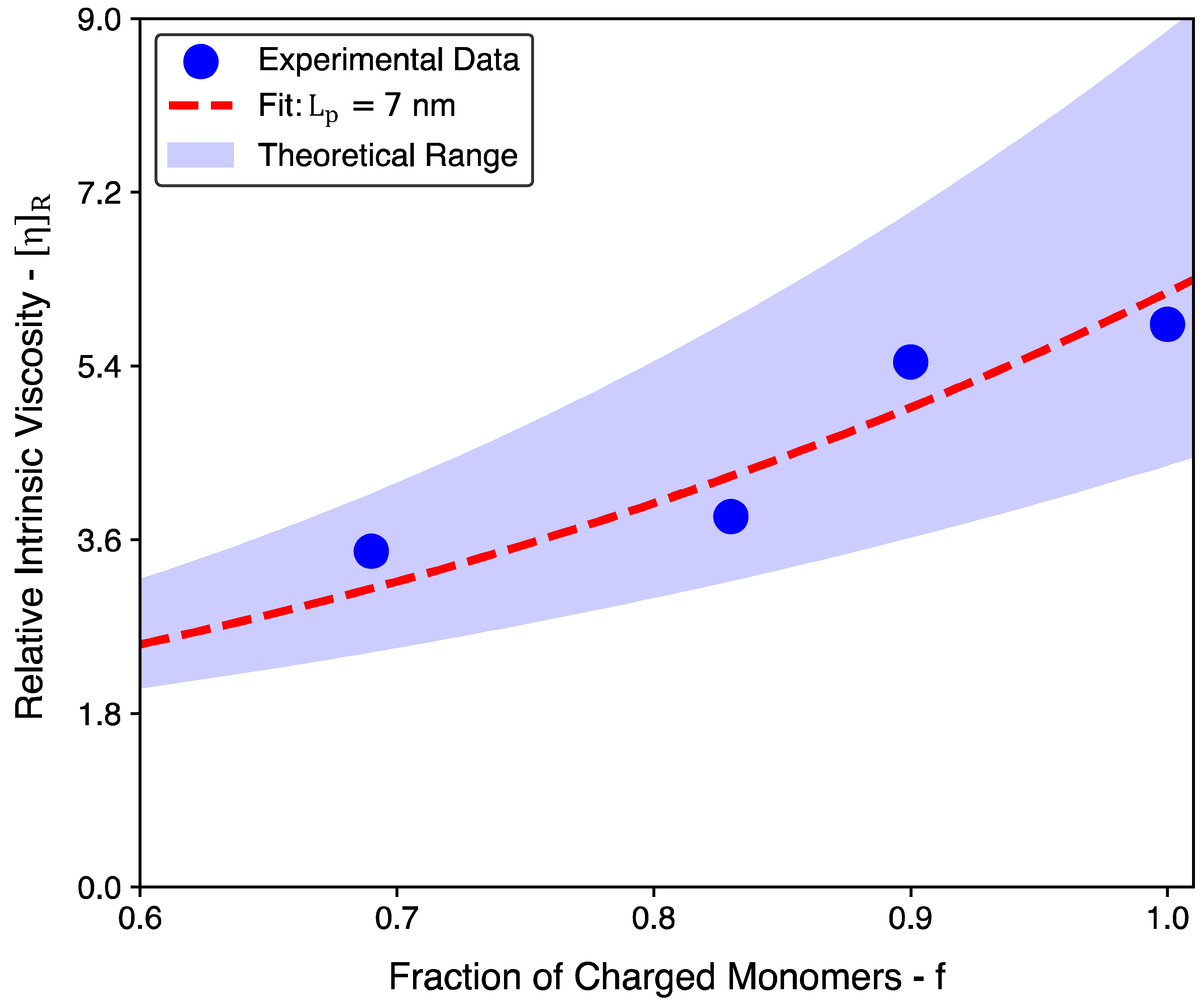
5. Discussion
5.1. Theory vs. Experiments
5.2. Molar Mass Dependence
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
Notations
| Symbol | Meaning | Units |
| Viscosity of solution | Pa·s | |
| Viscosity of solvent | Pa·s | |
| Volume fraction | - | |
| Intrinsic viscosity | m3/kg | |
| Mass concentration | kg/m3 | |
| End-to-end distance | nm | |
| Contour length | nm | |
| Persistence length | nm | |
| Number of repeating units | - | |
| Monomer size | nm | |
| Radius of gyration | nm | |
| Molar mass | kg or kg/mol | |
| Numerical function from Yamakawa—Fujii model | m3/2 or nm3/2 | |
| Weight-averaged molar mass | kg/mol | |
| Electrostatic persistence length | nm | |
| Persistence length of unscreened polymer chain | nm | |
| Bjerrum length | nm | |
| Fraction of charged monomers | - | |
| Inverse Debye length | nm−1 | |
| Intrinsic viscosity of unscreened biopolymer | m3/kg | |
| Intrinsic viscosity of screened biopolymer | m3/kg | |
| Relative intrinsic viscosity | - | |
| Expansion factor | - |
References
- Nelson, D.L.; Cox, M.M.; Hoskins, A.A. Lehninger Principles of Biochemistry, 8th ed.; Macmillan Learning: New York, NY, USA, 2021. [Google Scholar]
- Kontogiorgos, V. Pectin: Technological and Physiological Properties; Springer International: Cham, Switzerland, 2020. [Google Scholar]
- O’Mahony, J.; Fox, P. Milk Proteins: Introduction and Historical aspects. Advanced Dairy Chemistry: Volume 1A: Proteins: Basic Aspects, 4th ed.; Springer: New York, NY, USA, 2013; pp. 43–85. [Google Scholar]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- te Nijenhuis, K. On the nature of crosslinks in thermoreversible gels. Polym. Bull. 2007, 58, 27–42. [Google Scholar] [CrossRef]
- Picout, D.R.; Ross-Murphy, S.B. Rheology of biopolymer solutions and gels. Sci. World J. 2003, 3, 105–121. [Google Scholar] [CrossRef]
- Felz, S.; Al-Zuhairy, S.; Aarstad, O.A.; van Loosdrecht, M.C.; Lin, Y.M. Extraction of structural extracellular polymeric substances from aerobic granular sludge. JoVE J. Vis. Exp. 2016, 115, e54534. [Google Scholar]
- Some, D.; Amartely, H.; Tsadok, A.; Lebendiker, M. Characterization of proteins by size-exclusion chromatography coupled to multi-angle light scattering (SEC-MALS). JoVE J. Vis. Exp. 2019, 148, e59615. [Google Scholar]
- Farkas, N.; Kramar, J.A. Dynamic light scattering distributions by any means. J. Nanopart. Res. 2021, 23, 120. [Google Scholar] [CrossRef]
- Rubinstein, M.; Colby, R.H. Polymer Physics; Oxford University Press: Oxford, UK, 2003. [Google Scholar]
- Lopez, C.G.; Matsumoto, A.; Shen, A.Q. Dilute polyelectrolyte solutions: Recent progress and open questions. Soft Matter 2023, 20, 2635–2687. [Google Scholar] [CrossRef]
- Lopez, C.G. Entanglement of semiflexible polyelectrolytes: Crossover concentrations and entanglement density of sodium carboxymethyl cellulose. J. Rheol. 2020, 64, 191–204. [Google Scholar] [CrossRef]
- Yamakawa, H.; Fujii, M. Intrinsic viscosity of wormlike chains. Determination of the shift factor. Macromolecules 1974, 7, 128–135. [Google Scholar] [CrossRef]
- Sayko, R.; Jacobs, M.; Dobrynin, A.V. Quantifying properties of polysaccharide solutions. ACS Polym. Au 2021, 1, 196–205. [Google Scholar] [CrossRef]
- Dobrynin, A.V. Electrostatic persistence length of semiflexible and flexible polyelectrolytes. Macromolecules 2005, 38, 9304–9314. [Google Scholar] [CrossRef]
- Kamide, K.; Saito, M.; Suzuki, H. Persistence length of cellulose and cellulose derivatives in solution. Makromol. Chem. Rapid 2003, 4, 33–39. [Google Scholar] [CrossRef]
- Mackie, W.; Perez, S.; Rizzo, R.; Taravel, F.; Vignon, M. Aspects of the conformation of polyguluronate in the solid state and in solution. Int. J. Biol. Macromol. 1983, 5, 329–341. [Google Scholar] [CrossRef]
- Astbury, W. Structure of alginic acid. Nature 1945, 155, 667–668. [Google Scholar] [CrossRef]
- Dri, F.L.; Shang, S.; Hector, L.G.; Saxe, P.; Liu, Z.-K.; Moon, R.J.; Zavattieri, P.D. Anisotropy and temperature dependence of structural, thermodynamic, and elastic properties of crystalline cellulose Iβ: A first-principles investigation. Model. Simul. Mater. Sci. Eng. 2014, 22, 085012. [Google Scholar] [CrossRef]
- Robinson, R.A.; Stokes, R.H. Electrolyte Solutions; Dover Publication Inc.: Mineola, NY, USA, 2002. [Google Scholar]
- Bordi, F.; Colby, R.; Cametti, C.; De Lorenzo, L.; Gili, T. Electrical conductivity of polyelectrolyte solutions in the semidilute and concentrated regime: The role of counterion condensation. J Phys. Chem. B 2002, 106, 6887–6893. [Google Scholar] [CrossRef]
- Veis, A. The Macromolecular Chemistry of Gelatin; Academic Press: New York, NY, USA, 1964. [Google Scholar]
- Jiao, W.; Chen, W.; Mei, Y.; Yun, Y.; Wang, B.; Zhong, Q.; Chen, H.; Chen, W. Effects of molecular weight and guluronic acid/mannuronic acid ratio on the rheological behavior and stabilizing property of sodium alginate. Molecules 2019, 24, 4374. [Google Scholar] [CrossRef]
- Dobrynin, A.V.; Colby, R.H.; Rubinstein, M. Scaling theory of polyelectrolyte solutions. Macromolecules 1995, 28, 1859–1871. [Google Scholar] [CrossRef]
- Corey, R.B.; Pauling, L.C. Fundamental dimensions of polypeptide chains. Proc. R. Soc. B 1953, 141, 10–20. [Google Scholar]
- Bright, J.N.; Woolf, T.B.; Hoh, J.H. Predicting properties of intrinsically unstructured proteins. Prog. Biophys. Mol. Biol. 2001, 76, 131–173. [Google Scholar] [CrossRef]
- Masuelli, M.A.; Illanes, C.O. Review of the characterization of sodium alginate by intrinsic viscosity measurements: Comparative analysis between conventional and single point methods. Int. J. Biomat. Sci. Eng. 2014, 1, 1–11. [Google Scholar]
- Eremeeva, T.; Bykova, T. SEC of mono-carboxymethyl cellulose (CMC) in a wide range of pH; Mark–Houwink constants. Carbohydr. Polym. 1998, 36, 319–326. [Google Scholar] [CrossRef]
- Masuelli, M.A. Mark-Houwink parameters for aqueous-soluble polymers and biopolymers at various temperatures. J. Polym. Biopolym. Phys. Chem. 2014, 2, 37–43. [Google Scholar]
- Raja, A.; Wilfert, P.K.; Picken, S.J. Using the Herschel–Bulkley Consistency Index to Characterise Complex Biopolymer Systems—The Effect of Screening. Polymers 2024, 16, 2822. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer Name | Salient Properties |
|---|---|
| Sodium Alginate (Na—Alginate), CAS Number: 9005-38-3 | Polyanion: Carboxyl group, High mannuronic acid content. Linear copolymer. |
| Chitosan, CAS Number: 9012-76-4 | Polycation: Amine group, Linear homopolymer, ~50 to 190 kg/mol. |
| Sodium Carboxy Methyl Cellulose (Na—CMC), CAS Number: 9004-32-4 | Polyanion: Carboxyl group, Degree of Substitution = 0.9, Linearly substituted homopolymer. ~250 kg/mol. |
| Pectin from Citrus Peels (Pectin—Citrus), CAS Number: 9000-69-5 | Polyanion: Carboxyl group, Galacturonic acid ≥ 74.0%, degree of methylation ≥ 6.7%, Branched heteropolymer. |
| Gelatin from Porcine Skin (Porcine—Gelatin), CAS Number: 9000-70-8 | Polyampholyte: Carboxyl group and amine group, Linear—collagen derivative. |
| Solvent | (mPa·s) ± S.D. 1 |
|---|---|
| Water, 298 K—No Added Salt | 0.871 ± 0.005 |
| Water, 298 K—Added Salt | 0.880 ± 0.006 |
| Water, 338 K—Added Salt | 0.489 ± 0.002 |
| Biopolymer System | (m3/kg) ± S.D. 1 |
|---|---|
| Screened: Na-Alginate | 0.308 ± 0.065 |
| Screened: Chitosan | 0.795 ± 0.100 |
| Screened: Gelatin—Porcine, T = 338 K, pH = 6 | 0.102 ± 0.024 |
| Screened: Na-CMC | 1.034 ± 0.037 |
| Screened: Pectin—Citrus | 0.332 ± 0.064 |
| Unscreened: Na-Alginate | 1.183 ± 0.157 |
| Unscreened: Chitosan | 4.636 ± 0.379 |
| Unscreened: Na-CMC | 5.624 ± 0.626 |
| Unscreened: Pectin—Citrus | 0.175 |
| Screened: Gelatin—Porcine, T = 338 K, pH = 4 | 0.095 ± 0.017 |
| Screened: Gelatin—Porcine, T = 338 K, pH = 11 | 0.060 ± 0.017 |
| Biopolymer | Expt. | Expt. | Theory | Theory | |
|---|---|---|---|---|---|
| Sodium Alginate | 3.84 | 1.45 | 3.13–5.93 | 1.14–2.28 1 | 0.83 |
| Chitosan | 5.83 | 2.24 | 4.32–8.93 | 1.65–3.30 1 | 1.00 |
| Sodium Carboxy Methyl Cellulose | 5.44 | 2.09 | 3.58–7.05 | 1.34–2.68 1 | 0.90 |
| Pectin from Citrus Peels | 3.48 | 1.30 | 2.39–4.13 | 0.79–1.57 1 | 0.69 |
| Label | ||
|---|---|---|
| Experiment: Gelatin pH = 6/Gelatin pH = 4 | 1.07 | 0.05 |
| Experiment: Gelatin pH = 6/Gelatin pH = 11 | 1.71 | 0.43 |
| Theory: 0.05 | 1.25 | 0.16 |
| Theory: 0.10 | 2.11 | 0.65 |
| Theory: 0.25 | 11.3 | 4.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raja, A.; Wilfert, P.K.; Picken, S.J. Charge Mediated Changes to the Intrinsic Viscosity of Biopolymer Systems. Polymers 2024, 16, 2894. https://doi.org/10.3390/polym16202894
Raja A, Wilfert PK, Picken SJ. Charge Mediated Changes to the Intrinsic Viscosity of Biopolymer Systems. Polymers. 2024; 16(20):2894. https://doi.org/10.3390/polym16202894
Chicago/Turabian StyleRaja, Anand, Philipp K. Wilfert, and Stephen J. Picken. 2024. "Charge Mediated Changes to the Intrinsic Viscosity of Biopolymer Systems" Polymers 16, no. 20: 2894. https://doi.org/10.3390/polym16202894
APA StyleRaja, A., Wilfert, P. K., & Picken, S. J. (2024). Charge Mediated Changes to the Intrinsic Viscosity of Biopolymer Systems. Polymers, 16(20), 2894. https://doi.org/10.3390/polym16202894