
Poly(3-hydroxybutyrate) Modified with Thermoplastic Polyurethane and Microfibrillated Cellulose: Hydrolytic Degradation and Thermal and Mechanical Properties

 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of the Composites
2.3. Characterization
2.3.1. Thermogravimetric Analysis (TGA)
2.3.2. Differential Scanning Calorimetry (DSC)
2.3.3. Dynamic Mechanical Analysis (DMA)
2.3.4. Scanning Electron Microscopy (SEM) Analysis
2.3.5. X-Ray Diffraction (XRD)
2.3.6. Hydrolytic Degradation Tests
2.3.7. Fourier-Transform Infrared Spectroscopy (FTIR)
3. Results and Discussion
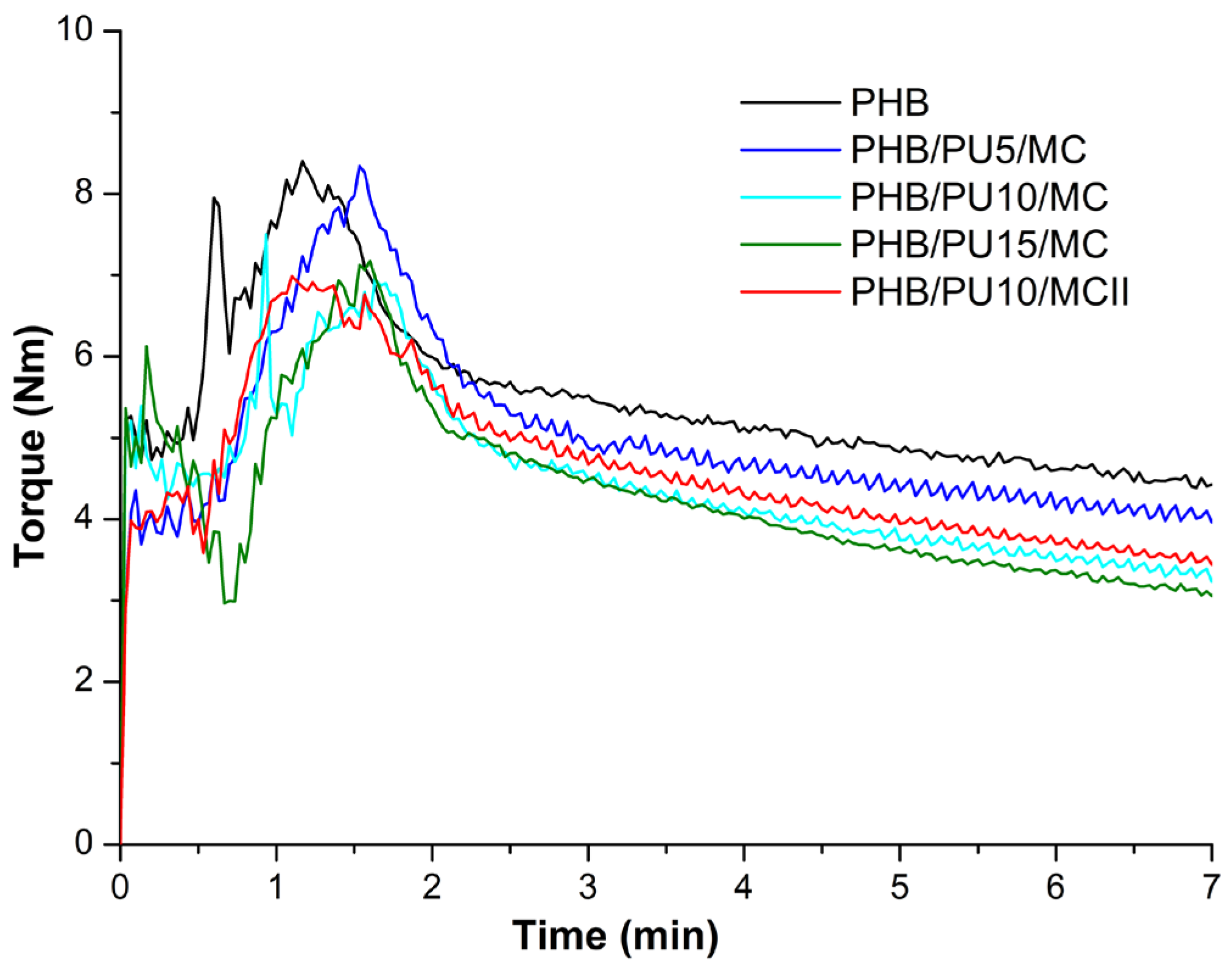
3.1. Melt Rheology
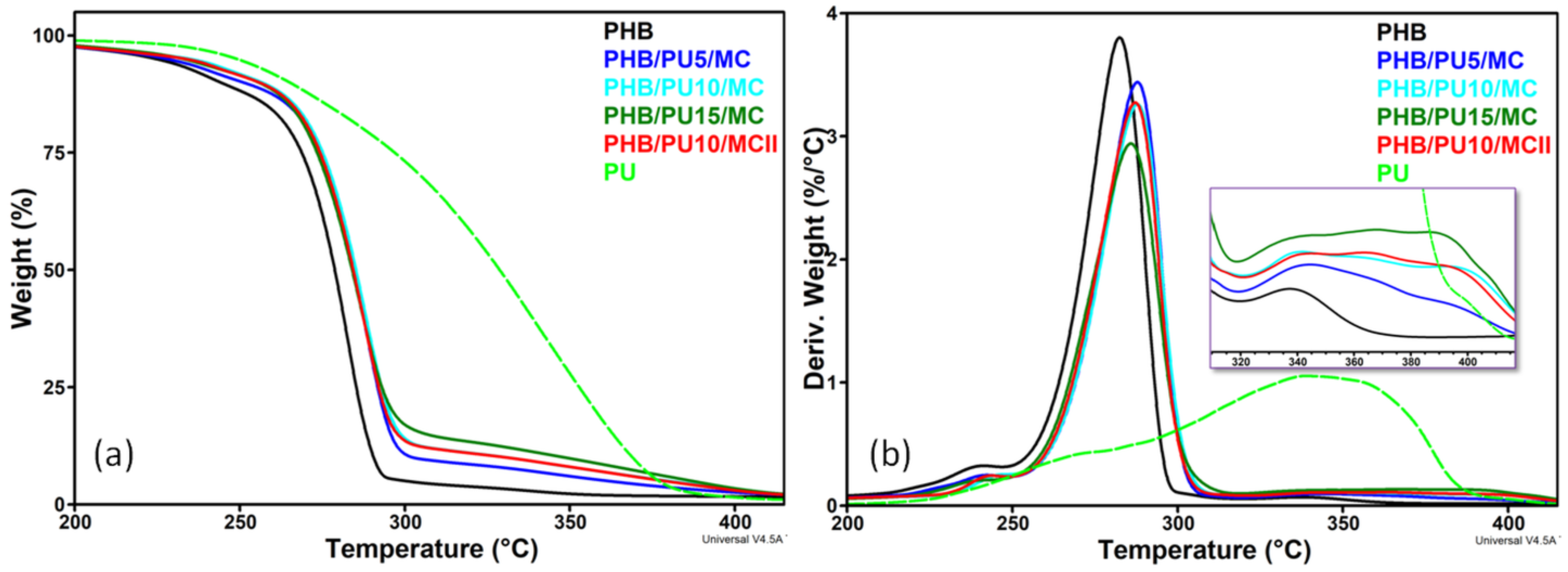
3.2. Thermal Stability of Composites
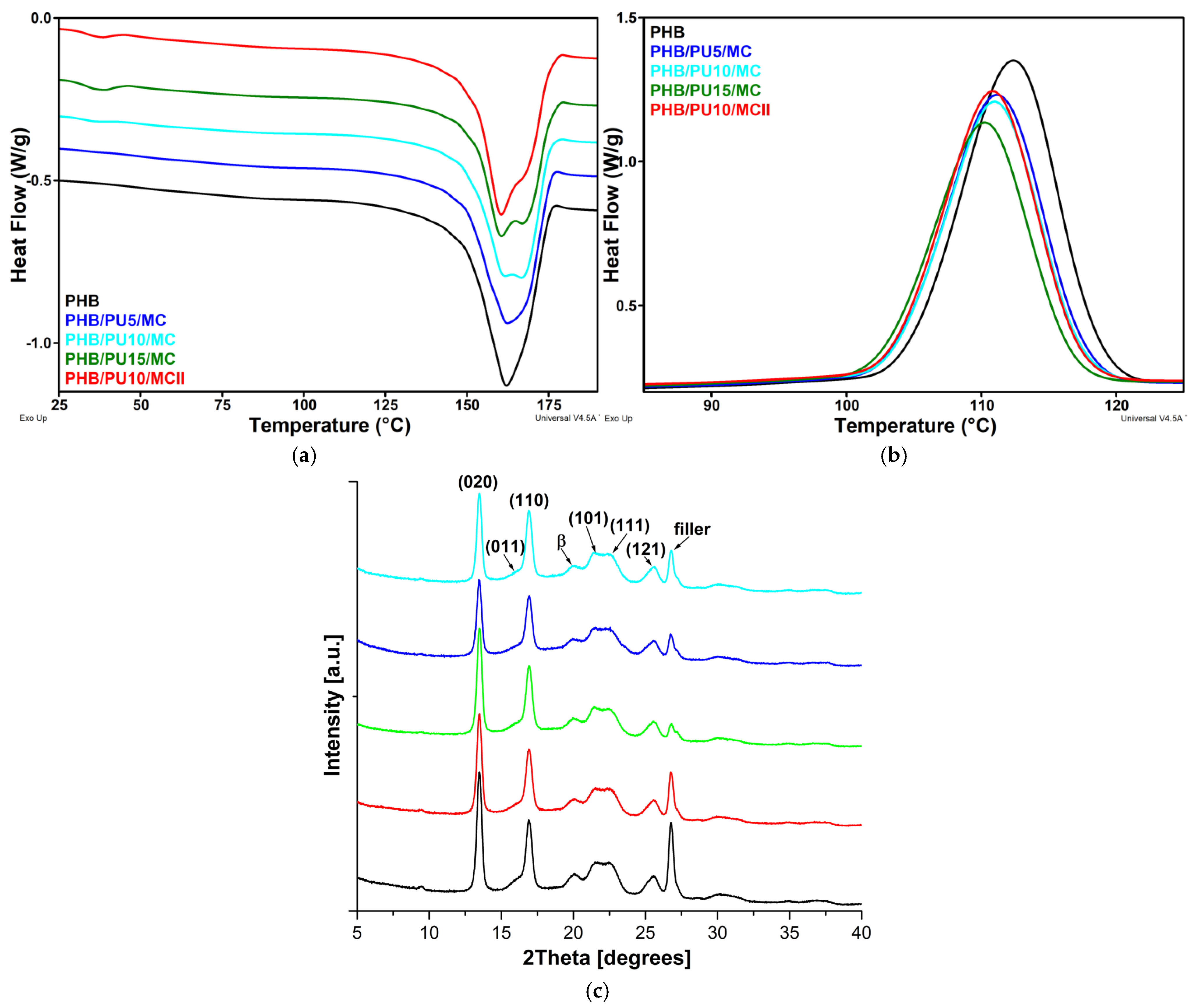
3.3. Melting and Crystallization Behaviors of the Composites
3.4. XRD Analysis of the Composites
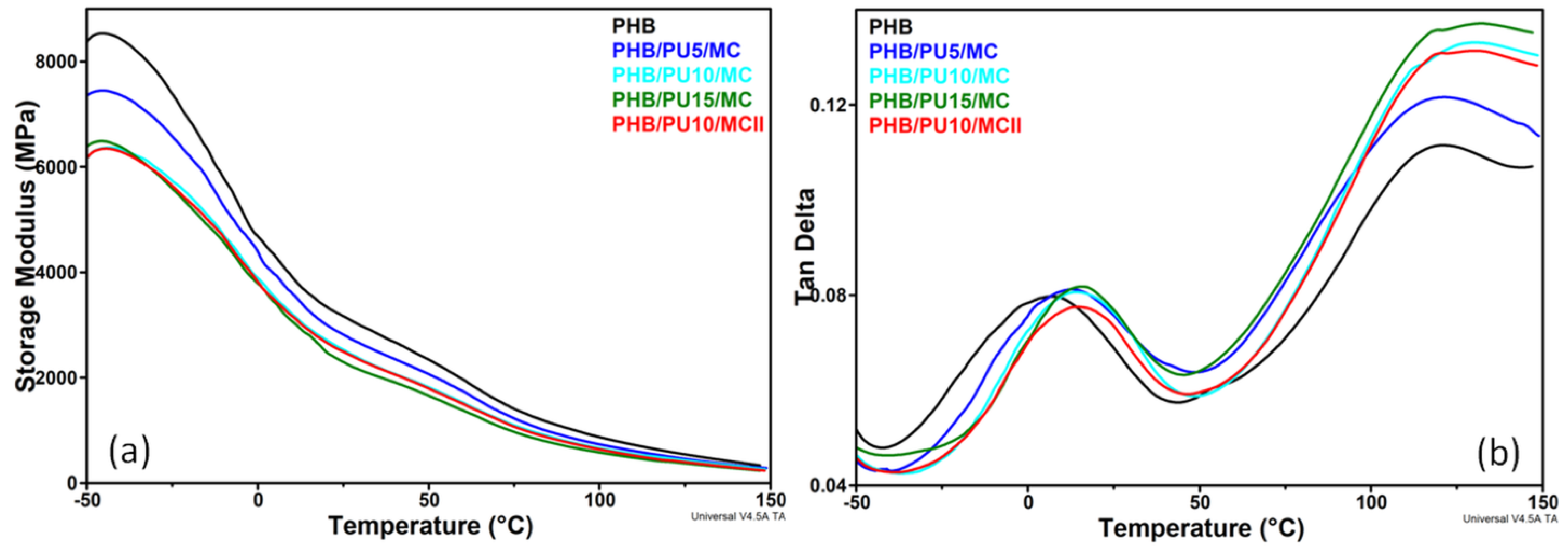
3.5. Dynamic Mechanical Analysis of the Composites
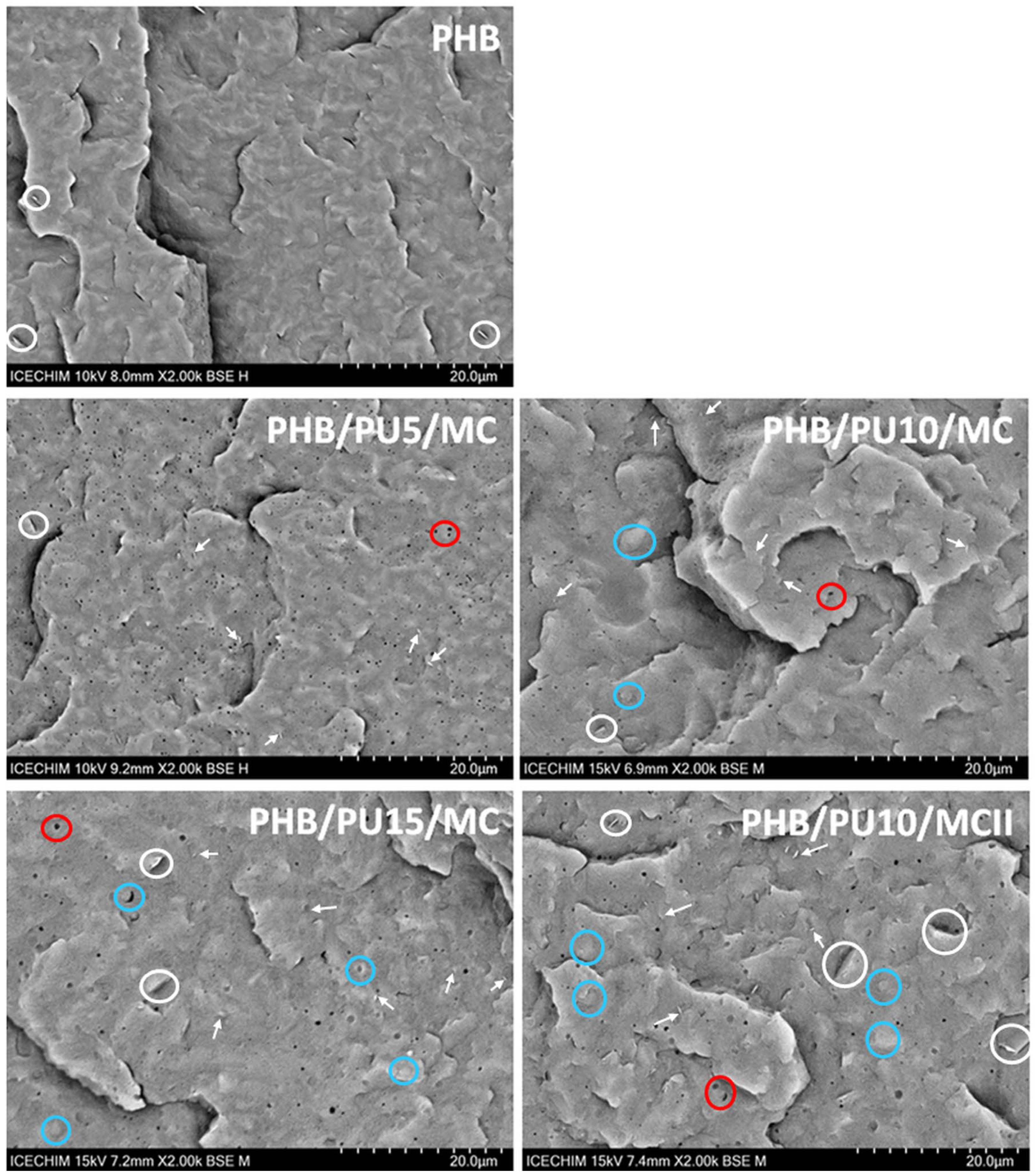
3.6. SEM Analysis of Composites
3.7. Hydrolytic Degradation of the Composites
3.7.1. Mass Loss During Hydrolytic Degradation
3.7.2. Thermal Stability of Degraded Samples
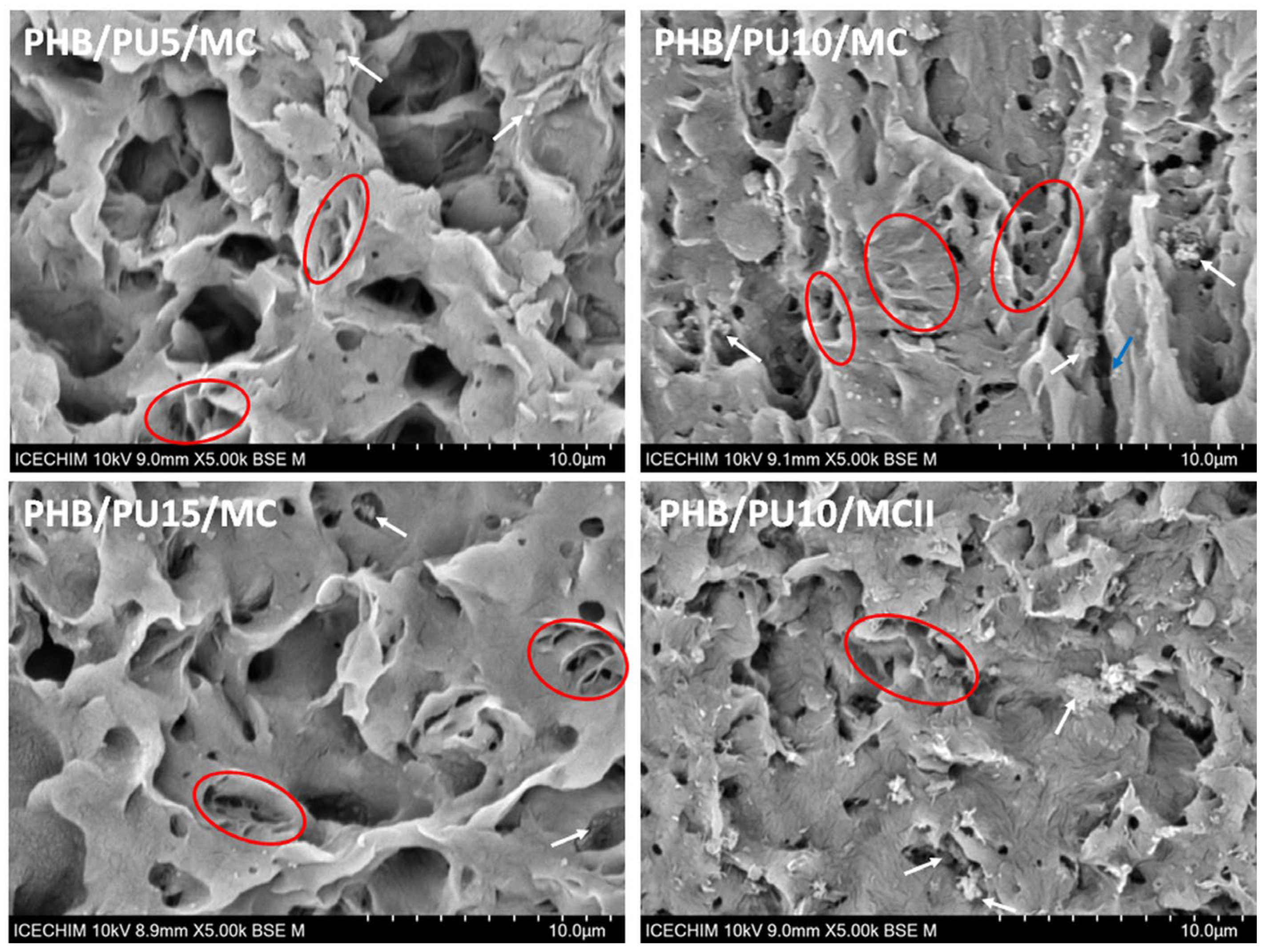
3.7.3. Morphological Analysis of the Degraded Samples
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tan, D.; Wang, Y.; Tong, Y.; Chen, G.-Q. Grand Challenges for Industrializing Polyhydroxyalkanoates (PHAs). Trends Biotechnol. 2021, 39, 953–963. [Google Scholar] [CrossRef]
- Popa, M.S.; Frone, A.N.; Panaitescu, D.M. Polyhydroxybutyrate Blends: A Solution for Biodegradable Packaging? Int. J. Biol. Macromol. 2022, 207, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.A.; Fitzgerald, A.V.L.; Jenkins, M.J. Control of the secondary crystallisation process in poly(hydroxybutyrate-co-hydroxyvalerate) through the incorporation of poly(ethylene glycol). Polym. Degrad. Stab. 2018, 148, 67–74. [Google Scholar] [CrossRef]
- Zembouai, I.; Kaci, M.; Bruzaud, S.; Benhamida, A.; Yves-Marie, C.; Grohens, Y. A Study of Morphological, Thermal, Rheological and Barrier Properties of Poly(3-Hydroxybutyrate-Co-3-Hydroxyvalerate)/Polylactide Blends Prepared by Melt Mixing. Polym. Test. 2013, 32, 842–851. [Google Scholar] [CrossRef]
- Panaitescu, D.M.; Frone, A.N.; Chiulan, I.; Nicolae, C.A.; Trusca, R.; Ghiurea, M.; Gabor, A.R.; Mihailescu, M.; Casarica, A.; Lupescu, I. Role of bacterial cellulose and poly (3-hydroxyhexanoate-co-3-hydroxyoctanoate) in poly (3-hydroxybutyrate) blends and composites. Cellulose 2018, 25, 5569–5591. [Google Scholar] [CrossRef]
- Frone, A.N.; Panaitescu, D.M.; Chiulan, I.; Gabor, A.R.; Nicolae, C.A.; Oprea, M.; Ghiurea, M.; Gavrilescu, D.; Puitel, A.C. Thermal and mechanical behavior of biodegradable polyester films containing cellulose nanofibers. J. Therm. Anal. Calorim. 2019, 138, 2387–2398. [Google Scholar] [CrossRef]
- Parulekar, Y.; Mohanty, A.K. Biodegradable toughened polymers from renewable resources: Blends of polyhydroxybutyrate with epoxidized natural rubber and maleated polybutadiene. Green Chem. 2006, 8, 206–213. [Google Scholar] [CrossRef]
- Wang, S.C.; Xiang, H.X.; Wang, R.L.; Peng, C.; Zhou, Z.; Zhu, M.F. Morphology and properties of renewable poly (3-hydroxybutyrate-co-3-hydroxyvalerate) blends with thermoplastic polyurethane. Polym. Eng. Sci. 2014, 54, 1113–1119. [Google Scholar] [CrossRef]
- Martínez-Abad, A.; González-Ausejo, J.; Lagarón, J.M.; Cabedo, L. Biodegradable poly(3-hydroxybutyrate-co-3-hydroxyvalerate)/thermoplastic polyurethane blends with improved mechanical and barrier performance. Polym. Degrad. Stab. 2016, 132, 52–61. [Google Scholar] [CrossRef]
- Panaitescu, D.M.; Melinte, V.; Frone, A.N.; Nicolae, C.A.; Gabor, A.R.; Capră, L. Influence of Biobased Polyurethane Structure on Thermal and Mechanical Properties of Poly(3-hydroxybutyrate-co-3-hydroxyvalerate)—Polyurethane Blends. J. Polym. Environ. 2023, 31, 1584. [Google Scholar] [CrossRef]
- González-Ausejo, J.; Sánchez-Safont, E.; Cabedo, L.; Gamez-Perez, J. Toughness Enhancement of Commercial Poly (Hydroxybutyrate-co-Valerate) (PHBV) by Blending with a Thermoplastic Polyurethane (TPU). J. Multiscale Model. 2016, 7, 1640008. [Google Scholar] [CrossRef]
- Sánchez-Safont, E.L.; Arrillaga, A.; Anakabe, J.; Gamez-Perez, J.; Cabedo, L. PHBV/TPU/cellulose compounds for compostable injection molded parts with improved thermal and mechanical performance. J. Appl. Polym. Sci. 2019, 136, 47257. [Google Scholar] [CrossRef]
- Delebecq, E.; Pascault, J.; Boutevin, B.; Lyon, F.G. On the Versatility of Urethane/Urea Bonds: Reversibility, Blocked Isocyanate, and Non-isocyanate Polyurethane. Chem. Rev. 2013, 113, 80–118. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; He, J.; Xue, R.; Xu, B.; Qian, X.; Xin, F.; Blank, L.M.; Zhou, J.; Wei, R.; Dong, W.; et al. Biodegradation and Up-Cycling of Polyurethanes: Progress, Challenges, and Prospects. Biotechnol. Adv. 2021, 48, 107730. [Google Scholar] [CrossRef] [PubMed]
- DIN EN 13432:2000-12; Packaging-Requirements for Packaging Recoverable Through Composting and Biodegradation-Test Scheme and Evaluation Criteria for the Final Acceptance of Packaging. German version EN 13432, 2000; Beuth Verlag GmbH: Berlin, Germany, 2000. [CrossRef]
- Magnin, A.; Pollet, E.; Phalip, V.; Avérous, L. Evaluation of biological degradation of polyurethanes. Biotechnol. Adv. 2020, 39, 107457. [Google Scholar] [CrossRef] [PubMed]
- Gogolewski, S.; Walpoth, B.; Rheiner, P. Polyurethane microporous membranes as pericardial substitutes. Colloid Polym. Sci. 1987, 265, 971–977. [Google Scholar] [CrossRef]
- Usurelu, C.D.; Badila, S.; Frone, A.N.; Panaitescu, D.M. Poly(3-hydroxybutyrate) Nanocomposites with Cellulose Nanocrystals. Polymers 2022, 14, 1974. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.; Phipps, J.; Greenwood, R.; Skuse, D.; Zhang, Z.J. The effect of pre-treatment and process conditions on the gas barrier properties of fibrillated cellulose films and coatings: A review. Carbohydr. Polym. 2024, 337, 122085. [Google Scholar] [CrossRef]
- Polyák, P.; Szemerszki, D.; Vörös, G.; Pukánszky, B. Mechanism and kinetics of the hydrolytic degradation of amorphous poly(3-hydroxybutyrate). Polym. Degrad. Stab. 2017, 140, 1–8. [Google Scholar] [CrossRef]
- Barham, P.J.; Keller, A.; Otun, E.L.; Holmer, P.A. Crystallization and morphology of a bacterial thermoplastic: Poly-3-hydroxybutyrate. J. Mater. Sci. 1984, 19, 2781–2794. [Google Scholar] [CrossRef]
- Keridou, I.; Franco, L.; del Valle, L.J.; Martínez, J.C.; Funk, L.; Turon, P.; Puiggalí, J. Hydrolytic and enzymatic degradation of biobased poly(4-hydroxybutyrate) films. Selective etching of spherulites. Polym. Degrad. Stab. 2021, 183, 109451. [Google Scholar] [CrossRef]
- Costa, A.R.M.; Reul, L.T.A.; Sousa, F.M.; Ito, E.N.; Carvalho, L.H.; Canedo, E.L. Degradation during processing of vegetable fiber compounds based on PBAT/PHB blends. Polym. Test. 2018, 69, 266–275. [Google Scholar] [CrossRef]
- Panaitescu, D.M.; Frone, A.N.; Nicolae, C.-A.; Gabor, A.R.; Miu, D.M.; Soare, M.-G.; Vasile, B.S.; Lupescu, I. Poly(3-hydroxybutyrate) nanocomposites modified with even and odd chain length polyhydroxyalkanoates. Int. J. Biol. Macromol. 2023, 244, 125324. [Google Scholar] [CrossRef] [PubMed]
- Gigante, V.; Canesi, I.; Cinelli, P.; Coltelli, M.B.; Lazzeri, A. Rubber toughening of polylactic acid (PLA) with poly(butylene adipate-co-terephthalate) (PBAT): Mechanical properties, fracture mechanics and analysis of ductile-to-brittle behavior while varying temperature and test speed. Eur. Polym. J. 2019, 115, 125–137. [Google Scholar] [CrossRef]
- Togliatti, E.; Lenzi, L.; Degli Esposti, M.; Castellano, M.; Milanese, D.; Sciancalepore, C.; Morselli, D.; Fabbri, P. Enhancing melt-processing and 3D printing suitability of polyhydroxybutyrate through compounding with a bioplasticizer derived from the valorization of levulinic acid and glycerol. Addit. Manuf. 2024, 89, 104290. [Google Scholar] [CrossRef]
- Nerkar, M.; Ramsay, J.A.; Ramsay, B.A.; Kontopoulou, M. Melt compounded blends of short and medium chain-length poly-3-hydroxyalkanoates. J. Polym. Environ. 2014, 22, 236–243. [Google Scholar] [CrossRef]
- Dorez, G.; Taguet, A.; Ferry, L.; Lopez-Cuesta, J.M. Thermal and fire behavior of natural fibers/PBS biocomposites. Polym. Degrad. Stab. 2013, 98, 87–95. [Google Scholar] [CrossRef]
- Panaitescu, D.M.; Nicolae, C.A.; Gabor, A.R.; Trusca, R. Thermal and mechanical properties of poly(3-hydroxybutyrate) reinforced with cellulose fibers from wood waste. Ind. Crops Prod. 2020, 145, 112071. [Google Scholar] [CrossRef]
- Yu, H.-Y.; Qin, Z.-Y.; Liu, Y.-N.; Chen, L.; Liu, N.; Zhou, Z. Simultaneous improvement of mechanical properties and thermal stability of bacterial polyester by cellulose nanocrystals. Carbohydr. Polym. 2012, 89, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Xiong, R.; Hameed, N.; Guo, Q. Cellulose/polycaprolactone blends regenerated from ionic liquid 1-butyl-3-methylimidazolium chloride. Carbohydr. Polym. 2012, 90, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Panaitescu, D.M.; Nicolae, C.A.; Melinte, V.; Scutaru, A.L.; Gabor, A.R.; Popa, M.S.; Oprea, M.; Buruiana, T. Influence of microfibrillated cellulose and soft biocomponent on the morphology and thermal properties of thermoplastic polyurethanes. J. Appl. Polym. Sci. 2021, 138, 50951. [Google Scholar] [CrossRef]
- Rodrigues, J.A.F.R.; Parra, D.F.; Lugao, A.B. Crystallization on films of PHB/PEG blends. J Therm. Anal. Calorim. 2005, 79, 379–381. [Google Scholar] [CrossRef]
- Frone, A.N.; Nicolae, C.A.; Eremia, M.C.; Tofan, V.; Ghiurea, M.; Chiulan, I.; Radu, E.; Damian, C.M.; Panaitescu, D.M. Low molecular weight and polymeric modifiers as toughening agents in poly(3-hydroxybutyrate) films. Polymers 2020, 12, 2446. [Google Scholar] [CrossRef] [PubMed]
- Nguyen Tri, P.; Prud’homme, R.E. Nanoscale Lamellar Assembly and Segregation Mechanism of Poly(3-hydroxybutyrate)/Poly(ethylene glycol) Blends. Macromolecules 2018, 51, 181–188. [Google Scholar] [CrossRef]
- You, J.-W.; Chiu, H.-J.; Don, T.-M. Spherulitic morphology and crystallization kinetics of melt-miscible blends of poly(3-hydroxybutyrate) with low molecular weight poly(ethylene oxide). Polymer 2003, 44, 4355–4362. [Google Scholar] [CrossRef]
- Valle Iulianelli, G.C.; David, G.D.S.; dos Santos, T.N.; Sebastião, P.J.O.; Tavares, M.I.B. Influence of TiO2 nanoparticle on the thermal, morphological and molecular characteristics of PHB matrix. Polym. Test. 2018, 65, 156–162. [Google Scholar] [CrossRef]
- Luo, H.; Shi, Z.M.; Wang, H.; Wang, W. The microstructure, phase transformation and sinterability of desert sand. Mater. Today Commun. 2023, 35, 105685. [Google Scholar] [CrossRef]
- Wojdyr, M. Fityk: A general-purpose peak fitting program. J. Appl. Crystallogr. 2010, 43, 1126–1128. [Google Scholar] [CrossRef]
- Patterson, A.L. The Scherrer formula for X-ray particle size determination. Phys. Rev. 1939, 56, 978–982. [Google Scholar] [CrossRef]
- Benetti, E.M.; Causin, V.; Marega, C.; Marigo, A.; Ferrara, G.; Ferraro, A.; Consalvi, M.; Fantinel, F. Morphological and structural characterization of polypropylene based nanocomposites. Polymer 2005, 46, 8275–8285. [Google Scholar] [CrossRef]
- Tang, X.; Yang, W.; Bao, R.; Shan, G.-F.; Xie, B.-H.; Yang, M.-B.; Hou, M. Effect of spatial confinement on the development of β-phase of polypropylene. Polymer 2009, 50, 4122–4127. [Google Scholar] [CrossRef]
- Brandolt, S.D.F.; Daitx, T.S.; Mauler, R.S.; Ornaghi Junior, H.L.; Crespo, J.S.; Carli, L.N. Synergistic effect between different clays and plasticizer on the properties of PHBV nanocomposites. Polym. Compos. 2019, 40, 3835–3843. [Google Scholar] [CrossRef]
- Ropers, S.; Kardos, M.; Osswald, T.A. A thermo-viscoelastic approach for the characterization and modeling of the bending behavior of thermoplastic composites. Compos.-A Appl. Sci. Manuf. 2016, 90, 22–32. [Google Scholar] [CrossRef]
- Gracia-Fernández, C.A.; Gómez-Barreiro, S.; López-Beceiro, J.; Tarrío Saavedra, J.; Naya, S.; Artiaga, R. Comparative study of the dynamic glass transition temperature by DMA and TMDSC. Polym. Test. 2010, 29, 1002–1006. [Google Scholar] [CrossRef]
- Patra, N.; Ramesh, P.; Ramanakumar, N. Oleic acid-induced changes in the dynamic viscoelastic behavior of poly(methyl methacrylate) polymer film. Colloids Surf. A 2024, 701, 134889. [Google Scholar] [CrossRef]
- Esmizadeh, E.; Gupta, A.; Asrat, S.; Mekonnen, T.H. Crystallization and performance evolution of PHBV nanocomposites through annealing: The role of surface modification of CNCs. Polymer 2024, 308, 127352. [Google Scholar] [CrossRef]
- Feijoo, P.; Samaniego-Aguilar, K.; Sánchez-Safont, E.; Torres-Giner, S.; Lagaron, J.M.; Gamez-Perez, J.; Cabedo, L. Development and characterization of fully renewable and biodegradable polyhydroxyalkanoate blends with improved thermoformability. Polymers 2022, 14, 2527. [Google Scholar] [CrossRef] [PubMed]
- Kovalcik, A.; Sangroniz, L.; Kalina, M.; Skopalova, K.; Humpolíček, P.; Omastova, M.; Mundigler, N.; Müller, A.J. Properties of scaffolds prepared by fused deposition modeling of poly(hydroxyalkanoates). Int. J. Biol. Macromol. 2020, 161, 364–376. [Google Scholar] [CrossRef]
- Silva, R.D.N.; Da Silva, L.R.C.; De Morais, A.C.L.; Alves, T.S.; Barbosa, R. Study of the hydrolytic degradation of poly-3-hydroxybutyrate in the development of blends and polymeric bionanocomposites. J. Thermoplast. Compos. Mater. 2021, 34, 884–901. [Google Scholar] [CrossRef]
- Parodi, A.; D’Ambrosio, M.; Mazzocchetti, L.; Martinez, G.A.; Samorì, C.; Torri, C.; Galletti, P. Chemical recycling of polyhydroxybutyrate (PHB) into bio-based solvents and their use in a circular PHB extraction. ACS Sustain. Chem. Eng. 2021, 9, 12575–12583. [Google Scholar] [CrossRef]
- Tarazona, N.A.; Machatschek, R.; Lendlein, A. Unraveling the interplay between abiotic hydrolytic degradation and crystallization of bacterial polyesters comprising short and medium side-chain-length polyhydroxyalkanoates. Biomacromolecules 2020, 21, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Foster, L.J.R.; Tighe, B.J. Centrifugally spun polyhydroxybutyrate fibres: Accelerated hydrolytic degradation studies. Polym. Degrad. Stab. 2005, 87, 1–10. [Google Scholar] [CrossRef]
- Polyak, P.; Dohovits, E.; Nagy, G.N.; Vertessy, B.G.; Voros, G.; Pukanszky, B. Enzymatic degradation of poly-[(R)-3-hydroxybutyrate]: Mechanism, kinetics, consequences. Int. J. Biol. Macromol. 2018, 112, 156–162. [Google Scholar] [CrossRef]
- Deroiné, M.; Le Duigou, A.; Corre, Y.-M.; Le Gac, P.-Y.; Davies, P.; César, G.; Bruzaud, S. Accelerated ageing and lifetime prediction of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) in distilled water. Polym. Test. 2014, 39, 70–78. [Google Scholar] [CrossRef]
- Saeki, T.; Tsukegi, T.; Tsuji, H.; Daimon, H.; Fujie, K. Hydrolytic degradation of poly[(R)-3-hydroxybutyric acid] in the melt. Polymer 2005, 46, 2157–2162. [Google Scholar] [CrossRef]
- Laycock, B.; Nikolić, M.; Colwell, J.M.; Gauthier, E.; Halley, P.; Bottle, S.; George, G. Lifetime prediction of biodegradable polymers. Prog. Polym. Sci. 2017, 71, 144–189. [Google Scholar] [CrossRef]
- Hernández, A.R.; Contreras, O.C.; Acevedo, J.C.; Moreno, L.G.N. Poly (ε-caprolactone) degradation under acidic and alkaline conditions. Am. J. Polym. Sci 2013, 3, 70–75. [Google Scholar] [CrossRef]
- Gil-Castell, O.; Badia, J.D.; Bou, J.; Ribes-Greus, A. Performance of polyester-based electrospun scaffolds under in vitro hydrolytic conditions: From short-term to long-term applications. Nanomaterials 2019, 9, 786. [Google Scholar] [CrossRef]
- Kupka, V.; Vojtova, L.; Fohlerova, Z.; Jancar, J. Solvent free synthesis and structural evaluation of polyurethane films based on poly(ethylene glycol) and poly(caprolactone). Express Polym. Lett. 2016, 10, 479–492. [Google Scholar] [CrossRef]
- Bartnikowski, M.; Dargaville, T.R.; Ivanovski, S.; Hutmacher, D.W. Degradation mechanisms of polycaprolactone in the context of chemistry, geometry and environment. Prog. Polym. Sci. 2019, 96, 1–20. [Google Scholar] [CrossRef]
- Zouari, M.; Devallance, D.B.; Marrot, L. Effect of biochar addition on mechanical properties, thermal stability, and water resistance of hemp-polylactic acid (PLA) composites. Materials 2022, 15, 2271. [Google Scholar] [CrossRef]
- Kaneko, J.J.; Harvey, W.J.; Bruss, M.L. Clinical Biochemistry of Domestic Animals, 6th ed.; Elsevier Academic Press: San Diego, CA, USA, 2008; ISBN 978-0-12-370491-7. [Google Scholar]
- Slaninova, E.; Obruca, S.; Kocherbitov, V.; Sedlacek, P. On the bioprotective effects of 3-hydroxybutyrate: Thermodynamic study of binary 3HB-water systems. Biophys. J. 2023, 122, 460–469. [Google Scholar] [CrossRef]
- Valapa, R.B.; Pugazhenthi, G.; Katiyar, V. Hydrolytic degradation behaviour of sucrose palmitate reinforced poly(lactic acid) nanocomposites. Int. J. Biol. Macromol. 2016, 89, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Tapadiya, A.; Vasanthan, N. Crystallization and alkaline hydrolysis of poly(3-hydroxybutyrate) films probed by thermal analysis and infrared spectroscopy. Int. J. Biol. Macromol. 2017, 102, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- Myung, J.; Strong, N.I.; Galega, W.M.; Sundstrom, E.R.; Flanagan, J.C.; Woo, S.G.; Waymouth, R.M.; Criddle, C.S. Disassembly and reassembly of polyhydroxyalkanoates: Recycling through abiotic depolymerization and biotic repolymerization. Bioresour. Technol. 2014, 170, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Barrera-Rivera, K.A.; Peponi, L.; Marcos-Fernández, Á.; Kenny, J.M.; Martínez-Richa, A. Synthesis, Characterization and Hydrolytic Degradation of Polyester-Urethanes Obtained by Lipase Biocatalysis. Polym. Degrad. Stab. 2014, 108, 188–194. [Google Scholar] [CrossRef]
- Mondal, S.; Martin, D. Hydrolytic Degradation of Segmented Polyurethane Copolymers for Biomedical Applications. Polym. Degrad. Stab. 2012, 97, 1553–1561. [Google Scholar] [CrossRef]
- Hong, J.H.; Jeon, H.J.; Yoo, J.H.; Yu, W.-R.; Youk, J.H. Synthesis and characterization of biodegradable poly(ɛ-caprolactone-co-β-butyrolactone)-based polyurethane. Polym. Degrad. Stabil. 2007, 92, 1186–1192. [Google Scholar] [CrossRef]
- Swensson, B.; Lages, S.; Berke, B.; Larsson, A.; Hasani, M. Scattering studies of the size and structure of cellulose dissolved in aqueous hydroxide base solvent. Carbohydr. Polym. 2021, 274, 118634. [Google Scholar] [CrossRef] [PubMed]
- Kamide, K.; Okajima, K.; Kowsaka, K. Dissolution of Natural Cellulose into Aqueous Alkali Solution: Role of Super-Molecular Structure of Cellulose. Polym. J. 1992, 24, 71–86. [Google Scholar] [CrossRef]
- Liu, L.-J.; Wang, X.-L.; Gu, Z.-Y.; Ren, X.-L.; Chang, T. Rapid hydrolysis of waste polyurethane and facile system separation. Sep. Purif. Technol. 2025, 354 Pt 4, 129068. [Google Scholar] [CrossRef]
- Yu, J.; Plackett, D.; Chen, L.X.L. Kinetics and mechanism of the monomeric products from abiotic hydrolysis of poly[(R)-3-hydroxybutyrate] under acidic and alkaline conditions. Polym. Degrad. Stab. 2005, 89, 289–299. [Google Scholar] [CrossRef]
- Zhijiang, C.; Chengwei, H.; Guang, Y. Crystallization behavior, thermal property and biodegradation of poly(3-hydroxybutyrate)/poly(ethylene glycol) grafting copolymer. Polym. Degrad. Stab. 2011, 96, 1602–1609. [Google Scholar] [CrossRef]
- Vaid, R.; Yildirim, E.; Pasquinelli, M.A.; King, M.W. Hydrolytic Degradation of Polylactic Acid Fibers as a Function of pH and Exposure Time. Molecules 2021, 26, 7554. [Google Scholar] [CrossRef]
- Kučera, F.; Petruš, J.; Jančář, J. The structure-hydrolysis relationship of poly(3-hydroxybutyrate). Polym. Test. 2019, 80, 106095. [Google Scholar] [CrossRef]
- Mousavioun, P.; George, G.A.; Doherty, W.O.S. Environmental degradation of lignin/poly(hydroxybutyrate) blends. Polym. Degrad. Stab. 2012, 97, 1114–1122. [Google Scholar] [CrossRef]
- Lee, M.-S.; Woo, E.M. Systematic probing into periodic lamellar assembly via induced cracks in crystallized polyesters. Polymer 2019, 166, 88–97. [Google Scholar] [CrossRef]
- Wu, C.-N.; Woo, E.M.; Nagarajan, S. Periodic crystal assembly of Poly(3-hydroxybutyric acid-co-3-hydroxyvaleric acid): From surface to interior microstructure. Polymer 2021, 228, 123866. [Google Scholar] [CrossRef]
- Wu, X.; Li, M.; Chen, X.; Xiong, P.; Gao, H.; Li, H.; Zhao, S.; Wang, Z.; Wang, Z. Fluorine-free liquid repellent coatings with fully self-healing and robust adhesion abilities. ACS Sustain. Chem. Eng. 2024, 12, 14319–14330. [Google Scholar] [CrossRef]
- Xiao, X.; Wang, J.; Cai, D.; Lou, L.; Xiao, F. A novel application of thermoplastic polyurethane/waste rubber powder blend for waterproof seal layer in high-speed railway. Transp. Geotech. 2021, 27, 100503. [Google Scholar] [CrossRef]
- Seoane, I.T.; Cerrutti, P.; Vazquez, A.; Manfredi, L.B.; Cyras, V.P. Polyhydroxybutyrate-based nanocomposites with cellulose nanocrystals and bacterial cellulose. J. Polym. Environ. 2017, 25, 586–598. [Google Scholar] [CrossRef]
- Mathew, A.P.; Oksman, K.; Sain, M. Mechanical properties of biodegradable composites from poly lactic acid (PLA) and microcrystalline cellulose (MCC). J. Appl. Polym. Sci. 2005, 97, 2014–2025. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composites | PHB | Masterbatch (PU/MC = 5/2) | PU | MC |
|---|---|---|---|---|
| PHB | 100 | - | - | - |
| PHB/PU5/MC | 93 | 7 | - | - |
| PHB/PU10/MC | 88 | 7 | 5 | - |
| PHB/PU15/MC | 83 | 7 | 10 | - |
| PHB/PU10/MCII * | 88 | - | 10 | 2 |
| Composites | T10% °C | Tmax °C | R700 % | TgPHB °C | TmPCL °C | Tm °C | Tc °C | XC % |
|---|---|---|---|---|---|---|---|---|
| PHB | 244.1 | 282.5 | 0.9 | −13.4 | - | 162.1 | 112.4 | 41.2 |
| PHB/PU5/MC | 251.0 | 287.7 | 1.1 | −13.2 | - | 162.4 | 111.1 | 40.2 |
| PHB/PU10/MC | 256.6 | 287.7 | 1.0 | −12.6 | 35.7 | 161.7 | 111.0 | 39.1 |
| PHB/PU15/MC | 255.6 | 285.8 | 1.0 | −8.9 | 38.1 | 160.6 | 110.2 | 39.3 |
| PHB/PU10/MCII | 255.8 | 287.1 | 1.0 | −11.1 | 37.9 | 160.5 | 110.9 | 41.5 |
| Composites | Miller Indices | β (Radian) | D (nm) | XXRD (%) |
|---|---|---|---|---|
| PHB | (020) (110) | 0.00680 0.00850 | 20.5 16.5 | 71.0 |
| PHB/PU5/MC | (020) (110) | 0.00668 0.00830 | 20.9 16.9 | 66.5 |
| PHB/PU10/MC | (020) (110) | 0.00660 0.00824 | 21.2 17.0 | 62.6 |
| PHB/PU15/MC | (020) (110) | 0.00660 0.00811 | 21.2 17.3 | 56.7 |
| PHB/PU10/MCII | (020) (110) | 0.00660 0.00823 | 21.2 17.0 | 63.1 |
| Samples | Tg, °C | tan δ | E′−25°C, MPa | E′25°C, MPa | E′100°C, MPa |
|---|---|---|---|---|---|
| PHB | 6.3 | 0.080 | 7388 | 3165 | 871 |
| PHB/PU5/MC | 10.6 | 0.081 | 6577 | 2816 | 734 |
| PHB/PU10/MC | 12.8 | 0.081 | 5735 | 2528 | 662 |
| PHB/PU15/MC | 15.2 | 0.082 | 5660 | 2512 | 659 |
| PHB/PU10/MCII | 14.5 | 0.078 | 5592 | 2296 | 582 |
| Composites | T10% °C | Tmax °C | R700 % |
|---|---|---|---|
| PHB/PU5/MC | 191.3 | 214.7 | 1.37 |
| PHB/PU10/MC | 191.4 | 211.6 | 1.51 |
| PHB/PU15/MC | 190.4 | 210.8 | 1.60 |
| PHB/PU10/MCII | 198.1 | 220.6 | 1.21 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frone, A.N.; Panaitescu, D.M.; Gabor, A.R.; Nicolae, C.-A.; Ghiurea, M.; Bradu, C. Poly(3-hydroxybutyrate) Modified with Thermoplastic Polyurethane and Microfibrillated Cellulose: Hydrolytic Degradation and Thermal and Mechanical Properties. Polymers 2024, 16, 3606. https://doi.org/10.3390/polym16243606
Frone AN, Panaitescu DM, Gabor AR, Nicolae C-A, Ghiurea M, Bradu C. Poly(3-hydroxybutyrate) Modified with Thermoplastic Polyurethane and Microfibrillated Cellulose: Hydrolytic Degradation and Thermal and Mechanical Properties. Polymers. 2024; 16(24):3606. https://doi.org/10.3390/polym16243606
Chicago/Turabian StyleFrone, Adriana Nicoleta, Denis Mihaela Panaitescu, Augusta Raluca Gabor, Cristian-Andi Nicolae, Marius Ghiurea, and Corina Bradu. 2024. "Poly(3-hydroxybutyrate) Modified with Thermoplastic Polyurethane and Microfibrillated Cellulose: Hydrolytic Degradation and Thermal and Mechanical Properties" Polymers 16, no. 24: 3606. https://doi.org/10.3390/polym16243606
APA StyleFrone, A. N., Panaitescu, D. M., Gabor, A. R., Nicolae, C.-A., Ghiurea, M., & Bradu, C. (2024). Poly(3-hydroxybutyrate) Modified with Thermoplastic Polyurethane and Microfibrillated Cellulose: Hydrolytic Degradation and Thermal and Mechanical Properties. Polymers, 16(24), 3606. https://doi.org/10.3390/polym16243606