


Current Positron Studies on the Modifications of the Molecular Packing in Green-Based Polymers Through Changes in the Synthesis Procedures or Environmental Conditions



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
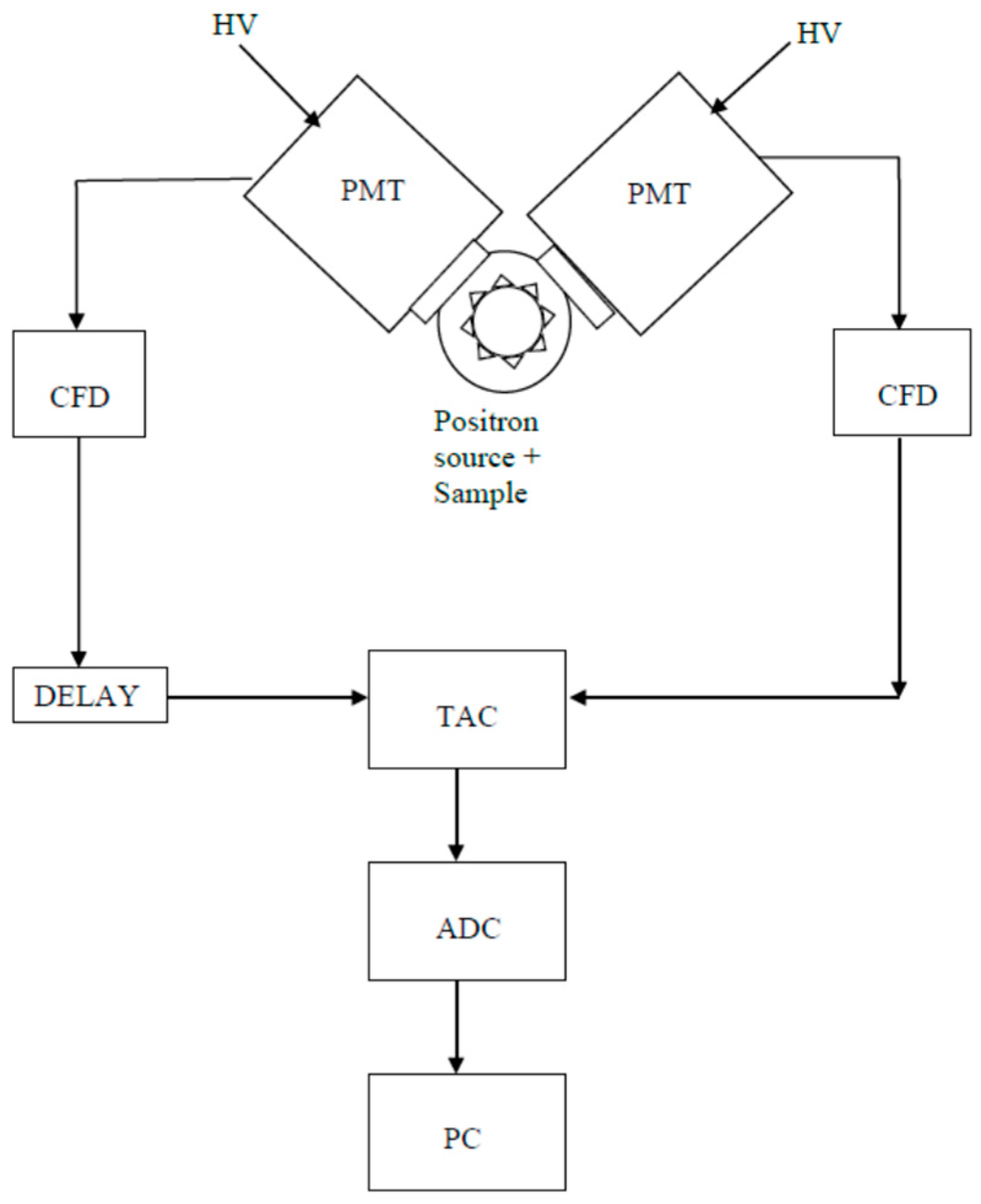
2. Experimental Setup
3. Results
3.1. Starch-Derived Systems
3.1.1. Maltodextrin-Based Matrices
3.1.2. Starch-Sucrose Blends
3.1.3. Thermoplastic Starch-Based Films
3.2. Chitosan
3.2.1. Pure Chitosan
3.2.2. Natural Chitosan-Based Polymers
3.2.3. Synthetic Chitosan-Based Polymers
3.3. Plant Oil-Based Polymers
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Hautojarvi, P. (Ed.) Positrons in Solids; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 1979. [Google Scholar] [CrossRef]
- Brandt, W.; Dupasquier, A. (Eds.) Positron Solid-State Physics; North-Holland: Amsterdam, The Netherlands; New York, NY, USA; Oxford, UK, 1983; ISBN 9780444865212. [Google Scholar]
- Salgueiro, W.; Somoza, A.; Cabrera, O.; Consolati, G. Porosity study on free mineral addition cement paste. Cem. Concr. Res. 2004, 34, 91–97. [Google Scholar] [CrossRef]
- Dupasquier, A.; Kögel, G.; Somoza, A. Studies of light alloys by positron annihilation techniques. Acta Mater. 2004, 52, 4707–4726. [Google Scholar] [CrossRef]
- Jean, Y. Positron annihilation spectroscopy for chemical analysis: A novel probe for microstructural analysis of polymers. Microchem. J. 1990, 42, 72–102. [Google Scholar] [CrossRef]
- Jean, Y.C. Positron annihilation in polymers. Mater. Sci. Forum 1995, 175–178, 59–70. [Google Scholar] [CrossRef]
- Dlubek, G.; Fretwell, H.M.; Alam, M.A. Positron/Positronium Annihilation as a Probe for the Chemical Environment of Free Volume Holes in Polymers. Macromolecules 2000, 33, 187–192. [Google Scholar] [CrossRef]
- Consolati, G.; Nichetti, D.; Quasso, F. Probing the free volume in polymers by means of positron annihilation lifetime spectroscopy. Polymers 2023, 15, 3128. [Google Scholar] [CrossRef]
- Mallon, P.E. Principles and Applications of Positron and Positronium Chemistry; Jean, Y.C., Mallon, P.E., Schrader, D.M., Eds.; World-Scientific: London, UK, 2003; pp. 253–280. ISBN 981-238-144-9. [Google Scholar]
- Saarinen, K.; Hautojärvi, P.; Corbel, C. Positron Annihilation Spectroscopy of Defects in Semiconductors. Semicond. Semimet. 1998, 51, 209–285. [Google Scholar] [CrossRef]
- Krause-Rehberg, R.; Leipner, H.S. Positron Annihilation in Semiconductors; Springer: Berlin/Heidelberg, Germany, 1999; ISBN 3-540-64371-0. [Google Scholar]
- Berko, S.; Pendleton, H.N. Positronium. Annu. Rev. Nucl. Part. Sci. 1980, 30, 543–581. [Google Scholar] [CrossRef]
- Tao, S.J. Positronium annihilation in molecular substances. J. Chem. Phys. 1972, 56, 5499–5510. [Google Scholar] [CrossRef]
- Eldrup, M.; Lightbody, D.; Sherwood, N.J. The temperature dependence of positron lifetimes in solid pivalic acid. Chem. Phys. 1981, 63, 51–58. [Google Scholar] [CrossRef]
- Nakanishi, H.; Jean, Y.C. Positrons and positronium in liquids. In Positron and Positronium Chemistry; Schrader, D.M., Jean, Y.C., Eds.; Elsevier: Amsterdam, The Netherlands, 1988; p. 159. [Google Scholar]
- Zaleski, R. Principles of positron porosimetry. Nukleonika 2015, 60, 795–800. [Google Scholar] [CrossRef]
- Gidley, D.W.; Frieze, W.E.; Dull, T.L.; Sun, J.; Yee, A.F.; Nguyen, C.V.; Yoon, D.Y. Determination of pore-size distribution in low-dielectric thin films. Appl. Phys. Lett. 2000, 76, 1282–1284. [Google Scholar] [CrossRef]
- Ciesielski, K.; Dawidowicz, A.L.; Goworek, T.; Jasińska, B.; Wawryszczuk, J. Positronium lifetimes in porous vycor glasses. Chem. Phys. Lett. 1998, 289, 41–45. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Zheng, W.; Meyer, E.F.; McGervey, J.D.; Jamieson, A.M.; Simha, R. Free volume and physical aging of poly(vinyl acetate) studied by positron annihilation. Macromolecules 1989, 22, 2302–2306. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Nakanishi, H.; Jean, Y.C.; Sandreczki, T.C. Positron annihilation in amine-cured epoxy polymers—Pressure dependence. J. Polym. Sci. Part B Polym. Phys. 1990, 28, 1431–1441. [Google Scholar] [CrossRef]
- Shantarovich, V.P. On the role of free volume in pick-off annihilation and positronium chemical reactions. J. Radioanal. Nucl. Chem. 1996, 210, 357–369. [Google Scholar] [CrossRef]
- Wang, C.L.; Hirade, T.; Maurer, F.J.H.; Eldrup, M.; Pedersen, N.J. Free-volume distribution and positronium formation in amorphous polymers: Temperature and positron-irradiation-time dependence. J. Chem. Phys. 1998, 108, 4654. [Google Scholar] [CrossRef]
- Mogensen, O.E. Positron Annihilation in Chemistry; Springer Series in Chemical Physics; Goldanskii, V.I., Ed.; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 1995; Volume 58. [Google Scholar]
- Hirade, T.; Maurer, F.H.; Eldrup, M. Positronium formation at low temperatures: The role of trapped electrons. Radiat. Phys. Chem. 2000, 58, 465. [Google Scholar] [CrossRef]
- Shantarovich, V.P.; Hirade, T.; Kevdina, I.B.; Gustov, V.W.; Oleinik, E.F. Comments on the effect of γ-irradiation on positronium formation in polymers at low temperatures. Acta Phys. Pol. A 2001, 99, 497–501. [Google Scholar] [CrossRef]
- Shantarovich, V.P. Positron annihilation and free volume studies in polymer glasses. J. Polym. Sci. Part B Polym. Phys. 2008, 46, 2485. [Google Scholar] [CrossRef]
- Zhang, H.J.; Sellaiyan, S.; Kakizaki, T.; Uedono, A.; Taniguchi, Y.; Hayashi, K. Effect of Free-Volume Holes on Dynamic Mechanical Properties of Epoxy Resins for Carbon-Fiber-Reinforced Polymers. Macromolecules 2017, 50, 3933–3942. [Google Scholar] [CrossRef]
- Sharma, J.; Tewari, K.; Arya, R.K. Diffusion in polymeric systems–A review on free volume theory. Prog. Org. Coat. 2017, 111, 83–92. [Google Scholar] [CrossRef]
- Merkel, T.C.; Freeman, B.D.; Spontak, R.J.; He, Z.; Pinnau, I.; Meakin, P.; Hill, A.J. Sorption, transport, and structural evidence for enhanced free volume in poly(4-methyl-2-pentyne)/fumed silica nanocomposite membranes. Chem. Mater. 2003, 15, 109–123. [Google Scholar] [CrossRef]
- Odegard, G.M.; Bandyopadhyay, A. Physical aging of epoxy polymers and their composites. J. Polym. Sci. Part B Polym. Phys. 2011, 49, 1695–1716. [Google Scholar] [CrossRef]
- Lamarre, L.; Sung, C.S.P. Studies of physical aging and molecular motion by azochromophoric labels attached to the main chains of amorphous polymers. Macromolecules 1983, 16, 1729–1736. [Google Scholar] [CrossRef]
- Victor, J.G.; Torkelson, J.M. On measuring the distribution of local free volume in glassy polymers by photochromic and fluorescence techniques. Macromolecules 1987, 20, 2241–2250. [Google Scholar] [CrossRef]
- Gregory, R.B. Free-volume and pore size distributions determined by numerical Laplace inversion of positron annihilation lifetime data. J. Appl. Phys. 1991, 70, 4665–4670. [Google Scholar] [CrossRef]
- Shukla, A.; Peter, M.; Hoffmann, L. Analysis of positron lifetime spectra using quantified maximum entropy and a general linear filter. Nucl. Instrum. Methods Phys. Res. Sect. A 1993, 335, 310–317. [Google Scholar] [CrossRef]
- Kansy, J. Microcomputer program for analysis of positron annihilation lifetime spectra. Nucl. Instrum. Methods Phys. Res. Sect. A 1996, 374, 235–244. [Google Scholar] [CrossRef]
- Okada, M. Chemical syntheses of biodegradable polymers. Prog. Polym. Sci. 2002, 27, 87–133. [Google Scholar] [CrossRef]
- Vroman, I.; Tighzert, L. Biodegradable Polymers. Materials 2009, 2, 307–344. [Google Scholar] [CrossRef]
- Doppalapudi, S.; Jain, A.; Khan, W.; Domb, A.J. Biodegradable polymers—An overview. Polym. Adv. Technol. 2014, 25, 427–435. [Google Scholar] [CrossRef]
- Wang, W.; Xue, C.; Mao, X. Chitosan: Structural modification, biological activity and application. Int. J. Biol. Macromol. 2020, 164, 4532–4546. [Google Scholar] [CrossRef]
- Borchert, K.B.L.; Boughanmi, R.; Reis, B.; Zimmermann, P.; Steinbach, C.; Graichen, P.; Svirepa, A.; Schwarz, J.; Boldt, R.; Schwarz, S.; et al. Removal of Lead, Cadmium, and Aluminum Sulfate from Simulated and Real Water with Native and Oxidized Starches. Polysaccharides 2021, 2, 429–453. [Google Scholar] [CrossRef]
- Weißpflog, J.; Steinbach, C.; Schwarz, D.; Schwarz, S. Simultaneous Adsorption of Iron and Sulfate Ions with Biopolymers. In Proceedings of the 2nd World Congress on Civil, Structural, and Environmental Engineering, Barcelona, Spain, 2–4 April 2017. [Google Scholar] [CrossRef]
- Gautam, R.K.; Sharma, S.K.; Mahiya, S.; Chattopadhyaya, M.C. Contamination of Heavy Metals in Aquatic Media: Transport, Toxicity and Technologies for Remediation. In Heavy Metals in Water: Presence, Removal and Safety; Sharma, S., Ed.; The Royal Society of Chemistry: London, UK, 2014; pp. 1–24. [Google Scholar] [CrossRef]
- Lligadas, G.; Ronda, J.C.; Galià, M.; Cádiz, V. Renewable polymeric materials from vegetable oils: A perspective. Mater. Today 2013, 16, 337–343. [Google Scholar] [CrossRef]
- Zhang, C.; Garrison, T.F.; Madbouly, S.A.; Kessler, M.R. Recent advances in vegetable oil-based polymers and their composites. Prog. Polym. Sci. 2017, 71, 91–143. [Google Scholar] [CrossRef]
- Güner, F.S.; Yağcı, Y.; Erciyes, A.T. Polymers from triglyceride oils. Prog. Polym. Sci. 2006, 31, 633–670. [Google Scholar] [CrossRef]
- Wallenberger, F.T.T.; Weston, N.E. Natural Fibers, Plastics and Composites; Springer Science+Business Media: New York, NY, USA, 2004. [Google Scholar] [CrossRef]
- Mosiewicki, M.A.; Aranguren, M.I. A short review on novel biocomposites based on plant oil precursors. Eur. Polym. J. 2013, 44, 1243–1256. [Google Scholar] [CrossRef]
- Coleman, G.P. Experimental techniques in positron spectroscopy. In Principles and Applications of Positron and Positronium Chemistry; Jean, Y.C., Mallon, P.E., Schrader, D.M., Eds.; World Scientific: London, UK, 2003; ISBN 981-238-144-9. [Google Scholar]
- Sharma, S.K.; Pujari, P.K. Role of free volume characteristics of polymer matrix in bulk physical properties of polymer nanocomposites: A review of positron annihilation lifetime studies. Prog. Polym. Sci. 2017, 75, 31–47. [Google Scholar] [CrossRef]
- Saito, H.; Nagashima, Y.; Kurihara, T.; Hyodo, T. A new positron lifetime spectrometer using a fast digital oscilloscope and BaF2 scintillators. Nucl. Instrum. Methods Phys. Res. Sect. A 2002, 487, 612–617. [Google Scholar] [CrossRef]
- Rytsölä, K.; Nissilä, J.; Kokkonen, J.; Laakso, A.; Aavikko, R.; Saarinen, K. Digital measurement of positron lifetime. Appl. Surf. Sci. 2002, 194, 260–263. [Google Scholar] [CrossRef]
- Bečvář, F. Methodology of positron lifetime spectroscopy: Present status and perspectives. Nucl. Instrum. Methods Phys. Res. Sect. B 2007, 261, 871–874. [Google Scholar] [CrossRef]
- Panzarasa, G.; Aghion, S.; Soliveri, G.; Consolati, G.; Ferragut, R. Positron Annihilation Spectroscopy: A New Frontier for Understanding Nanoparticle-Loaded Polymer Brushes. Nanotechnology 2016, 27, 02LT03. [Google Scholar] [CrossRef] [PubMed]
- Algers, J.; Suzuki, R.; Ohdaira, T.; Maurer, F.H. Characterization of free volume and density gradients of polystyrene surfaces by low-energy positron lifetime measurements. Polymer 2004, 45, 4533. [Google Scholar] [CrossRef]
- Panzarasa, G.; Aghion, S.; Marra, G.; Wagner, A.; Liedke, M.O.; Elsayed, M.; Krause-Rehberg, R.; Ferragut, R.; Consolati, G. Probing the Impact of the Initiator Layer on Grafted-from Polymer Brushes: A Positron Annihilation Spectroscopy Study. Macromolecules 2017, 50, 5574. [Google Scholar] [CrossRef]
- Gabriel, F.; Gippner, P.; Grosse, E.; Janssen, D.; Michel, P.; Prade, H.; Schamlott, A.; Seidel, W.; Wolf, A.; Wünsch, R. The Rossendorf Radiation Source ELBE and Its FEL Projects. Nucl. Instrum. Methods Phys. Res. Sect. B 2000, 161–163, 1143–1147. [Google Scholar] [CrossRef]
- Olsen, J.V.; Kirkegaard, P.; Pedersen, N.J.; Eldrup, M. PALSfit: A new program for the evaluation of positron lifetime spectra. Phys. Status Solidi C 2007, 4, 4004–4006. [Google Scholar] [CrossRef]
- Kilburn, D.; Claude, J.; Mezzenga, R.; Dlubek, G.; Alam, A.; Ubbink, J. Water in Glassy Carbohydrates: Opening It Up at the Nanolevel. J. Phys. Chem. B 2004, 108, 12436–12441. [Google Scholar] [CrossRef]
- Kilburn, D.; Claude, J.; Schweizer, T.; Alam, A.; Ubbink, J. Carbohydrate Polymers in Amorphous States: An Integrated Thermodynamic and Nanostructural Investigation. Biomacromolecules 2005, 6, 864–879. [Google Scholar] [CrossRef]
- Townrow, S.; Kilburn, D.; Alam, A.; Ubbink, J. Molecular Packing in Amorphous Carbohydrate Matrixes. J. Phys. Chem. B 2007, 111, 12643–12648. [Google Scholar] [CrossRef] [PubMed]
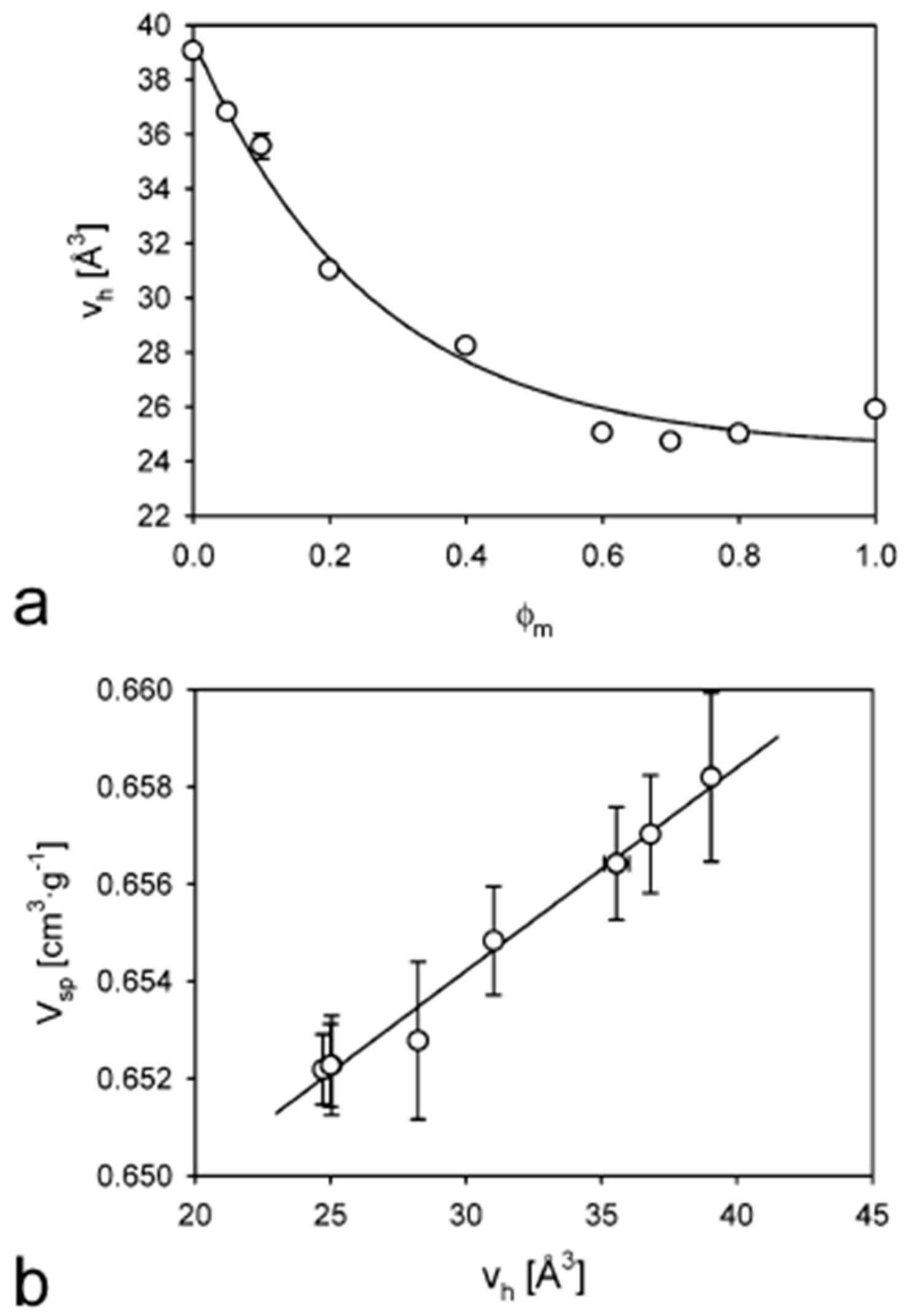
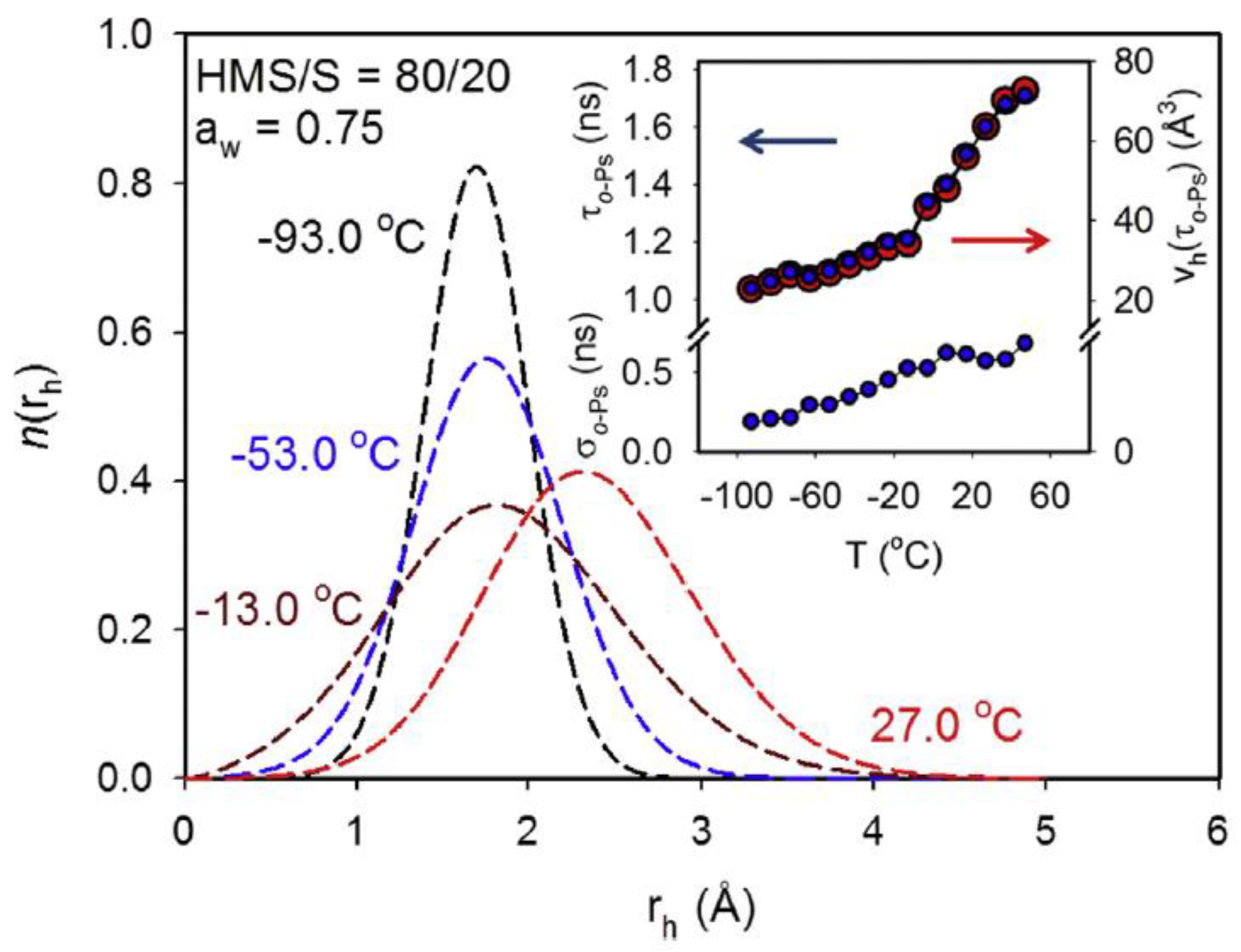
- Townrow, S.; Roussenova, M.; Giardiello, M.-I.; Alam, A.; Ubbink, J. Specific Volume−Hole Volume Correlations in Amorphous Carbohydrates: Effect of Temperature, Molecular Weight, and Water Content. J. Phys. Chem. B 2010, 114, 1568–1578. [Google Scholar] [CrossRef]
- Roussenova, M.; Murith, M.; Alam, A.; Ubbink, J. Plasticization, Antiplasticization, and Molecular Packing in Amorphous Carbohydrate-Glycerol Matrices. Biomacromolecules 2010, 11, 3237–3247. [Google Scholar] [CrossRef] [PubMed]
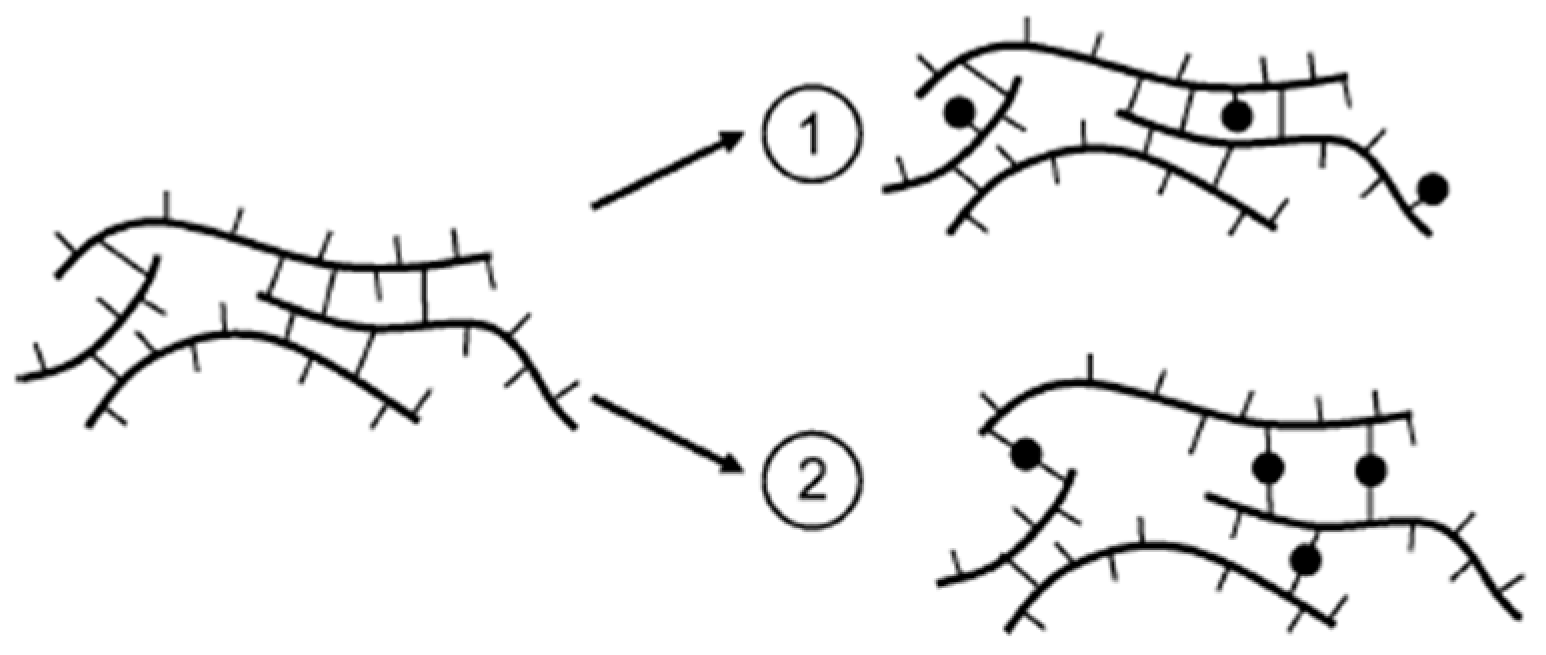
- Hughes, D.; Tedeschi, C.; Leuenberger, B.; Roussenova, M.; Coveney, A.; Richardson, R.; Bönisch, G.B.; Alam, M.A.; Ubbink, J. Amorphous-amorphous phase separation in hydrophobically-modified starch–sucrose blends II. Crystallinity and local free volume investigation using wide-angle X-ray scattering and positron annihilation lifetime spectroscopy. Food Hydrocoll. 2016, 58, 316–323. [Google Scholar] [CrossRef]
- Roudaut, G.; Duplâtre, G. Positronium as a probe in natural polymers: Decomposition in starch. Phys. Chem. Chem. Phys. 2009, 11, 9556–9561. [Google Scholar] [CrossRef]
- Sharma, S.K.; Roudaut, G.; Fabing, I.; Duplâtre, G. Characterization of a sucrose/starch matrix through positron annihilation lifetime spectroscopy: Unravelling the decomposition and glass transition processes. Phys. Chem. Chem. Phys. 2010, 12, 14278–14284. [Google Scholar] [CrossRef]
- Liu, H.; Chaudhary, D.; Roberts, J.; Weed, R.; Sullivan, J.; Buckman, S. The interaction in sorbitol-plasticized starch bionanocomposites via positron annihilation lifetime spectroscopy and small angle X-ray scattering. Carbohydr. Polym. 2012, 88, 1172–1176. [Google Scholar] [CrossRef]
- Yan, B.; Shen, H.; Fan, D.; Tao, Y.; Wu, Y.; Wang, M.; Zhao, J.; Zhang, H. Microwave treatment regulates the free volume of rice starch. Sci. Rep. 2019, 9, 3876. [Google Scholar] [CrossRef] [PubMed]
- Martini, F.; Hughes, D.J.; Bönisch, G.B.; Zwick, T.; Schäfer, C.; Geppi, M.; Alam, M.A.; Ubbink, J. Antiplasticization and phase behavior in phase-separated modified starch-sucrose blends: A positron lifetime and solid-state NMR study. Carbohydr. Polym. 2020, 250, 116931. [Google Scholar] [CrossRef]
- Liu, H.; Chaudhary, D.; Campbell, C.; Roberts, J.; Buckman, S.; Sullivan, J. Investigations into the free-volume changes within starch/plasticizer/nanoclay systems using Positron Annihilation Lifetime Spectroscopy. Mater. Chem. Phys. 2014, 148, 349–355. [Google Scholar] [CrossRef]
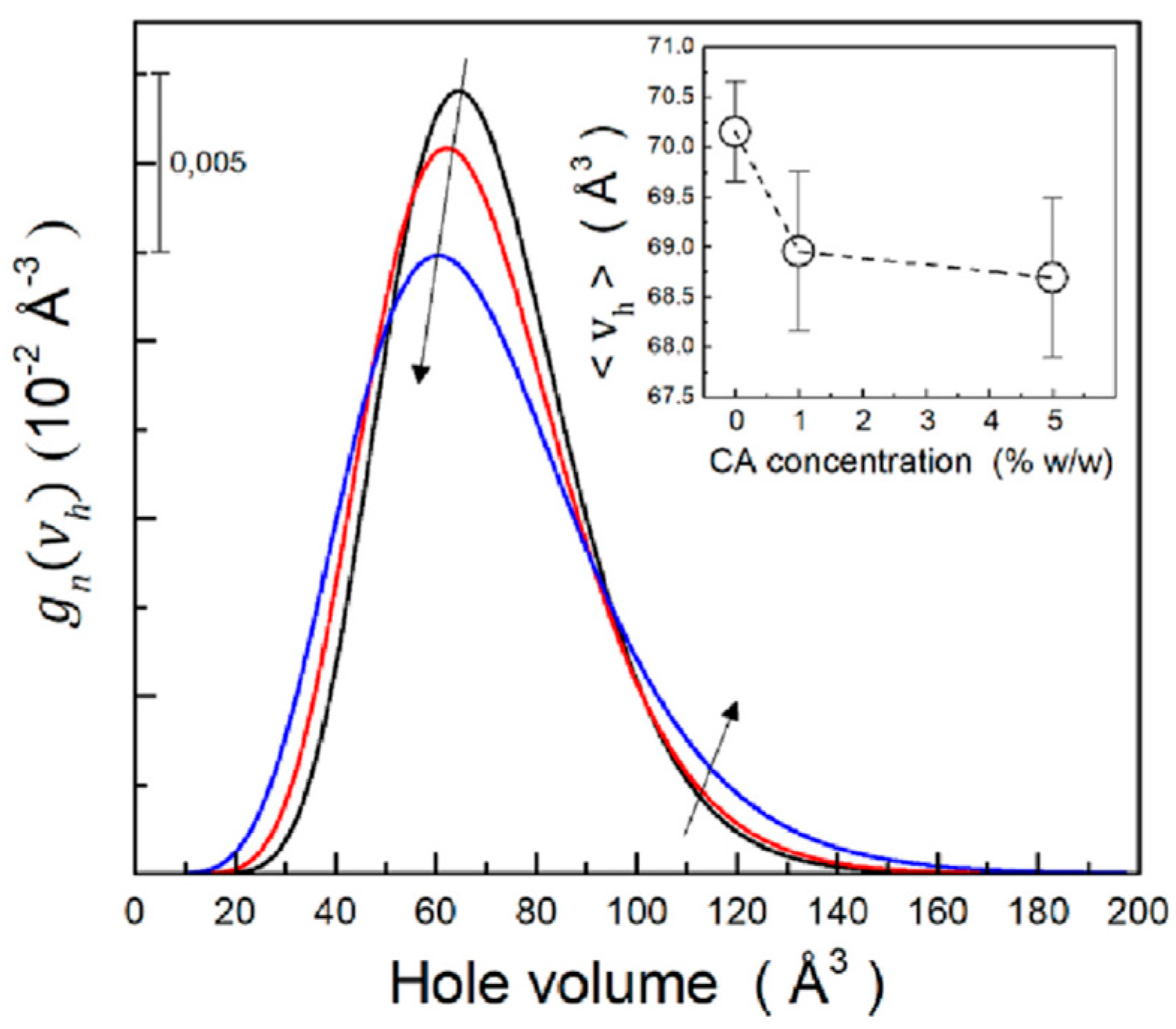
- Estevez-Areco, S.; Macchi, C.; Guz, L.; Goyanes, S.; Somoza, A. Evolution of the free volume during water desorption in thermoplastic starch/citric acid films: In situ positron annihilation studies. Carbohydr. Polym. 2023, 310, 120739. [Google Scholar] [CrossRef]
- Levine, H. (Ed.) Amorphous Food and Pharmaceutical Systems; Royal Society of Chemistry: London, UK, 2002. [Google Scholar] [CrossRef]
- Ubbink, J. Structural Advances in the Understanding of Carbohydrated Glasses. In Modern Biopolymer Science, 1st ed; Kasapis, S., Norton, I.T., Ubbink, J., Eds.; Academic Press: New York, NY, USA, 2009; pp. 277–293. ISBN 9780123741950. [Google Scholar]
- van der Sman, R.G.M. Phase separation, antiplasticization and moisture sorption in ternary systems containing polysaccharides and polyols. Food Hydrocoll. 2019, 87, 360–370. [Google Scholar] [CrossRef]
- Gomaa, M.M.; Hugenschmidt, C.; Dickmann, M.; Abdel-Hady, E.E.; Mohamed, H.F.M.; Abdel-Hamed, M.O. Crosslinked PVA/SSA proton exchange membranes: Correlation between physiochemical properties and free volume determined by positron annihilation spectroscopy. Phys. Chem. Chem. Phys. 2018, 20, 28287–28299. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sellaiyan, S.; Sako, K.; Uedono, A.; Taniguchi, Y.; Hayashi, K. Effect of free-volume holes on static mechanical properties of epoxy resins studied by positron annihilation and PVT experiments. Polymer 2020, 190, 122225. [Google Scholar] [CrossRef]
- Minfeng, Z.; Xudong, S.; Yun, W.; Xiandong, Y.; Huiquan, X.; Baoyi, W.; Chenze, Q. Correlations between the free-volume properties and the miscibility of chitosan/polar polymers blend membranes. Radiat. Phys. Chem. 2008, 77, 1062–1068. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, M.; Wu, H.; Yin, X.; Chen, J.; Jiang, Z. Mussel-inspired fabrication of structurally stable chitosan/polyacrylonitrile composite membrane for pervaporation dehydration. J. Membr. Sci. 2010, 348, 150–159. [Google Scholar] [CrossRef]
- El Wahab, M.A.; Abdou, E.S. Influence of Starch and Glycerol on the Properties of Chitosan by Positron Annihilation Spectroscopy. J. Appl. Polym. Sci. 2010, 116, 2874–2883. [Google Scholar] [CrossRef]
- Lin, B.; Du, Y.; Li, Y.; Liang, X.; Wang, X.; Deng, W.; Wang, X.; Li, L.; Kennedy, J.F. The effect of moist heat treatment on the characteristic of starch-based composite materials coating with chitosan. Carbohydr. Polym. 2010, 81, 554–559. [Google Scholar] [CrossRef]
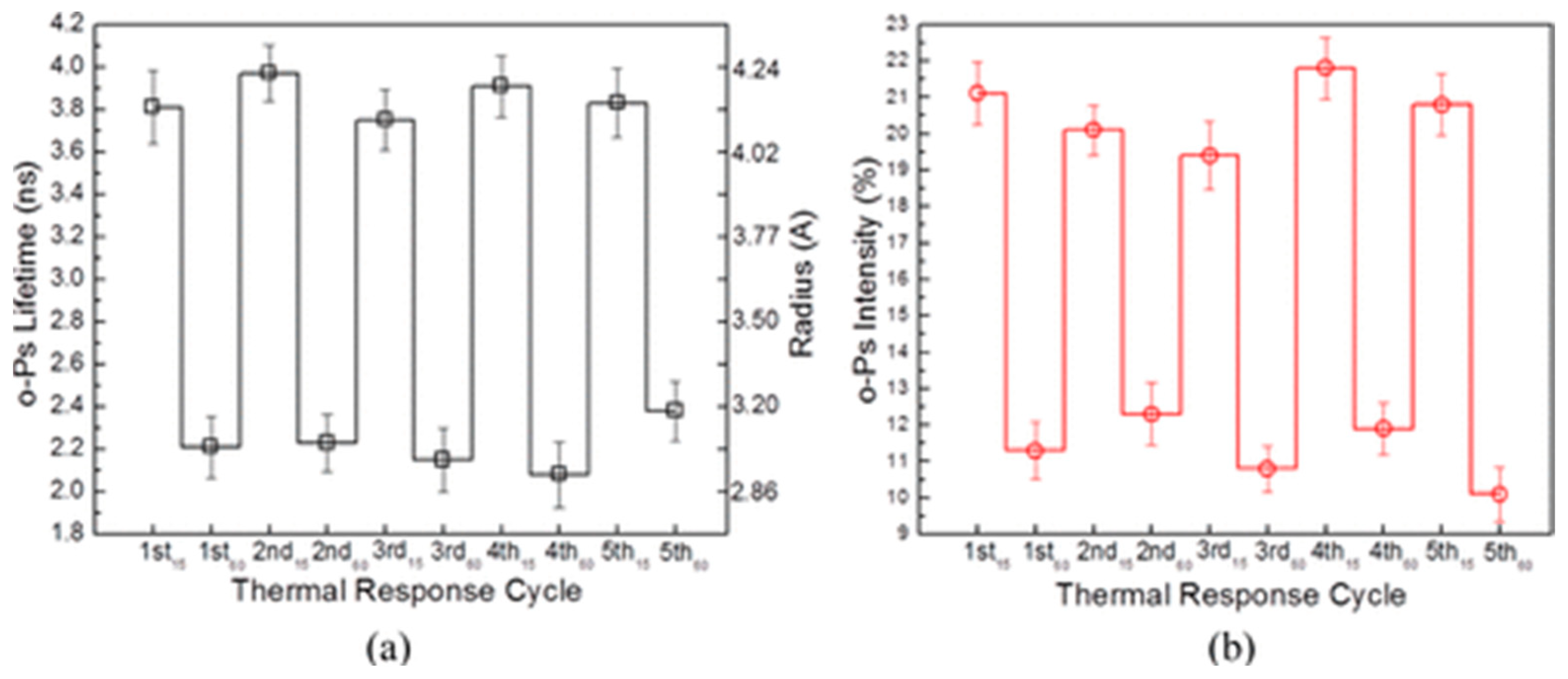
- Lecaros, R.L.G.; Syu, Z.-C.; Chiao, Y.-H.; Wickramasinghe, S.R.; Ji, Y.-L.; An, Q.-F.; Hung, W.-S.; Hu, C.-C.; Lee, K.-R.; Lai, J.-Y. Characterization of a Thermoresponsive Chitosan Derivative as a Potential Draw Solute for Forward Osmosis. Environ. Sci. Technol. 2016, 50, 11935–11942. [Google Scholar] [CrossRef]
- Anbinder, P.; Macchi, C.; Amalvy, J.; Somoza, A. Chitosan-graft-poly(n-butyl acrylate) copolymer: Synthesis and characterization of a natural/synthetic hybrid material. Carbohydr. Polym. 2016, 145, 86–94. [Google Scholar] [CrossRef]
- Xia, R.; Cao, X.; Gao, M.; Zhang, P.; Zeng, M.; Wang, B.; Wei, L. Probing sub-nano level molecular packing and correlated positron annihilation characteristics of ionic cross-linked chitosan membranes using positron annihilation spectroscopy. Phys. Chem. Chem. Phys. 2016, 19, 3616–3626. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Xu, M.; Zhao, J.; Wang, Y.; Qi, C.; Zeng, M.; Xia, R.; Cao, X.; Wang, B. Insightful understanding of the correlations of the microstructure and catalytic performances of Pd@chitosan membrane catalysts studied by positron annihilation spectroscopy. RSC Adv. 2018, 8, 3225–3236. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Han, T.; Wang, J.; Shao, L.; Qi, C.; Zhang, X.M.; Liu, C.; Liu, X. Removal of heavy metal chromium using cross-linked chitosan composite nanofiber mats. Int. J. Biol. Macromol. 2018, 120, 213–221. [Google Scholar] [CrossRef]
- Anbinder, P.S.; Macchi, C.; Amalvy, J.; Somoza, A. A study of the structural changes in a chitosan matrix produced by the adsorption of copper and chromium ions. Carbohydr. Polym. 2019, 222, 114987. [Google Scholar] [CrossRef]
- Subramanian, M.; Suganthi, S.; Raj, V.; Baghavathy, S.; Alwarappan, S. Methodical Exploration on Silicotungstic Acid Tailored Hybrid Bio-polymeric Proton-Exchange Membrane Electrolytes: Correlating Positron Annihilation Characteristics with Electrochemical Selectivity for Direct Methanol Fuel Cells. J. Electrochem. Soc. 2020, 167, 064511. [Google Scholar] [CrossRef]
- Shelly, M.; Raghavendra, M.; Ravikumar, H.B.; Francis, T. Structural and free-hole volume characterization of high-density polyethylene-chitosan composites plasticized with palm oil. Polym. Eng. Sci. 2021, 61, 3060–3068. [Google Scholar] [CrossRef]
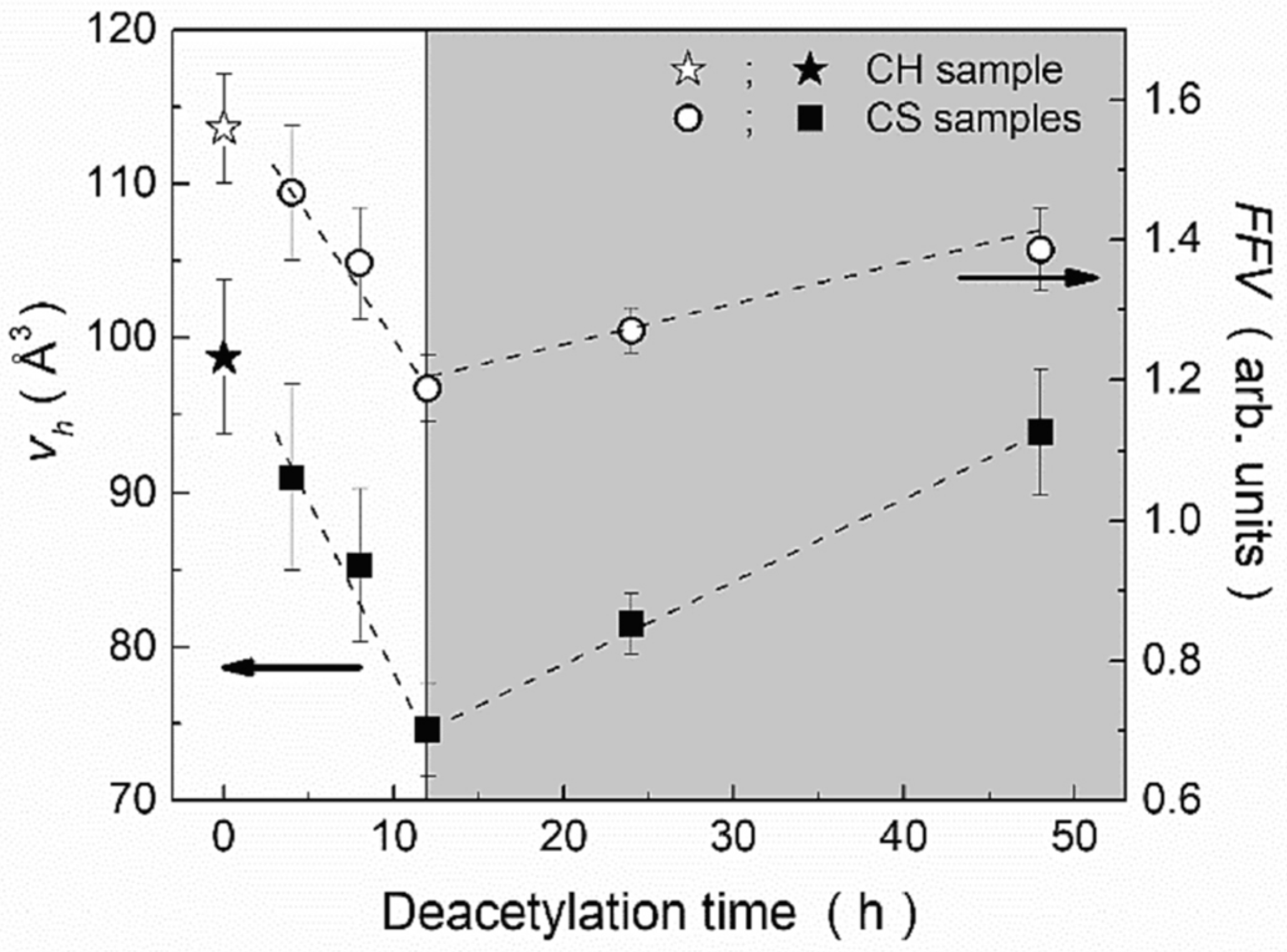
- Lucanera, E.; Anbinder, S.; Macchi, C.; Somoza, A. Tailoring nanohole sizes through the deacetylation process in chitosan powders obtained from squid pens. Carbohydr. Polym. 2022, 297, 120026. [Google Scholar] [CrossRef]
- Yao, Y.; Deng, Y.; Liang, Y.; Li, X.; Tang, X.; Lin, M.; Xu, C.; Fu, L.; Lin, B. Convenient, nondestructive monitoring and sustained-release of ethephon/chitosan film for on-demand of fruit ripening. Int. J. Biol. Macromol. 2022, 214, 338–347. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, S.; Zhang, T.; Xu, M.; Zhao, J.; Zeng, M.; Sun, K.; Feng, R.; Yang, Z.; Zhang, P.; et al. Positron annihilation study of chitosan and its derived carbon/pillared montmorillonite clay stabilized Pd species nanocomposites. Polym. Test. 2022, 114, 107689. [Google Scholar] [CrossRef]
- Abdel-Hady, E.E.; Hafez, S.H.M.; Mohamed, H.F.M.; Elsharkawy, M.R.M. Characterization and application of LDH with chitosan composites investigated by positron annihilation lifetime spectroscopy and surface texture for the adsorption of methyl orange. Sci. Rep. 2024, 14, 16501. [Google Scholar] [CrossRef]
- Kumar, B.V.S.; Siddaramaiah; Shayan, M.B.; Manjula, K.S.; Ranganathaiah, C.; Rao, G.V.N.; Basavalingu, B.; Byrappa, K. Effect of zeolite particulate filler on the properties of polyurethane composites. J. Polym. Res. 2010, 17, 135–142. [Google Scholar] [CrossRef]
- Macchi, C.; Meiorin, C.; Mosiewicki, M.A.; Aranguren, M.I.; Somoza, A. Effect of the composition and chemical aging in tung oil-styrene networks: Free volume and dynamic-mechanical properties. Eur. Polym. J. 2017, 87, 231–240. [Google Scholar] [CrossRef]
- Kavetskyy, T.; Smutok, O.; Demkiv, O.; Kasetaite, S.; Ostrauskaite, J.; Švajdlenková, H.; Šauša, O.; Zubrytska, K.; Hoivanovych, N.; Gonchar, M. Dependence of operational parameters of laccase-based biosensors on structure of photocross-linked polymers as holding matrixes. Eur. Polym. J. 2019, 115, 391–398. [Google Scholar] [CrossRef]
- Anbinder, P.S.; Meiorin, C.; Macchi, C.; Mosiewicki, M.A.; Aranguren, M.I.; Somoza, A. Structural properties of vegetable oil thermosets: Effect of crosslinkers, modifiers and oxidative aging. Eur. Polym. J. 2020, 124, 109470. [Google Scholar] [CrossRef]



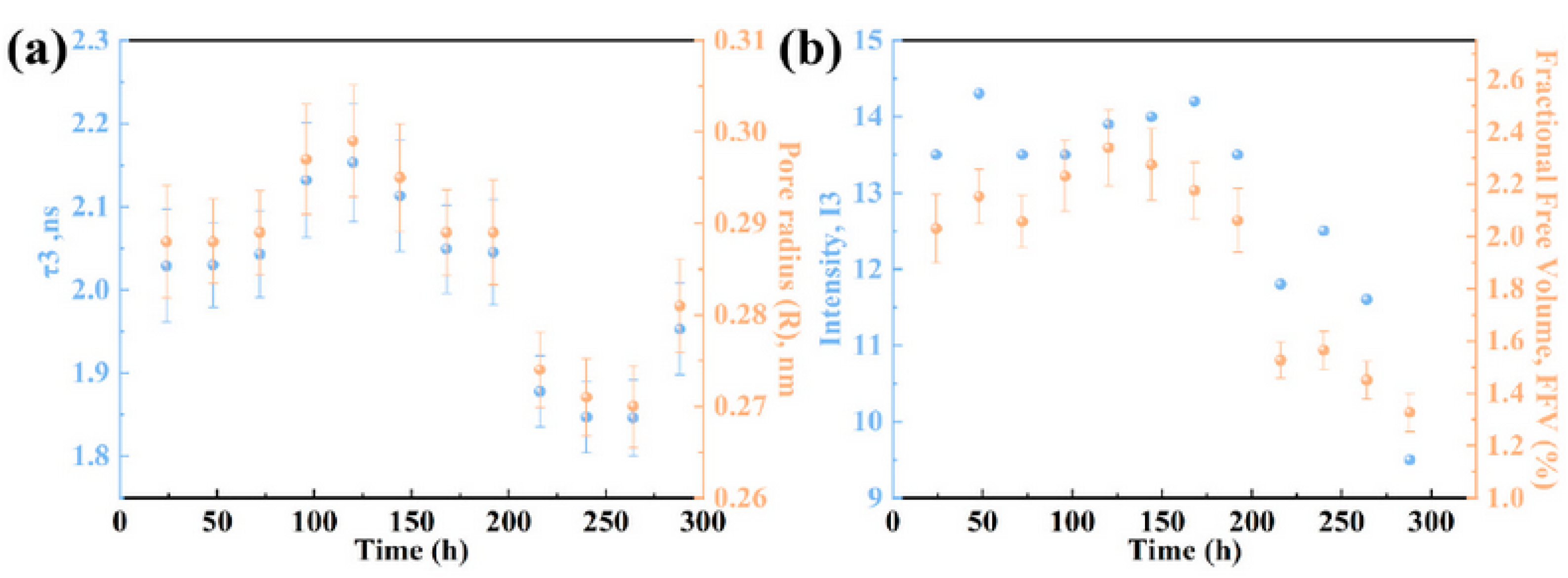
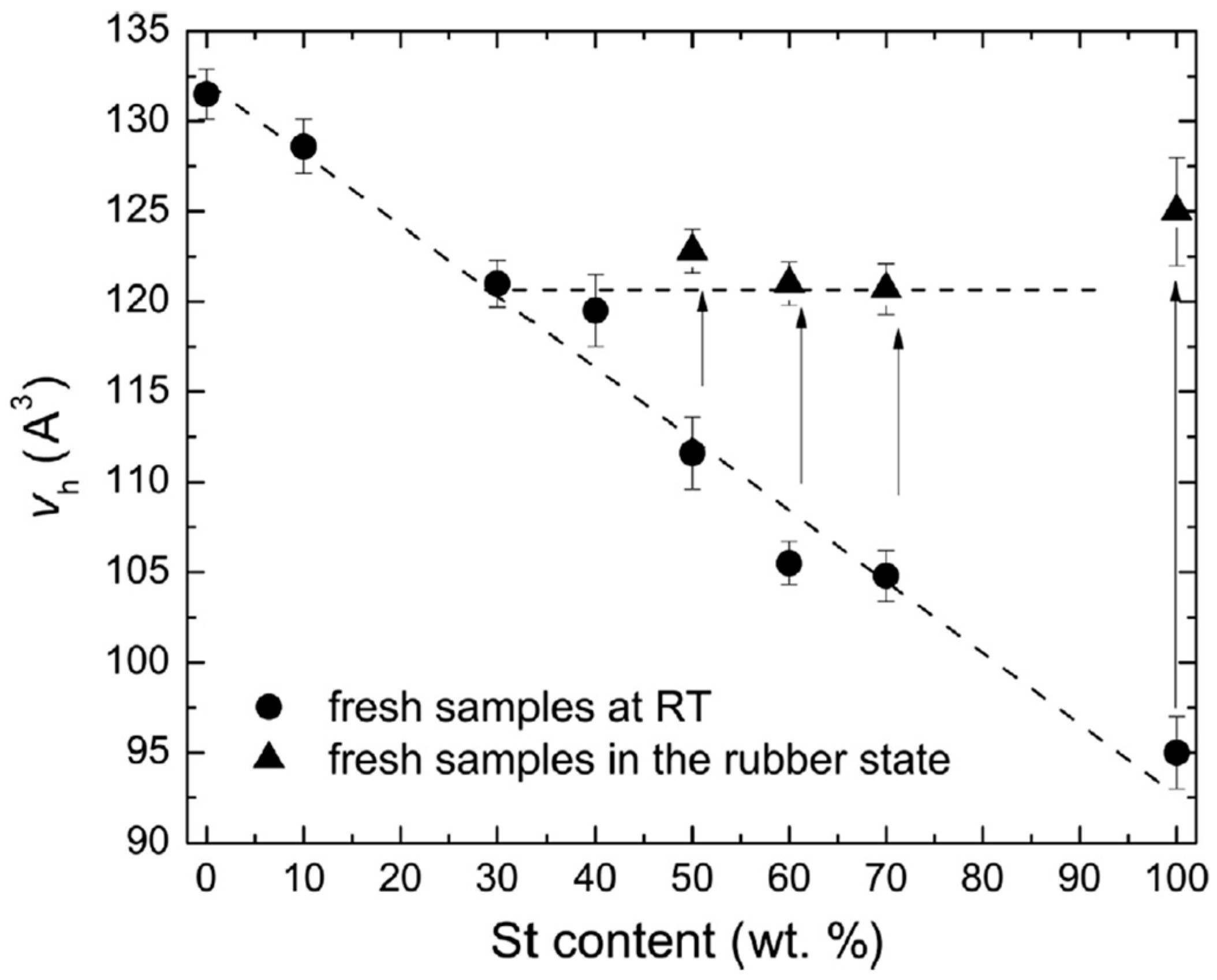
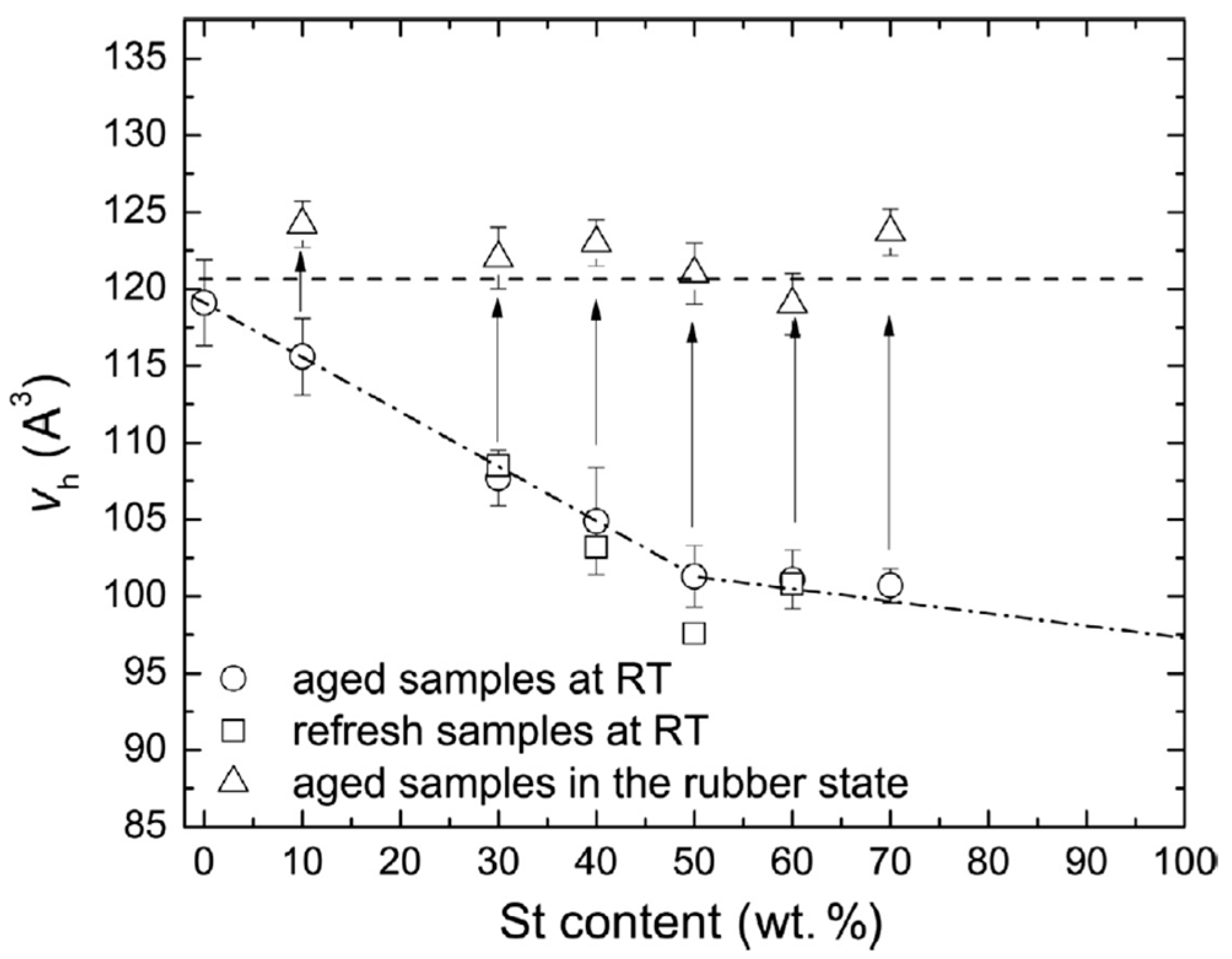

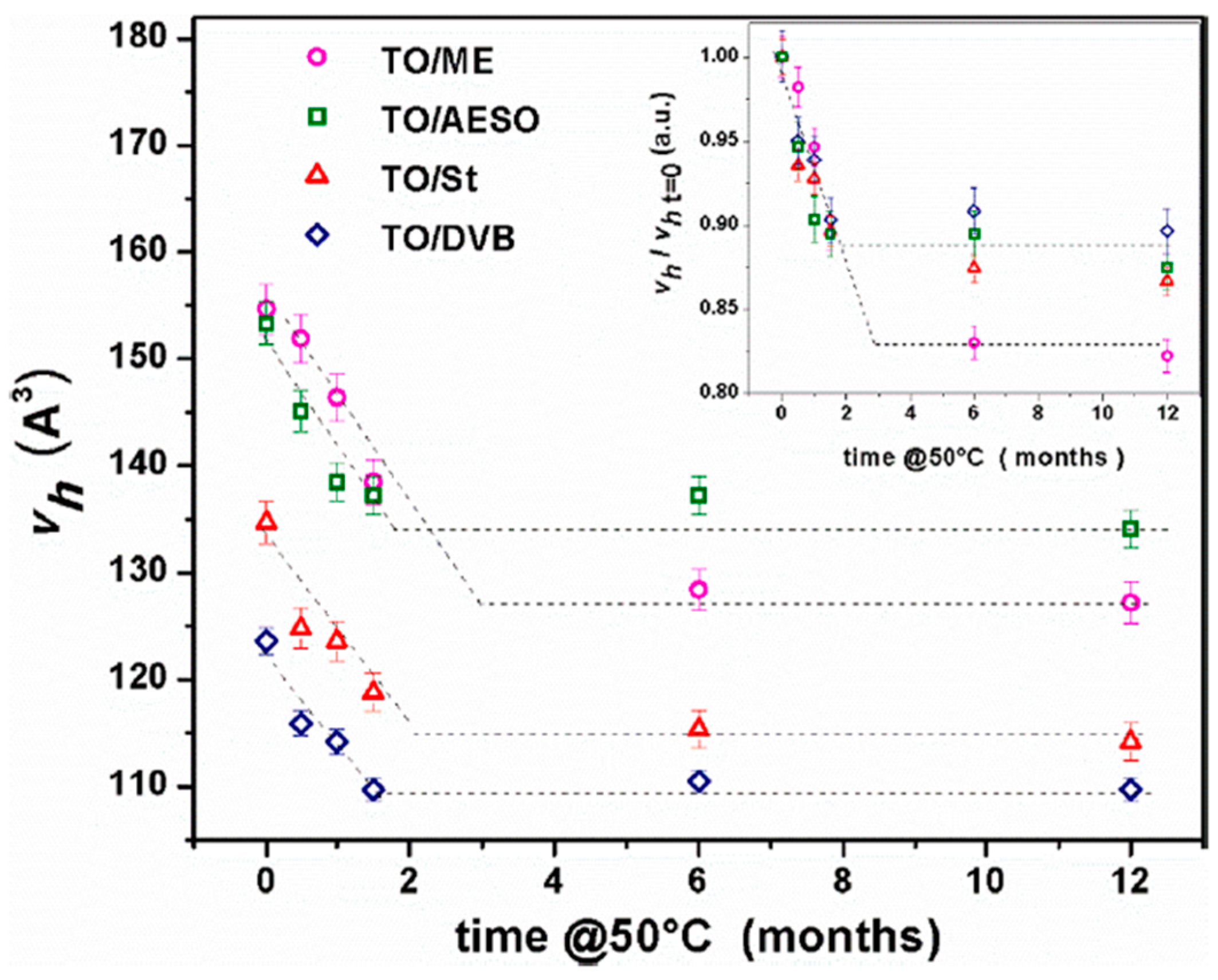
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Consolati, G.; Macchi, C.; Somoza, A. Current Positron Studies on the Modifications of the Molecular Packing in Green-Based Polymers Through Changes in the Synthesis Procedures or Environmental Conditions. Polymers 2024, 16, 3611. https://doi.org/10.3390/polym16243611
Consolati G, Macchi C, Somoza A. Current Positron Studies on the Modifications of the Molecular Packing in Green-Based Polymers Through Changes in the Synthesis Procedures or Environmental Conditions. Polymers. 2024; 16(24):3611. https://doi.org/10.3390/polym16243611
Chicago/Turabian StyleConsolati, Giovanni, Carlos Macchi, and Alberto Somoza. 2024. "Current Positron Studies on the Modifications of the Molecular Packing in Green-Based Polymers Through Changes in the Synthesis Procedures or Environmental Conditions" Polymers 16, no. 24: 3611. https://doi.org/10.3390/polym16243611
APA StyleConsolati, G., Macchi, C., & Somoza, A. (2024). Current Positron Studies on the Modifications of the Molecular Packing in Green-Based Polymers Through Changes in the Synthesis Procedures or Environmental Conditions. Polymers, 16(24), 3611. https://doi.org/10.3390/polym16243611