
Nickel-Catalyzed Ethylene Copolymerization with Vinylalkoxysilanes: A Computational Study

 ,
,
Abstract
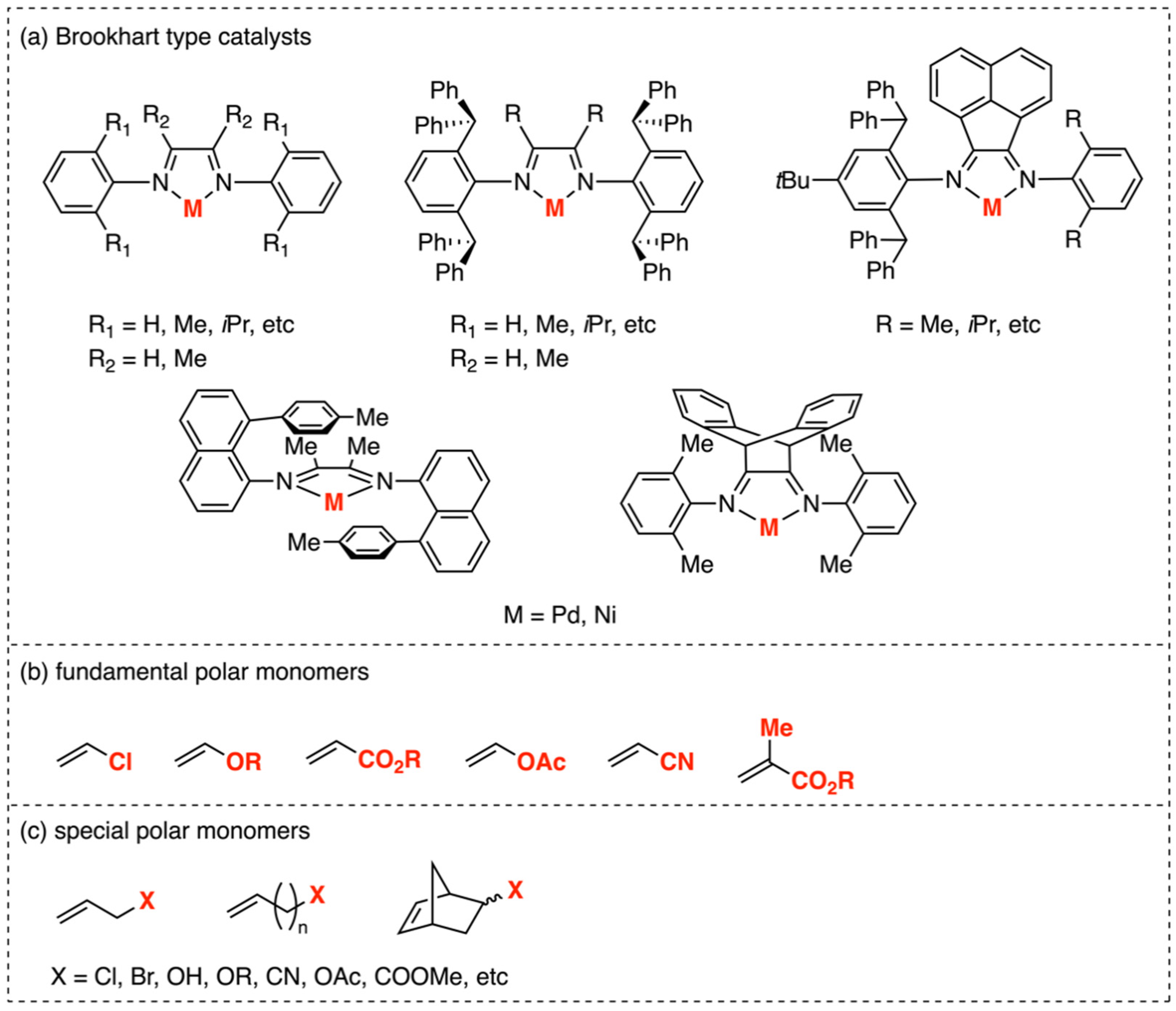
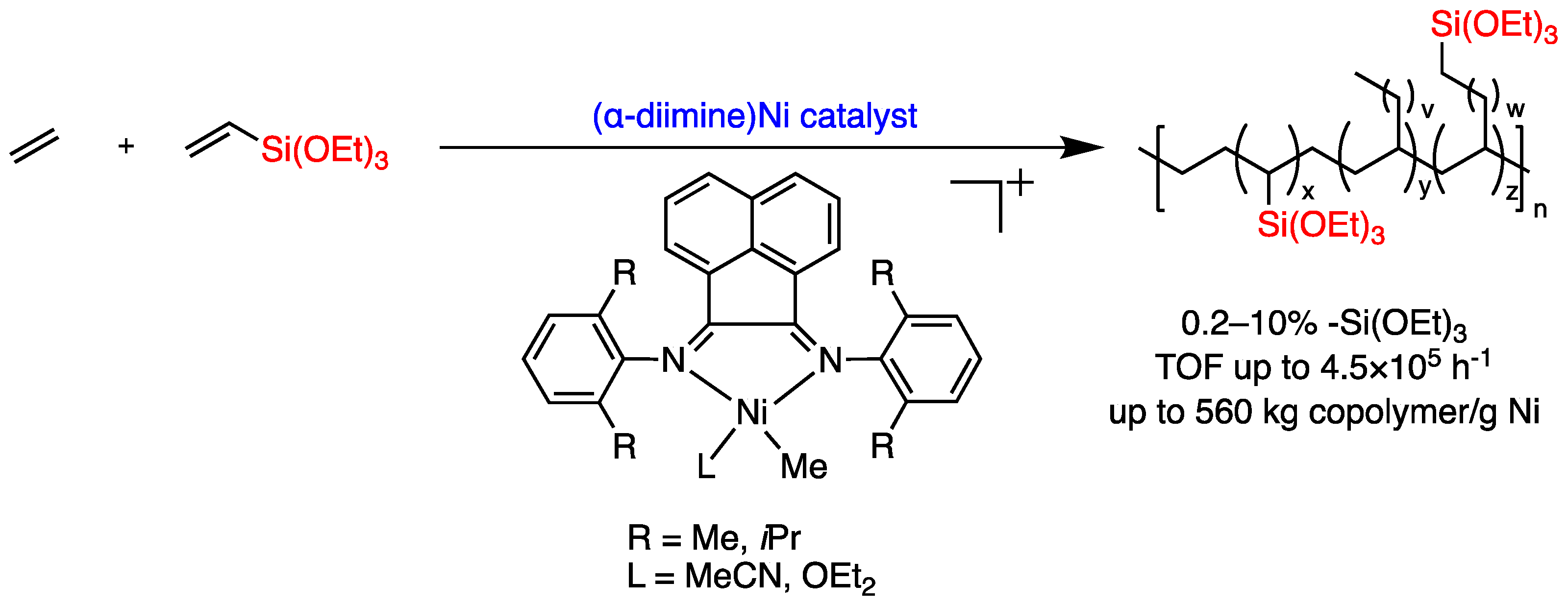
1. Introduction
2. Computational Methods
3. Results and Discussion
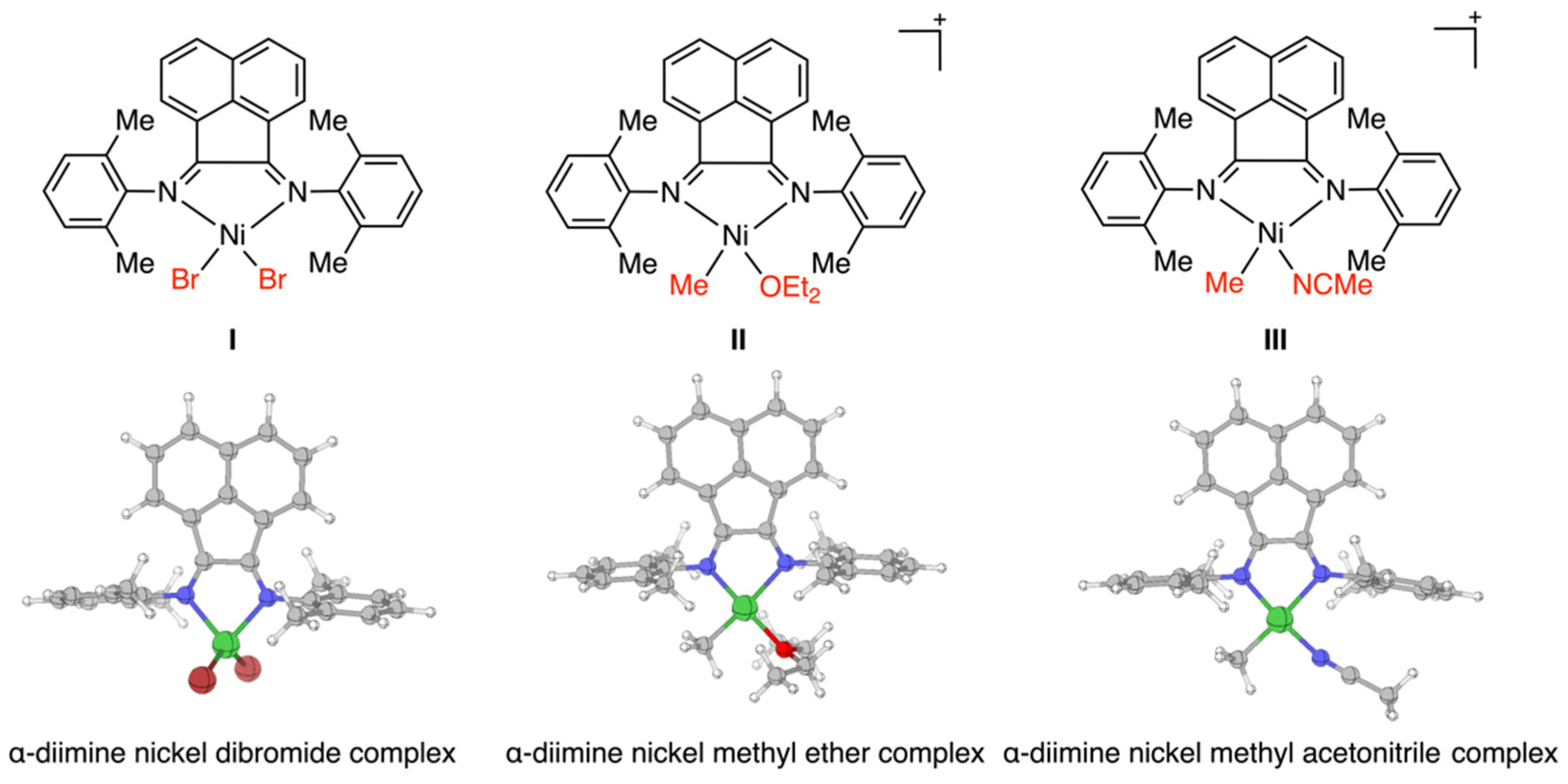
3.1. Nickel Complexes and Cationic Active Species
3.2. Chain Initiation and Chain Propagation (Ethylene Insertion)
3.3. Chain Propagation (Vinylalkoxysilane Insertion) and Chain Termination
3.4. Energetic Analysis of In-Chain and Chain-End Silane Enchainment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Annual Production of Plastics Worldwide from 1950 to 2021. Available online: https://www.statista.com/statistics/282732/global-production-of-plastics-since-1950/ (accessed on 11 October 2023).
- Dong, J.-Y.; Hu, Y. Design and synthesis of structurally well-defined functional polyolefins via transition metal-mediated olefin polymerization chemistry. Coord. Chem. Rev. 2006, 250, 47–65. [Google Scholar] [CrossRef]
- Rünzi, T.; Mecking, S. Saturated Polar- Substituted Polyethylene Elastomers from Insertion Polymerization. Adv. Funct. Mater. 2014, 24, 387–395. [Google Scholar] [CrossRef]
- Dai, S.Y.; Chen, C.L. Palladium-Catalyzed Direct Synthesis of Various Branched, Carboxylic Acid-Functionalized Polyolefins: Characterization, Derivatization, and Properties. Macromolecules 2018, 51, 6818–6824. [Google Scholar] [CrossRef]
- Na, Y.N.; Dai, S.Y.; Chen, C.L. Direct Synthesis of Polar-Functionalized Linear Low-Density Polyethylene (LLDPE) and Low-Density Polyethylene (LDPE). Macromolecules 2018, 51, 4040–4048. [Google Scholar] [CrossRef]
- Sui, X.; Hong, C.; Pang, W.; Chen, C. Unsymmetrical α-diimine palladium catalysts and their properties in olefin (co)polymerization. Mater. Chem. Front. 2017, 1, 967–972. [Google Scholar] [CrossRef]
- Chen, E.Y.X. Coordination Polymerization of Polar Vinyl Monomers by Single-Site Metal Catalysts. Chem. Rev. 2009, 109, 5157–5214. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gao, Y.; Marks, T.J. Early Transition Metal Catalysis for Olefin-Polar Monomer Copolymerization. Angew. Chem. Int. Ed. 2020, 59, 14726–14735. [Google Scholar] [CrossRef]
- Song, Z.; Wang, S.; Gao, R.; Wang, Y.; Gou, Q.; Zheng, G.; Feng, H.; Fan, G.; Lai, J. Recent Advancements in Mechanistic Studies of Palladium- and Nickel-Catalyzed Ethylene Copolymerization with Polar Monomers. Polymers 2023, 15, 4343. [Google Scholar] [CrossRef]
- Johnson, L.K.; Killian, C.M.; Brookhart, M. New Pd(II)-Based and Ni(II)-Based Catalysts for Polymerization of Ethylene and alpha-Olefins. J. Am. Chem. Soc. 1995, 117, 6414–6415. [Google Scholar] [CrossRef]
- Johnson, L.K.; Mecking, S.; Brookhart, M. Copolymerization of ethylene and propylene with functionalized vinyl monomers by palladium(II) catalysts. J. Am. Chem. Soc. 1996, 118, 267–268. [Google Scholar] [CrossRef]
- Dai, S.Y.; Sui, X.L.; Chen, C.L. Highly Robust Palladium(II) α-Diimine Catalysts for Slow-Chain-Walking Polymerization of Ethylene and Copolymerization with Methyl Acrylate. Angew. Chem. Int. Ed. 2015, 54, 9948–9953. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Li, S.; Gong, Q.; Zhang, S.; Liu, B.; Dai, S. Systematic Investigations of Ligand Steric Effects on α-Diimine Nickel Catalyzed Olefin Polymerization and Copolymerization. Organometallics 2019, 38, 2919–2926. [Google Scholar] [CrossRef]
- Luo, S.; Jordan, R.F. Copolymerization of silyl vinyl ethers with olefins by (α-diimine)PdR+. J. Am. Chem. Soc. 2006, 128, 12072–12073. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Luo, S.; Jordan, R.F. Cationic Polymerization and Insertion Chemistry in the Reactions of Vinyl Ethers with (α-Diimine)PdMe+ Species. J. Am. Chem. Soc. 2010, 132, 5273–5284. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, W.; Daugulis, O.; Brookhart, M. Mechanistic studies of Pd(II)-catalyzed copolymerization of ethylene and vinylalkoxysilanes: Evidence for a β-silyl elimination chain transfer mechanism. J. Am. Chem. Soc. 2016, 138, 16120–16129. [Google Scholar] [CrossRef]
- Camacho, D.H.; Guan, Z. Designing late-transition metal catalysts for olefin insertion polymerization and copolymerization. Chem. Commun. 2010, 46, 7879–7893. [Google Scholar] [CrossRef]
- Dong, Z.; Ye, Z. Hyperbranched polyethylenes by chain walking polymerization: Synthesis, properties, functionalization, and applications. Polym. Chem. 2012, 3, 286–301. [Google Scholar] [CrossRef]
- Takeuchi, D. Stereo-controlled synthesis of polyolefins with cycloalkane groups by using late transition metals. Polym. J. 2012, 44, 919–928. [Google Scholar] [CrossRef]
- Tan, C.; Chen, C. Emerging Palladium and Nickel Catalysts for Copolymerization of Olefins with Polar Monomers. Angew. Chem. Int. Ed. 2019, 58, 7192–7200. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, L.; Yu, H.; Zhao, Y.; Sun, R.; Jing, G.; Huang, J.; Khalid, H.; Abbasi, N.M.; Akram, M. Synthesis and application of polyethylene-based functionalized hyperbranched polymers. Prog. Polym. Sci. 2015, 45, 23–43. [Google Scholar] [CrossRef]
- Guo, L.; Chen, C. (α-Diimine)palladium catalyzed ethylene polymerization and (co)polymerization with polar comonomers. Sci. China Chem. 2015, 58, 1663–1673. [Google Scholar] [CrossRef]
- Guo, L.; Dai, S.; Sui, X.; Chen, C. Palladium and Nickel Catalyzed Chain Walking Olefin Polymerization and Copolymerization. ACS Catal. 2016, 6, 428–441. [Google Scholar] [CrossRef]
- Dai, S.; Chen, C. Direct Synthesis of Functionalized High-Molecular-Weight Polyethylene by Copolymerization of Ethylene with Polar Monomers. Angew. Chem. Int. Ed. 2016, 55, 13281–13285. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Liu, W.; Chen, C. Late transition metal catalyzed α-olefin polymerization and copolymerization with polar monomers. Mater. Chem. Front. 2017, 1, 2487–2494. [Google Scholar] [CrossRef]
- Johnson, L.; Wang, L.; McLain, S.; Bennett, A.; Dobbs, K.; Hauptman, E.; Ionkin, A.; Ittel, S.; Kunitsky, K.; Marshall, W.; et al. Copolymerization of ethylene and acrylates by nickel catalysts. In Beyond Metallocenes: Next Generation Polmerization Catalysts; ACS Publications: Washington, DC, USA, 2003; Volume 857, pp. 131–142. [Google Scholar]
- Long, B.K.; Eagan, J.M.; Mulzer, M.; Coates, G.W. Semi-Crystalline Polar Polyethylene: Ester-Functionalized Linear Polyolefins Enabled by a Functional-Group-Tolerant, Cationic Nickel Catalyst. Angew. Chem. Int. Ed. 2016, 55, 7106–7110. [Google Scholar] [CrossRef]
- Chen, Z.; Leatherman, M.D.; Daugulis, O.; Brookhart, M. Nickel-Catalyzed Copolymerization of Ethylene and Vinyltrialkoxysilanes: Catalytic Production of Cross-Linkable Polyethylene and Elucidation of the Chain-Growth Mechanism. J. Am. Chem. Soc. 2017, 139, 16013–16022. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, P.; Baione, F.; Guerra, G.; Martinotto, L.; Albizzati, E. Polyethylene unit cell and crystallinity variations as a consequence of different cross-linking processes. Macromolecules 2001, 34, 5175–5179. [Google Scholar] [CrossRef]
- Khonakdar, H.A.; Morshedian, J.; Wagenknecht, U.; Jafari, S.H. An investigation of chemical crosslinking effect on properties of high-density polyethylene. Polymer 2003, 44, 4301–4309. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Grimme, S. Accurate description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S. Density functional theory with London dispersion corrections. WIREs Comput. Mol. Sci. 2011, 1, 211–228. [Google Scholar] [CrossRef]
- Ehrlich, S.; Moellmann, J.; Grimme, S. Dispersion-Corrected Density Functional Theory for Aromatic Interactions in Complex Systems. Acc. Chem. Res. 2013, 46, 916–926. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Performance of SM6, SM8, and SMD on the SAMPL1 Test Set for the Prediction of Small-Molecule Solvation Free Energies. J. Phys. Chem. B 2009, 113, 4538–4543. [Google Scholar] [CrossRef]
- Legault, C.Y. CYLview20. Available online: http://www.cylview.org (accessed on 31 October 2020).
- Sun, J.; Chen, M.; Luo, G.; Chen, C.; Luo, Y. Diphosphazane-monoxide and Phosphine-sulfonate Palladium Catalyzed Ethylene Copolymerization with Polar Monomers: A Computational Study. Organometallics 2019, 38, 638–646. [Google Scholar] [CrossRef]
- Yan, M.; Kang, X.; Li, S.; Xu, X.; Luo, Y.; He, S.; Chen, C. Mechanistic Studies on Nickel-Catalyzed Ethylene Polymerization: Ligand Effects and Quantitative Structure–Activity Relationship Model. Organometallics 2022, 41, 3212–3218. [Google Scholar] [CrossRef]
- Nozaki, K.; Kusumoto, S.; Noda, S.; Kochi, T.; Chung, L.W.; Morokuma, K. Why Did Incorporation of Acrylonitrile to a Linear Polyethylene Become Possible? Comparison of Phosphine−Sulfonate Ligand with Diphosphine and Imine−Phenolate Ligands in the Pd-Catalyzed Ethylene/Acrylonitrile Copolymerization. J. Am. Chem. Soc. 2010, 132, 16030–16042. [Google Scholar] [CrossRef]
- Li, M.; Wang, X.; Luo, Y.; Chen, C. A Second-Coordination-Sphere Strategy to Modulate Nickel- and Palladium-Catalyzed Olefin Polymerization and Copolymerization. Angew. Chem. Int. Ed. 2017, 56, 11604–11609. [Google Scholar] [CrossRef]
- Mehmood, A.; Xu, X.W.; Raza, W.; Kim, K.H.; Luo, Y. Mechanistic Studies for Palladium Catalyzed Copolymerization of Ethylene with Vinyl Ethers. Polymers 2020, 12, 2401. [Google Scholar] [CrossRef]
- Chen, M.; Chen, C. A Versatile Ligand Platform for Palladium- and Nickel-Catalyzed Ethylene Copolymerization with Polar Monomers. Angew. Chem. Int. Ed. 2018, 57, 3094–3098. [Google Scholar] [CrossRef]
- Rünzi, T.; Guironnet, D.; Göttker-Schnetmann, I.; Mecking, S. Reactivity of methacrylates in insertion polymerization. J. Am. Chem. Soc. 2010, 132, 16623–16630. [Google Scholar] [CrossRef]
- Williams, B.S.; Leatherman, M.D.; White, P.S.; Brookhart, M. Reactions of Vinyl Acetate and Vinyl Trifluoroacetate with Cationic Diimine Pd(II) and Ni(II) Alkyl Complexes: Identification of Problems Connected with Copolymerizations of These Monomers with Ethylene. J. Am. Chem. Soc. 2005, 127, 5132–5146. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Z.; Gao, R.; Wu, C.; Gou, Q.; Zheng, G.; Liu, J.; Yang, S.; Feng, H. Nickel-Catalyzed Ethylene Copolymerization with Vinylalkoxysilanes: A Computational Study. Polymers 2024, 16, 762. https://doi.org/10.3390/polym16060762
Song Z, Gao R, Wu C, Gou Q, Zheng G, Liu J, Yang S, Feng H. Nickel-Catalyzed Ethylene Copolymerization with Vinylalkoxysilanes: A Computational Study. Polymers. 2024; 16(6):762. https://doi.org/10.3390/polym16060762
Chicago/Turabian StyleSong, Zhihui, Rong Gao, Changjiang Wu, Qingqiang Gou, Gang Zheng, Junjie Liu, Shifang Yang, and Huasheng Feng. 2024. "Nickel-Catalyzed Ethylene Copolymerization with Vinylalkoxysilanes: A Computational Study" Polymers 16, no. 6: 762. https://doi.org/10.3390/polym16060762
APA StyleSong, Z., Gao, R., Wu, C., Gou, Q., Zheng, G., Liu, J., Yang, S., & Feng, H. (2024). Nickel-Catalyzed Ethylene Copolymerization with Vinylalkoxysilanes: A Computational Study. Polymers, 16(6), 762. https://doi.org/10.3390/polym16060762