

Coarse-Grained Simulations on Polyethylene Crystal Network Formation and Microstructure Analysis




Abstract
1. Introduction
2. Simulation Methodology
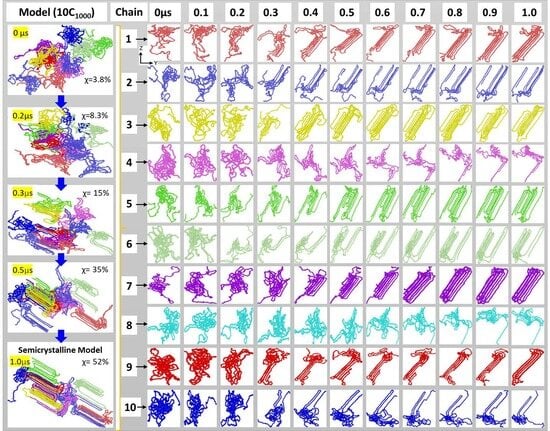
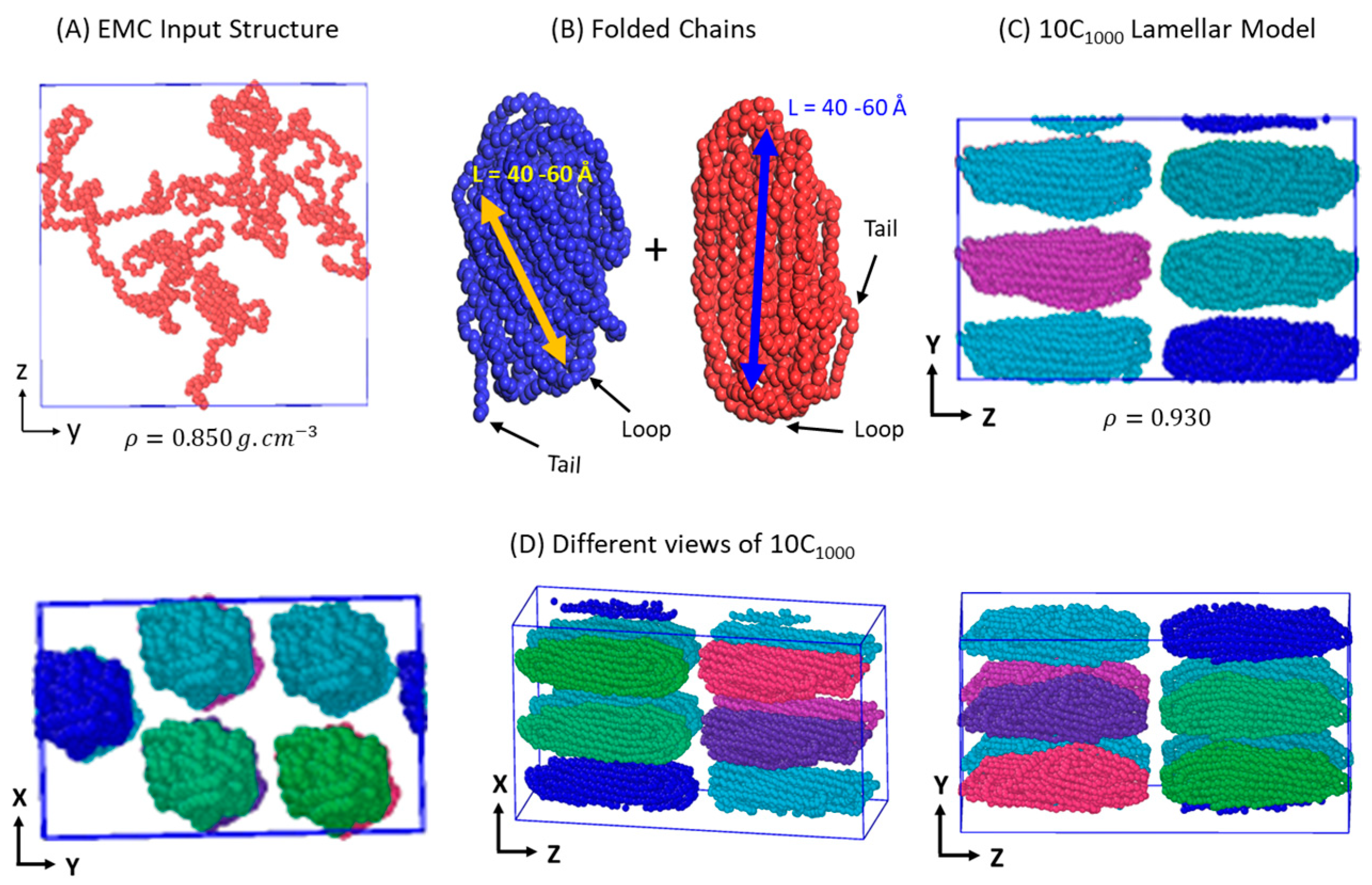
2.1. Preparation and Packing of Multi-Folded HDPE Chains
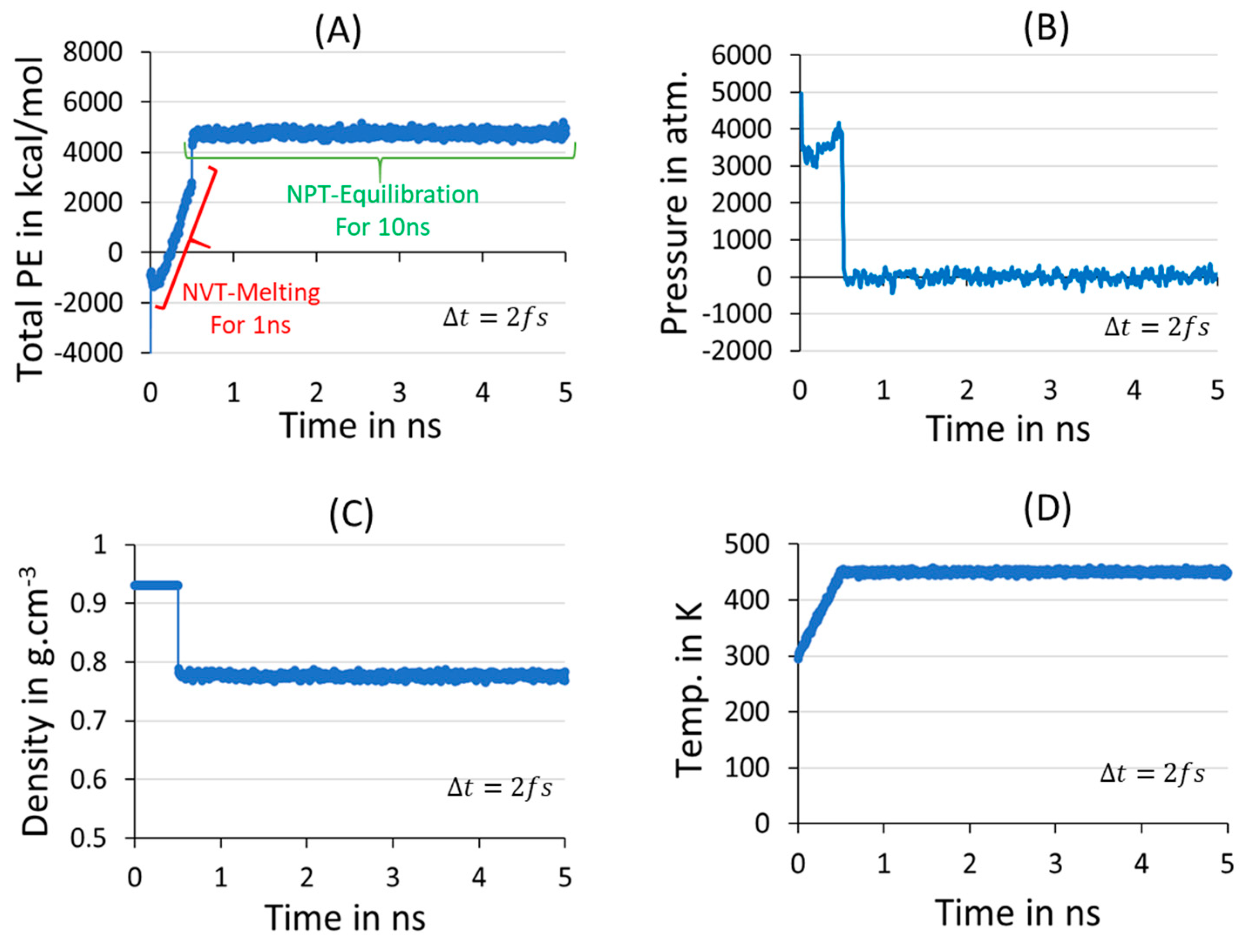
2.2. Melting, Equilibration, and Isothermal Crystallization
2.3. Analysis of the Model
2.3.1. One-Dimensional Density
2.3.2. Degree of Crystallinity ()
2.3.3. Mechanical Properties
3. Results and Discussion
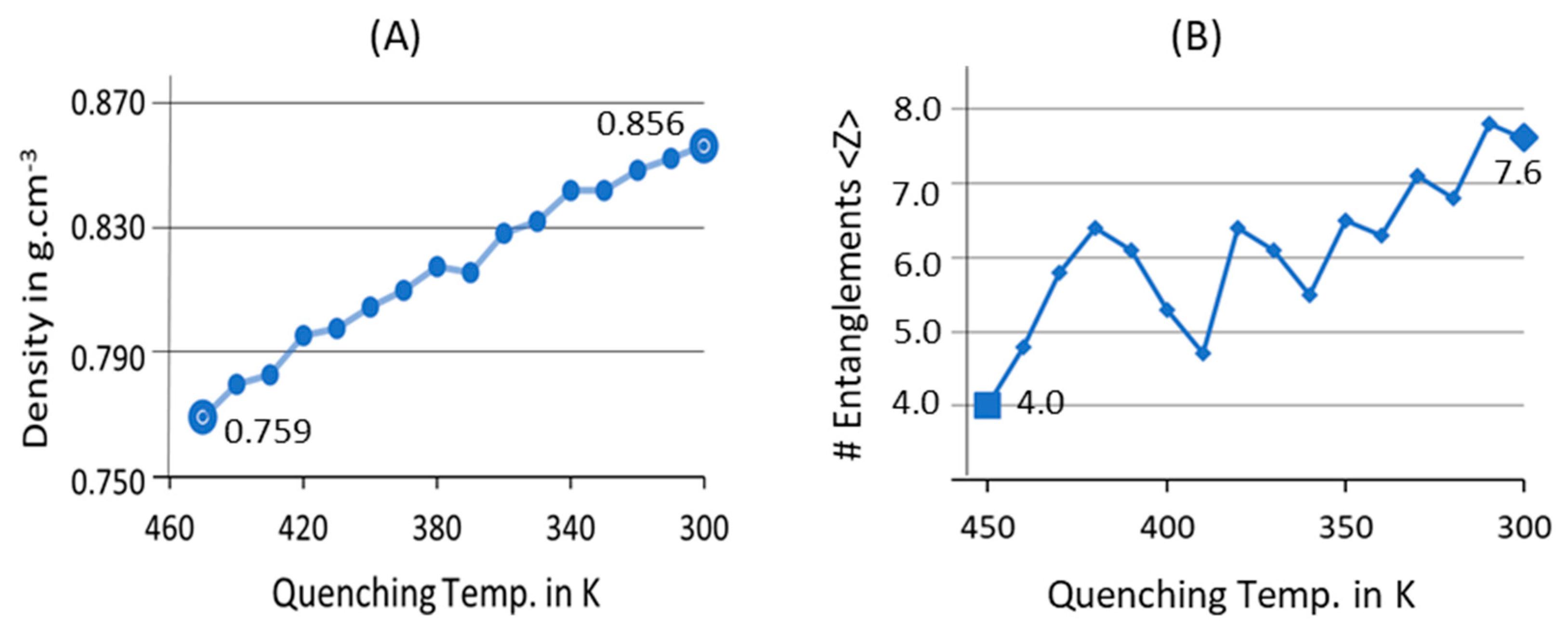
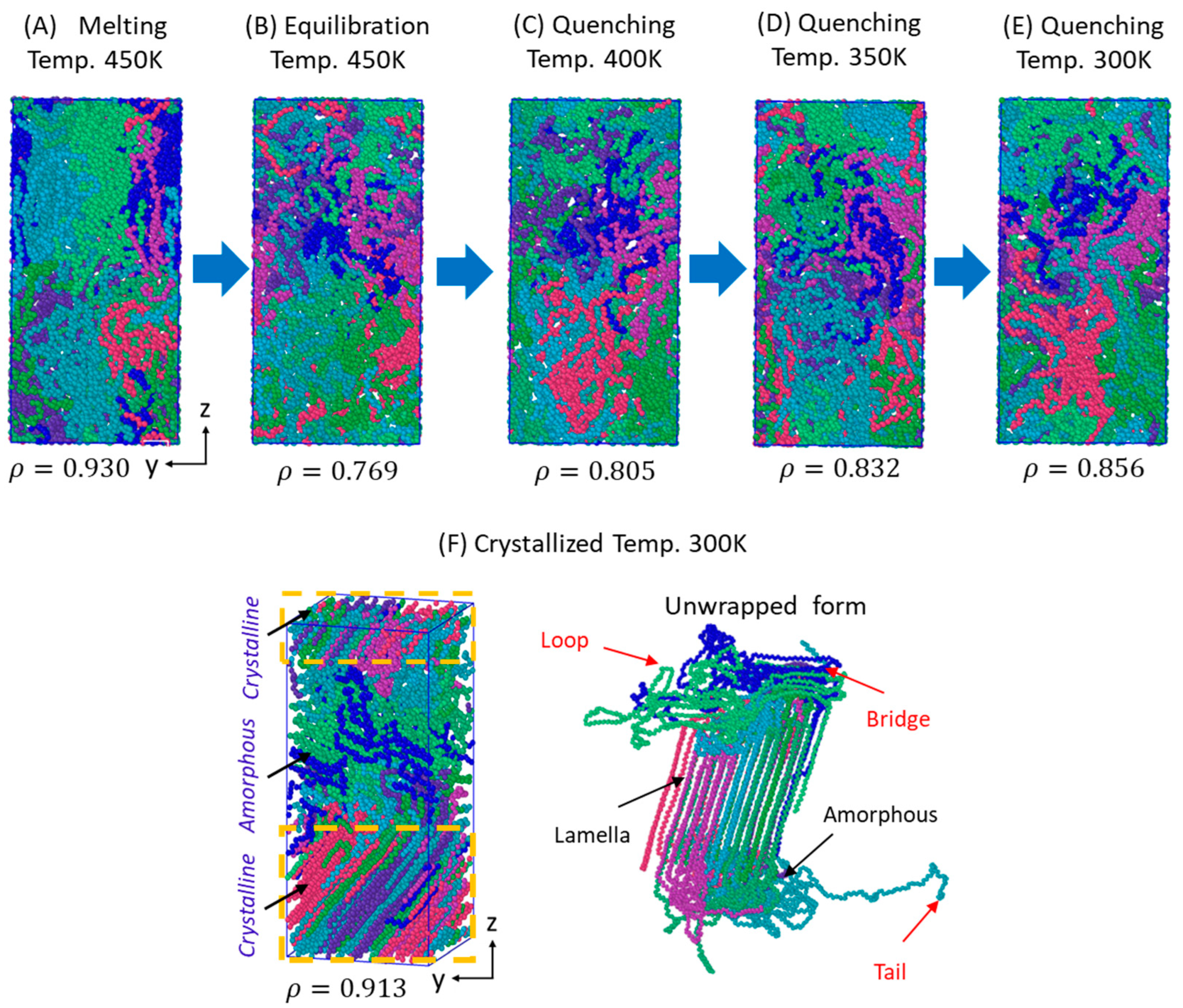
3.1. Melt-Equilibration and Deep Quenching
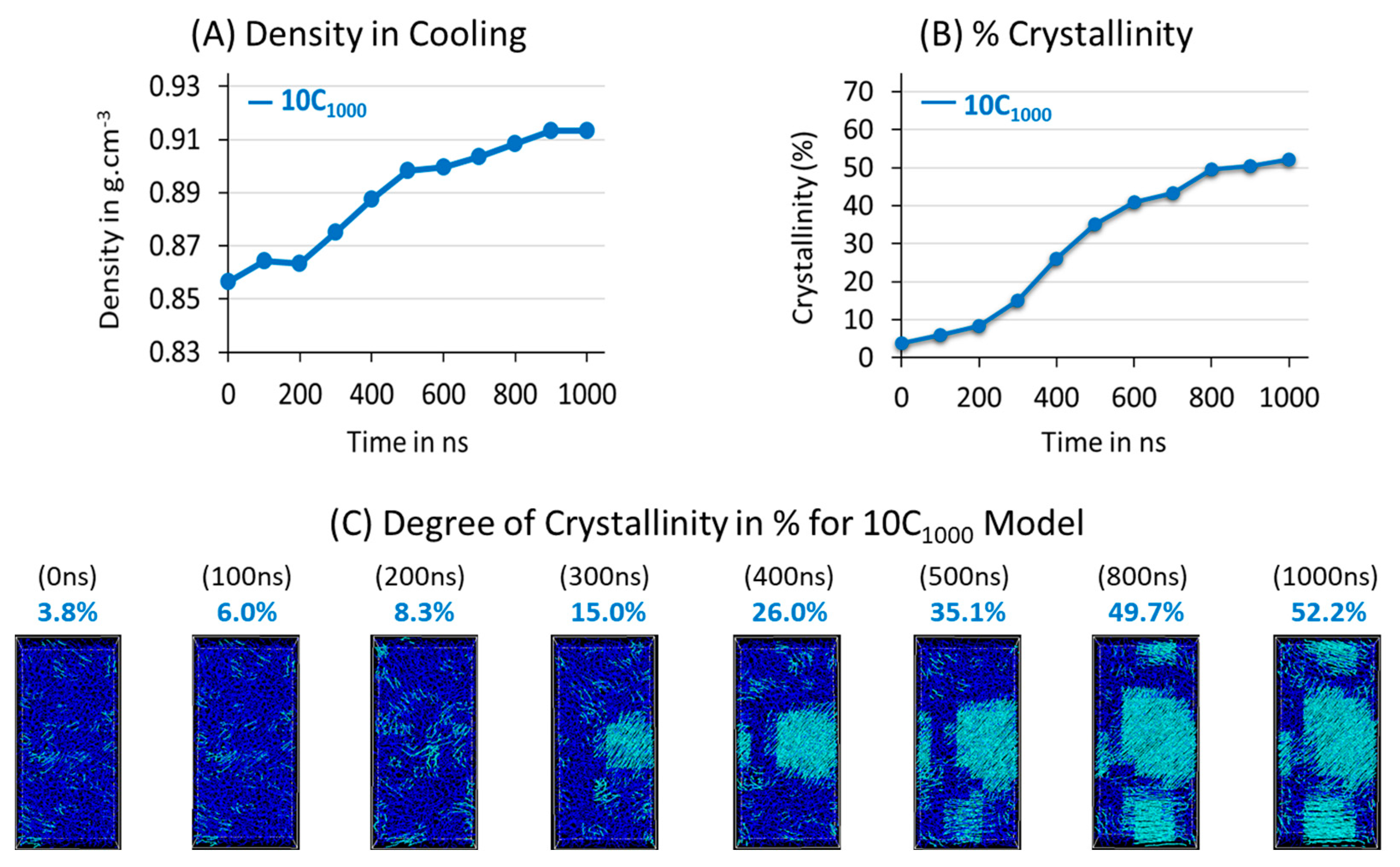
3.2. Isothermal Crystallization from Melt
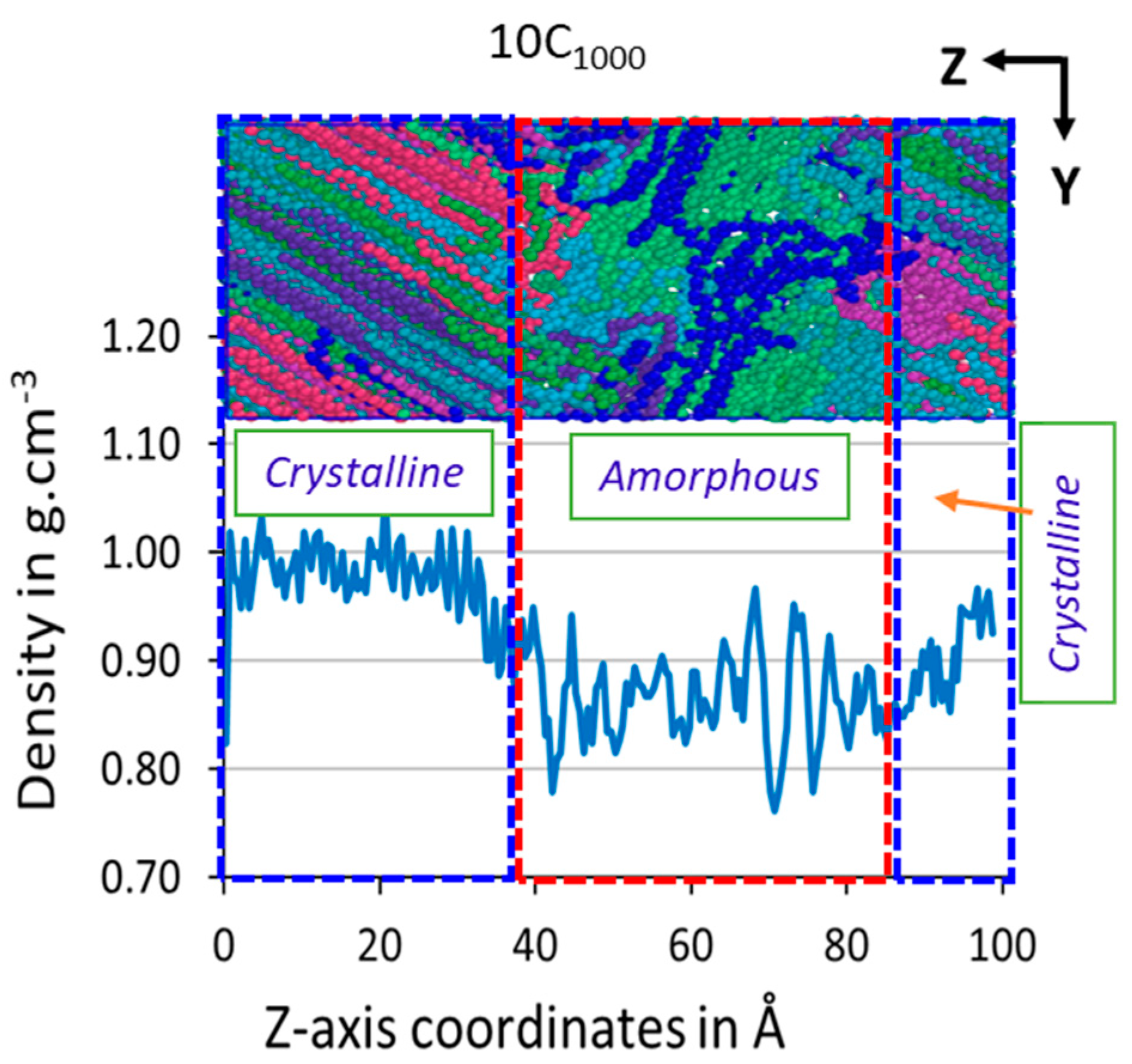
3.3. Z-Axis Density {ρ(z)}
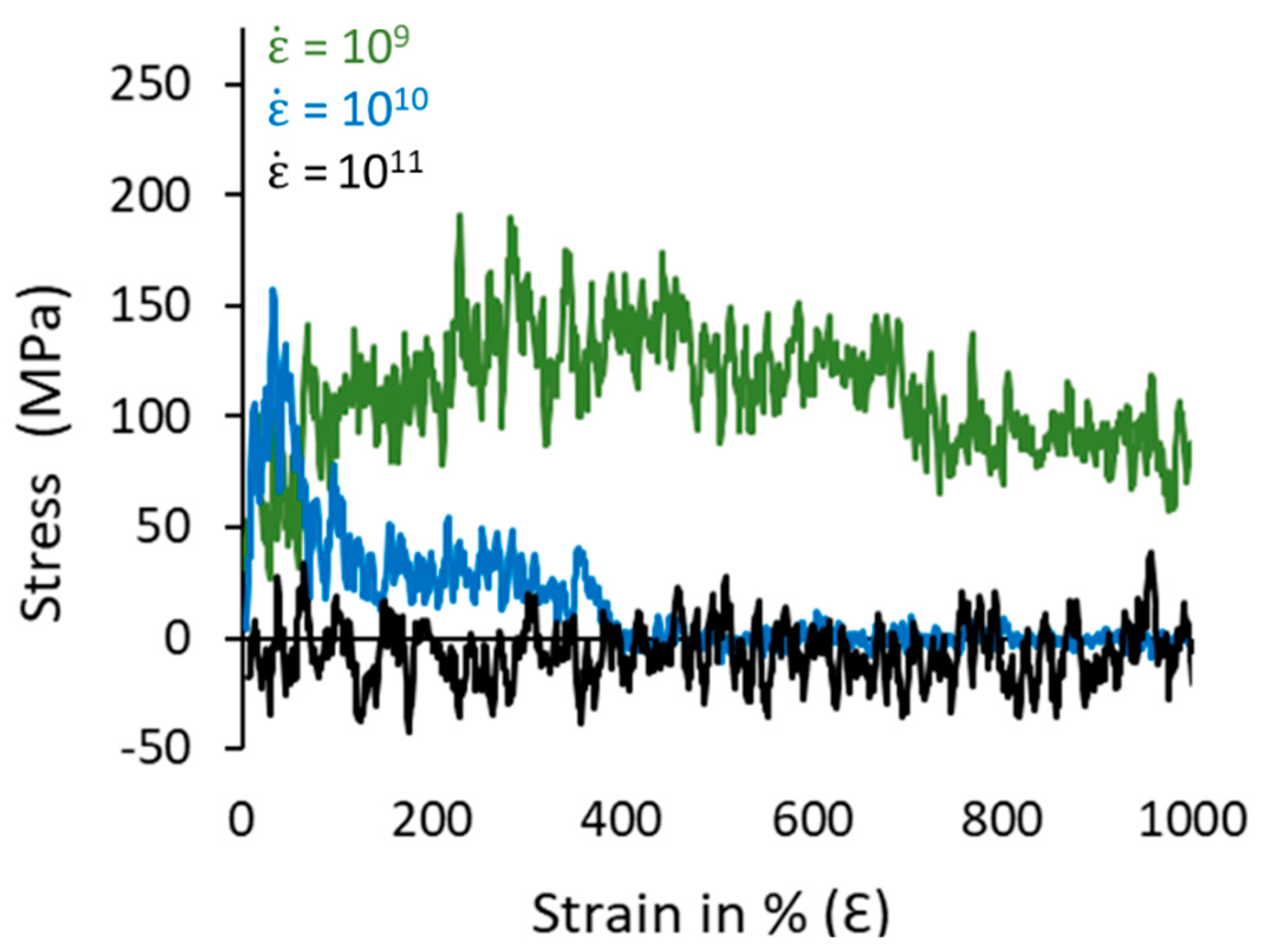
3.4. Mechanical Properties and Microstructure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ministry of the Environment. Resource Circulation Strategy for Plastics. Available online: https://www.env.go.jp/en/focus/jeq/issue/pdf/Volume20January2018.pdf (accessed on 16 July 2021).
- Gareiou, Z.; Chroni, C.; Kontoleon, K.; El Bachawati, M.; Saba, M.; Herrero Martin, R.; Zervas, E. Awareness of Citizens for the Single-Use Plastics: Comparison between a High-Income and an Upper-Middle-Income Economy of the Easter Mediterranean Region, Greece and Lebanon. Sustainability 2022, 14, 1912. [Google Scholar] [CrossRef]
- Hussain, M.A.; Yamamoto, T.; Adil, S.F.; Yao, S. Preparation and Characterization of High-Density Polyethylene with Alternating Lamellar Stems Using Molecular Dynamics Simulations. Polymers 2024, 16, 304. [Google Scholar] [CrossRef]
- Yamamoto, T.; Hussain, M.A.; Yao, S. Material Property Recovery by Controlling the Melt Memory Effects on Recrystallization and on Crystal Deformation: An Approach by the Molecular Dynamics Simulation for Polyethylene. Polymers 2022, 14, 3082. [Google Scholar] [CrossRef] [PubMed]
- Kavassalis, T.A.; Sundararajan, P.R. A molecular-dynamics study of polyethylene crystallization. Macromolecules 1993, 26, 4144–4150. [Google Scholar] [CrossRef]
- Taherzadehboroujeni, M.; Case, S.W. Mechanical Characterization and Numerical Modeling of High-Density Polyethylene Pipes. In Dynamic Substructures, Volume 4: Proceedings of the 37th IMAC, A Conference and Exposition on Structural Dynamics 2019; Conference Proceedings of the Society for Experimental Mechanics Series; Linderholt, A., Allen, M., Mayes, R., Rixen, D., Eds.; Springer: Cham, Switzerland, 2020; Volume 4. [Google Scholar] [CrossRef]
- da Costa-Haveroth, T.C.; Haveroth, G.A.; Kühl, A.; Boldrini, J.L.; Bittencourt, M.L.; Sasse, F.D.; Polak, M.A.; Muñoz-Rojas, P.A. Aspects on viscoelasticity modeling of HDPE using fractional derivatives: Interpolation procedures and efficient numerical scheme. Mech. Adv. Mater. Struct. 2021, 29, 4343–4358. [Google Scholar] [CrossRef]
- Qi, Z.; Hu, N.; Li, G.; Zeng, D.; Su, X. Constitutive modeling for the elastic-viscoplastic behavior of high-density polyethylene under cyclic loading. Int. J. Plast. 2019, 113, 125–144. [Google Scholar] [CrossRef]
- Gedde, U.W.; Hedenqvist, M.S. Fundamental Polymer Science, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar] [CrossRef]
- Pandey, A.; Champouret, Y.; Rastogi, S. Heterogeneity in the Distribution of Entanglement Density during Polymerization in Disentangled Ultrahigh Molecular Weight Polyethylene. Macromolecules 2011, 44, 4952–4960. [Google Scholar] [CrossRef]
- Pawlak, A.; Krajenta, J.; Galeski, A. The Crystallization of Polypylene with Reduced Density of Entanglements. J. Polym. Sci. B Polym. Phys. 2017, 55, 748–756. [Google Scholar] [CrossRef]
- Lippits, D.R.; Rastogi, S.; Höhne, G.W.H.; Mezari, B.; Magusin, P.C.M.M. Heterogeneous Distribution of Entanglements in the Polymer Melt and Its Influence on Crystallization. Macromolecules 2007, 40, 1004–1010. [Google Scholar] [CrossRef]
- Ajji, A.; Carriau, P.J.; Schriber, H.P.; Rudin, A. Chain Entanglement and Viscoelastic Properties of Molten Polymers. J. Polym. Sci. B Polym. Phys. 1986, 24, 1983–1990. [Google Scholar] [CrossRef]
- Sharma, G.K. Easily Processable Ultra-High Molecular Weight Polyethylene with Narrow Molecular Weight Distribution. Ph.D. Thesis, Eindhoven University of Technology, Eindhoven, The Netherlands, 2005. [Google Scholar]
- Lippits, D.R. Controlling the Melting Kinetics of Polymers; a Route to a New Melt State. Ph.D. Thesis, Eindhoven University of Technology, Eindhoven, The Netherlands, 2007. [Google Scholar]
- Yi, P.; Locker, C.R.; Rutledge, G.C. Molecular Dynamics Simulation of Homogeneous Crystal Nucleation in Polyethylene. Macromolecules 2013, 46, 4723–4733. [Google Scholar] [CrossRef]
- Sreenivas, K.; Kumaraswamy, G. Large amplitude oscillatory shear induces crystal chain orientation in velocity gradient direction. ACS Macro Lett. 2013, 3, 6–9. [Google Scholar] [CrossRef]
- Wang, B.; Cavallo, D.; Zhang, X.; Zhang, B.; Chen, J. Evolution of chain entanglements under large amplitude oscillatory shear flow and its effect on crystallization of isotactic polypropylene. Polymer 2020, 186, 121899. [Google Scholar] [CrossRef]
- Chandran, S.; Baschnagel, J.; Cangialosi, D.; Fukao, K.; Glynos, E.; Janssen, L.M.C.; Müller, M.; Muthukumar, M.; Steiner, U.; Xu, J.; et al. Processing pathways decide polymer properties at the molecular level. Macromolecules 2019, 52, 7146–7156. [Google Scholar] [CrossRef]
- Kaneyasu, H.; Phanthong, P.; Okubo, H.; Yao, S. Investigation of Degradation Mechanism from Shear Deformation and the Relationship with Mechanical Properties, Lamellar Size, and Morphology of High-Density Polyethylene. Appl. Sci. 2021, 11, 8436. [Google Scholar] [CrossRef]
- Yadegari, A.; Morshedian, J.; Khonakdar, H.A.; Wagenknecht, U.; Heinrich, G.; Malanin, M.; Eichhorn, K.J.; Fischer, D. Correlation of crystal alignment with interphase content in oriented high-density polyethylene cast films. CrystEngComm 2016, 18, 2337–2346. [Google Scholar] [CrossRef]
- Liu, Y.; Gao, S.; Hsiao, B.S.; Norman, A.; Tous, A.H.; Throckmorton, J.; Doufas, A.; Zhang, Y. Shear-induced crystallization of bimodal and unimodal high-density polyethylene. Polymer 2018, 153, 223–231. [Google Scholar] [CrossRef]
- Gartner, T.E., III; Jayaraman, A. Modeling and Simulations of Polymers: A Roadmap. Macromolecules 2019, 52, 755–786. [Google Scholar] [CrossRef]
- Graham, R.S. Understanding flow-induced Crystallization in Polymers: A Perspective on the Role of Molecular Simulations. J. Rheol. 2019, 63, 203–214. [Google Scholar] [CrossRef]
- Baig, C.; Edwards, B.J.; Keffer, D.J.; Cochran, H.D. A proper Approach for Nonequilibrium Molecular Dynamics Simulations of Planar Elongational Flow. J. Chem. Phys. 2005, 122, 114103. [Google Scholar] [CrossRef]
- Grommes, D.; Schenk, M.R.; Bruch, O.; Reith, D. Initial Crystallization Effects in Coarse-Grained Polyethylene Systems after Uni- and Biaxial Stretching in Blow-Molding Cooling Scenarios. Polymers 2022, 14, 5144. [Google Scholar] [CrossRef] [PubMed]
- Hagita, K.; Yamamoto, T.; Saito, H.; Abe, E. Chain-Level Analysis of Reinforced Polyethylene through Stretch-Induced Crystallization. ACS Macro Lett. 2024, 13, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Yamamoro, T. Molecular Dynamics Simulation of Stretch-Induced Crystallization in Polyethylene: Emergence of Fiber Structure and Molecular Network. Macromolecules 2019, 52, 1695–1706. [Google Scholar] [CrossRef]
- Chen, R.; Jiang, S.; Luo, C. Reconstruction of the entanglement network and its effect on the crystallization and mechanical strength of sintered polymers. Macromolecules 2023, 56, 5237–5247. [Google Scholar] [CrossRef]
- Olsson, P.A.; in’t Veld, P.J.; Andreasson, E.; Bergvall, E.; Jutemar, E.P.; Petersson, V.; Rutledge, G.C.; Kroon, M. All-atomic and coarse-grained molecular dynamics investigation of deformation in semi-crystalline lamellar polyethylene. Polymer 2018, 153, 305–316. [Google Scholar] [CrossRef]
- Yeh, I.C.; Lenhart, J.L.; Rutledge, G.C.; Andzelm, J.W. Molecular Dynamics Simulation of the Effects of Layer Thickness and Chain Tilt on Tensile Deformation Mechanisms of Semi-crystalline Polyethylene. Macromolecules 2017, 50, 1700–1712. [Google Scholar] [CrossRef]
- Kim, J.M.; Locker, R.; Rutledge, G.C. Plastic deformation of semi-crystalline polyethylene under extension, compression, and shear using molecular dynamics simulation. Macromolecules 2014, 47, 2515–2528. [Google Scholar] [CrossRef]
- Che, J.; Locker, C.R.; Lee, S.; Rutledge, G.C.; Hsiao, B.S.; Tsou, A.H. Plastic deformation of semi-crystalline polyethylene by X-ray scattering: Comparison with atomistic simulations. Macromolecules 2013, 46, 5279–5289. [Google Scholar] [CrossRef]
- Ghazavizadeh, A.; Rutledge, G.C.; Atai, A.A.; Ahzi, S.; Rémond, Y.; Soltani, N. Micromechanical characterization of the interphase layer in semi-crystalline polyethylene. J. Polym. Sci. Part B Polym. Phys. 2013, 51, 1228–1243. [Google Scholar] [CrossRef]
- Martin, M.G.; Siepmann, J.I. Transferable Potentials for Phase Equilibria. 1. United Atom Description of n-Alkanes. J. Phys. Chem. B 1998, 102, 2569–2577. [Google Scholar] [CrossRef]
- Ramos, J.; Vega, J.F.; Martínez-Salazar, J. Predicting Experimental Results for Polyethylene by Computer Simulation. Eur. Polym. J. 2018, 99, 298–331. [Google Scholar] [CrossRef]
- Paul, W.; Yoon, D.Y.; Smith, G.D. An Optimized United Atom Model for Simulations of Polymethylene Melts. J. Chem. Phys. 1995, 103, 1702–1709. [Google Scholar] [CrossRef]
- Waheed, N.; Lavine, M.S.; Rutledge, G.C. Molecular simulation of crystal growth in n-eicosane. J. Chem. Phys. 2002, 116, 2301–2309. [Google Scholar] [CrossRef]
- Ko, M.J.; Waheed, N.; Lavine, M.S.; Rutledge, G.C. Characterization of polyethylene crystallization from an oriented melt by molecular dynamics simulation. J. Chem. Phys. 2004, 121, 2823–2832. [Google Scholar] [CrossRef] [PubMed]
- in‘t Veld, P.J.; Rutledge, G.C. Temperature-dependent elasticity of a semi-crystalline interphase composed of freely rotating chains. Macromolecules 2003, 36, 7358–7365. [Google Scholar] [CrossRef]
- Gavartin, J.; Heine, T.; Romero, A.H. Forcite: A powerful molecular dynamics platform for simulations of materials and biological systems. J. Chem. Theory Comput. 2018, 14, 4811–4824. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Yamamoto, T. Molecular dynamics of crystallization in n-alkane mixtures; texture, compatibility, and diffusion in crystals. Polymer 2016, 99, 721–733. [Google Scholar] [CrossRef]
- Adhikari, S.; Muthukumar, M. Theory of statics of ties, loops, and tails in semi-crystalline polymers. J. Chem. Phys. 2019, 151, 114905. [Google Scholar] [CrossRef]
- Greathouse, J.A. Building LAMMPS Data Files with car/mdf Files and the msi2lmp Utility; Sandia National Laboratories: Albuquerque, NM, USA, 2010. [Google Scholar]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Integrated Simulation System for Polymeric Material Design (OCTA), Nagoya University Doi Project, Japan. Available online: http://octa.jp/index.html (accessed on 20 July 2022).
- Yi, P.; Rutledge, G.C. Molecular simulation of bundle-like crystal nucleation from n-eicosane melts. J. Chem. Phys. 2011, 135, 024903. [Google Scholar] [CrossRef] [PubMed]
- Hossain, D.; Tschopp, M.A.; Ward, D.K.; Bouvard, J.L.; Horstemeyer, M.F. Molecular dynamics simulations of deformation mechanisms of amorphous polyethylene. Polymer 2010, 51, 6071–6083. [Google Scholar] [CrossRef]
- Evans, D.J.; Holian, B.L. The Nosé_Hoover thermostat. J. Chem. Phys. 1985, 83, 4069–4074. [Google Scholar] [CrossRef]
- Kröger, M. Shortest multiple disconnected path for the analysis of entanglements in two- and three-dimensional polymeric systems. Comput. Phys. Commun. 2005, 168, 209–232. [Google Scholar] [CrossRef]
- Kröger, M.; Dietz, J.D.; Hoy, R.S.; Luap, C. The Z1+ package: Shortest multiple disconnected path for the analysis of entanglements in macromolecular systems. Comput. Phys. Commun. 2023, 283, 108567. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO—The Open Visualization Tool. Modell. Simul. Mater. Sci. Eng. 2010, 18, 015012. [Google Scholar] [CrossRef]
- Dong, X.; McDowell, D.L.; Kalidindi, S.R.; Jacob, K.I. Dependence of mechanical properties son crystal orientation of semi-crystalline polyethylene structures. Polymer 2014, 55, 4248–4257. [Google Scholar] [CrossRef]
- Jabbari-Faroujji, S.; Rottler, J.; Lame, O.; Makke, A.; Perez, M.; Barrat, J.L. Plastic Deformation Mechanisms of Semi-crystalline and Amorphous Polymers. ACS Macro Lett. 2015, 4, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Fall, W.S.; Baschnagel, J.; Benzerara, O.; Lhost, O.; Meyer, H. Molecular simulations of controlled polymer crystallization in polyethylene. ACS Macro Lett. 2023, 12, 808–813. [Google Scholar] [CrossRef]
- Ranganathan, R.; Kumar, V.; Nrayton, A.L.; Kröger, M.; Rutledge, G.C. Atomistic modeling of plastic deformation in semi-crystalline polyethylene: Role of interphase topology, entanglements, and chain dynamics. Macromolecules 2020, 53, 4605–4617. [Google Scholar] [CrossRef]
- Yeh, I.; Andzelm, J.W.; Rutledge, G.C. Mechanical and structural characterization of semi-crystalline polyethylene under tensile deformation by molecular dynamics simulations. Macromolecules 2015, 48, 4228–4239. [Google Scholar] [CrossRef]
- Belin, B.; Yiannourakou, M.; Lachet, V.; Rousseau, B. Modeling method for semi-crystalline polymers controlling aspects of the morphology at the molecular scale for the study of mechanical and physicochemical properties. J. Phys. Chem. B 2022, 126, 9673–9685. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Tian, Y.; Wu, H.; Li, J. Revealing the deformation mechanism of amorphous polyethylene subjected to cycle loading via molecular dynamics simulations. RSC Adv. 2018, 8, 32377–32386. [Google Scholar] [CrossRef] [PubMed]
- Hedeisiu, C.E. Structure-Property Relationships in Polyolefins. Ph.D. Thesis, RWTH Aachen University, Aachen, Germany, 2007. [Google Scholar]
- Hall, K.W.; Sirk, T.W.; Klein, M.L.; Shinode, W. A coarse-grain model for entangled polyethylene melts and polyethylene crystallization. J. Chem. Phys. 2019, 150, 24901. [Google Scholar] [CrossRef]
- Grommes, D.; Schenk, M.R.; Bruch, O.; Reith, D. Investigation of Crystallization and Relaxation effects in Coarse-Grained Polyethylene Systems after Uniaxial Stretching. Polymers 2021, 13, 4466. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bonded Terms | Non-Bonded Terms | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Bond | Angle | Dihedral Angle | Lennar-Jones (12-6 Potential) | |||||||
| Kb | req | Kθ | θeq | C0 | C1 | C2 | C3 | ε | σ | Cut-off (rcut) |
| 350.0 | 1.53 | 60.0 | 109.0 | 1.57 | −4.05 | 0.86 | 6.48 | 0.112 | 4.01 | 9.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain, M.A.; Yamamoto, T.; Adil, S.F.; Yao, S. Coarse-Grained Simulations on Polyethylene Crystal Network Formation and Microstructure Analysis. Polymers 2024, 16, 1007. https://doi.org/10.3390/polym16071007
Hussain MA, Yamamoto T, Adil SF, Yao S. Coarse-Grained Simulations on Polyethylene Crystal Network Formation and Microstructure Analysis. Polymers. 2024; 16(7):1007. https://doi.org/10.3390/polym16071007
Chicago/Turabian StyleHussain, Mohammed Althaf, Takashi Yamamoto, Syed Farooq Adil, and Shigeru Yao. 2024. "Coarse-Grained Simulations on Polyethylene Crystal Network Formation and Microstructure Analysis" Polymers 16, no. 7: 1007. https://doi.org/10.3390/polym16071007
APA StyleHussain, M. A., Yamamoto, T., Adil, S. F., & Yao, S. (2024). Coarse-Grained Simulations on Polyethylene Crystal Network Formation and Microstructure Analysis. Polymers, 16(7), 1007. https://doi.org/10.3390/polym16071007