
Performance of Recycled Polylactic Acid/Amorphous Polyhydroxyalkanoate Blends


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Extrusion and Molding of the Blends
2.3. Characterization of the Blends
3. Results and Discussion
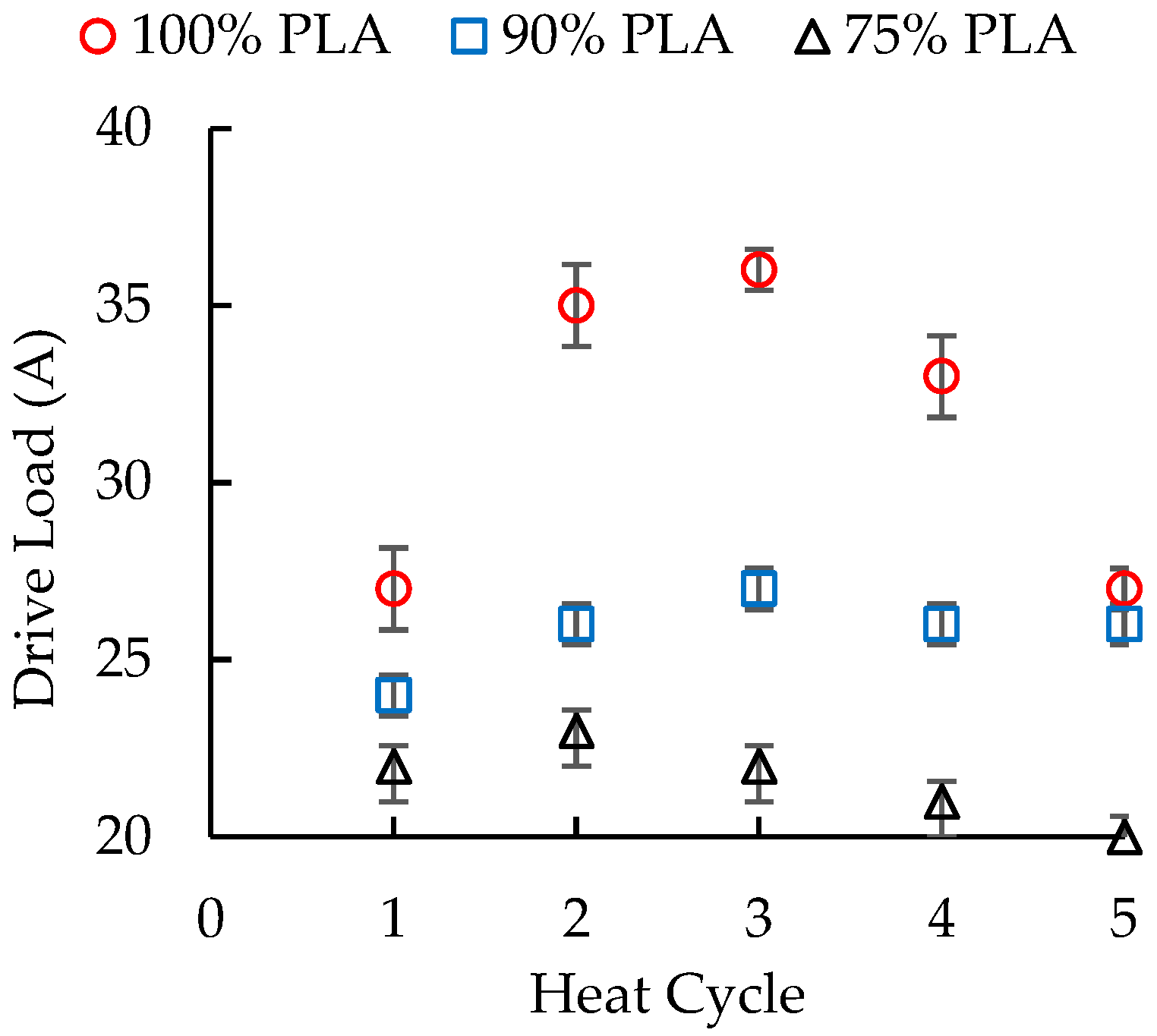
3.1. Performance during Extrusion and Injection Molding
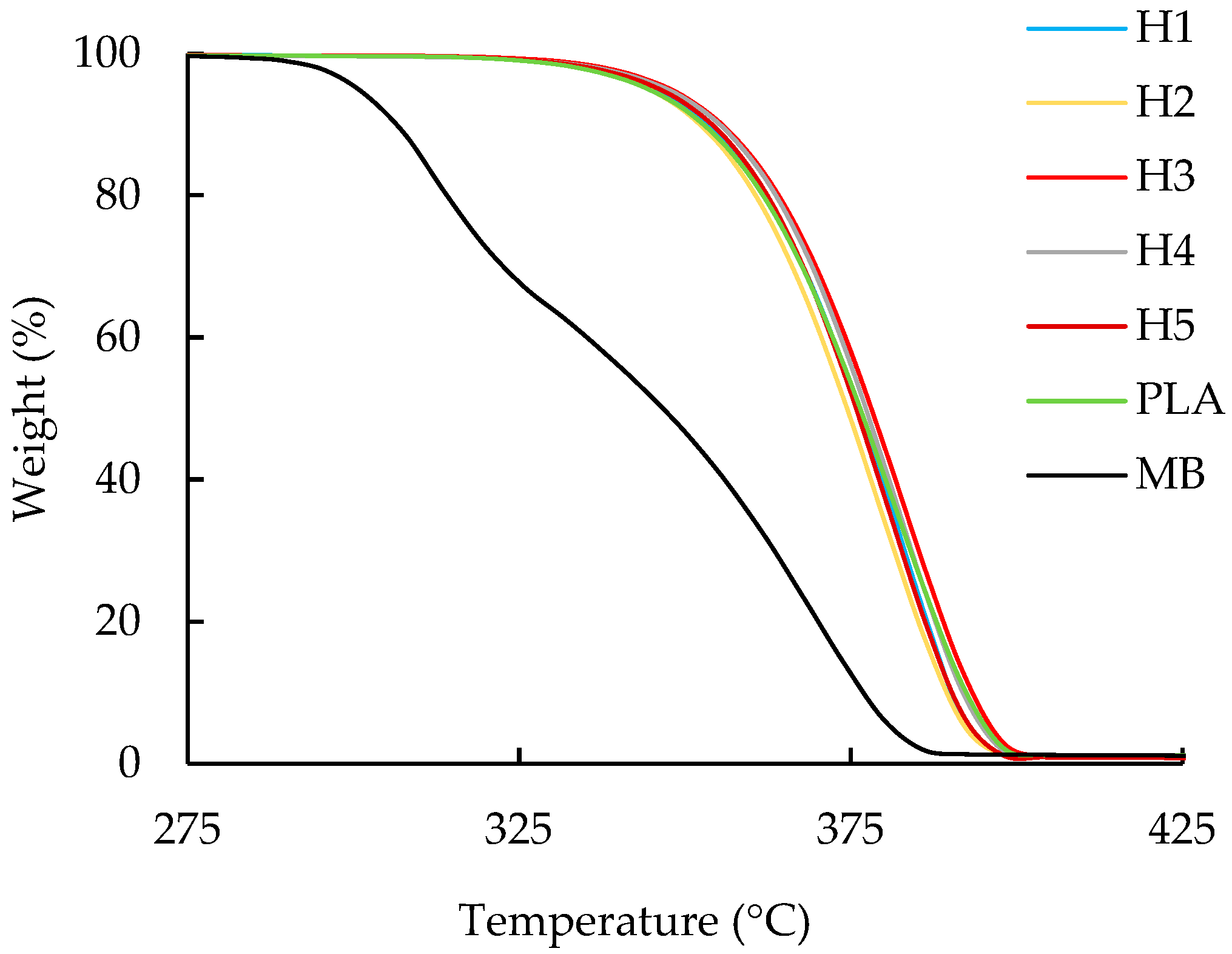
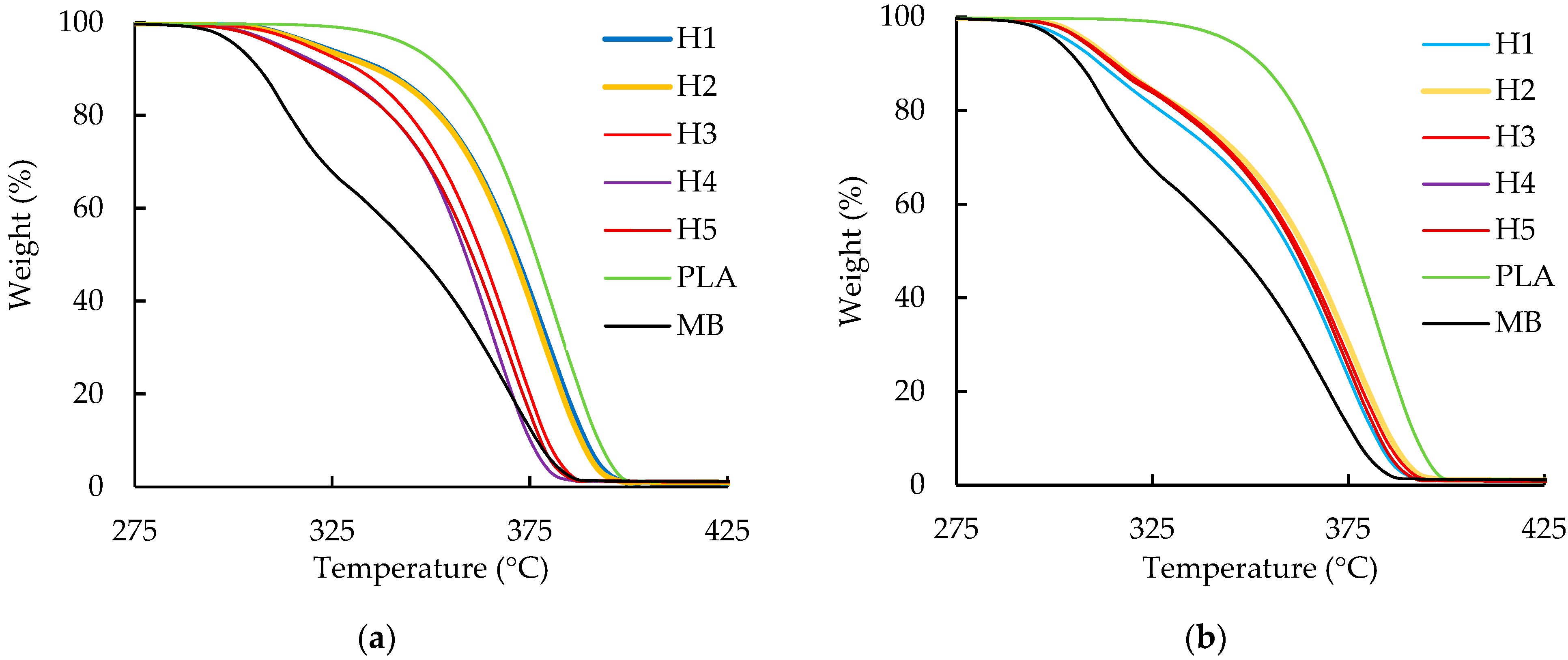
3.2. Thermogravimetric Analysis
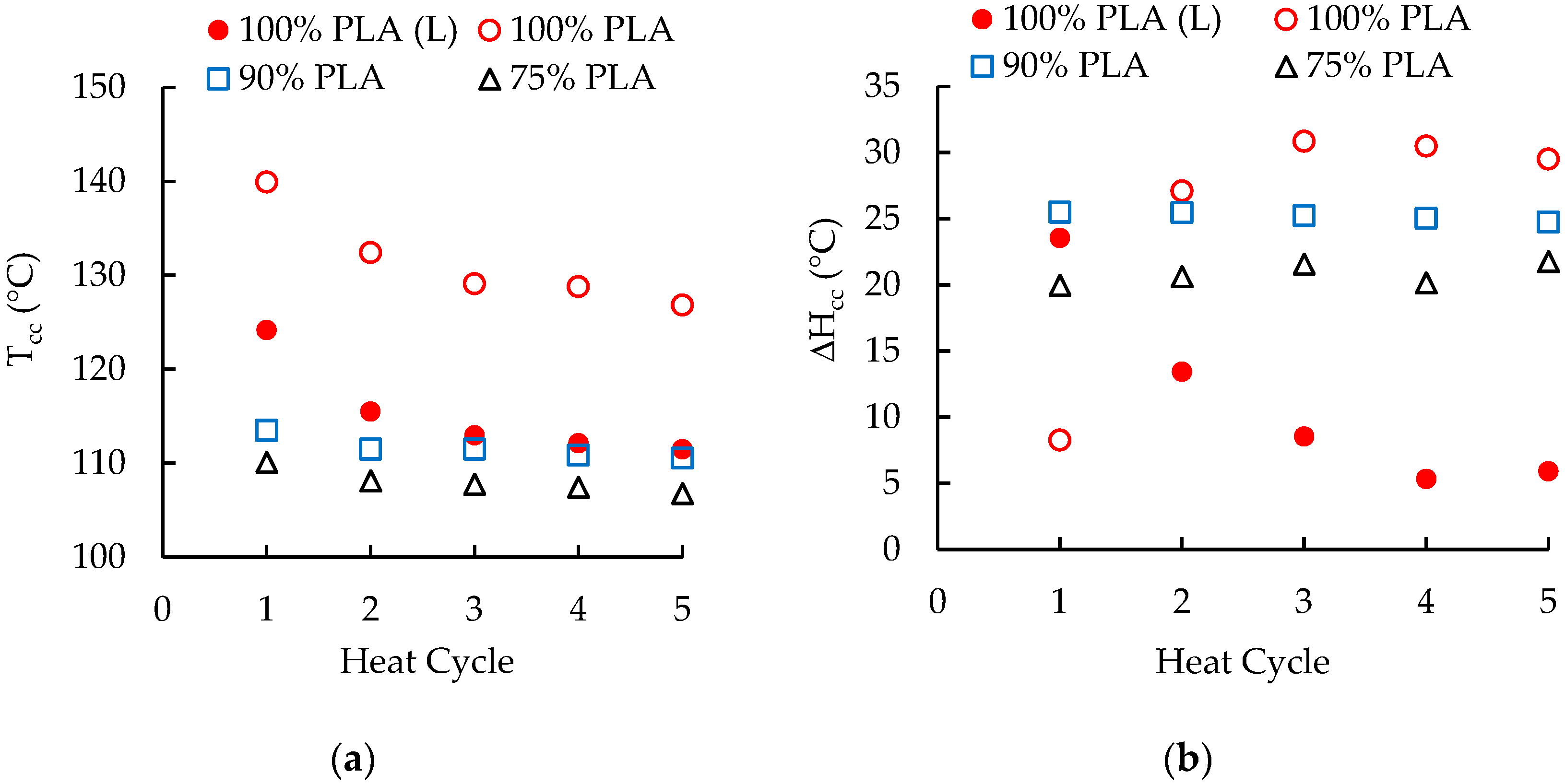
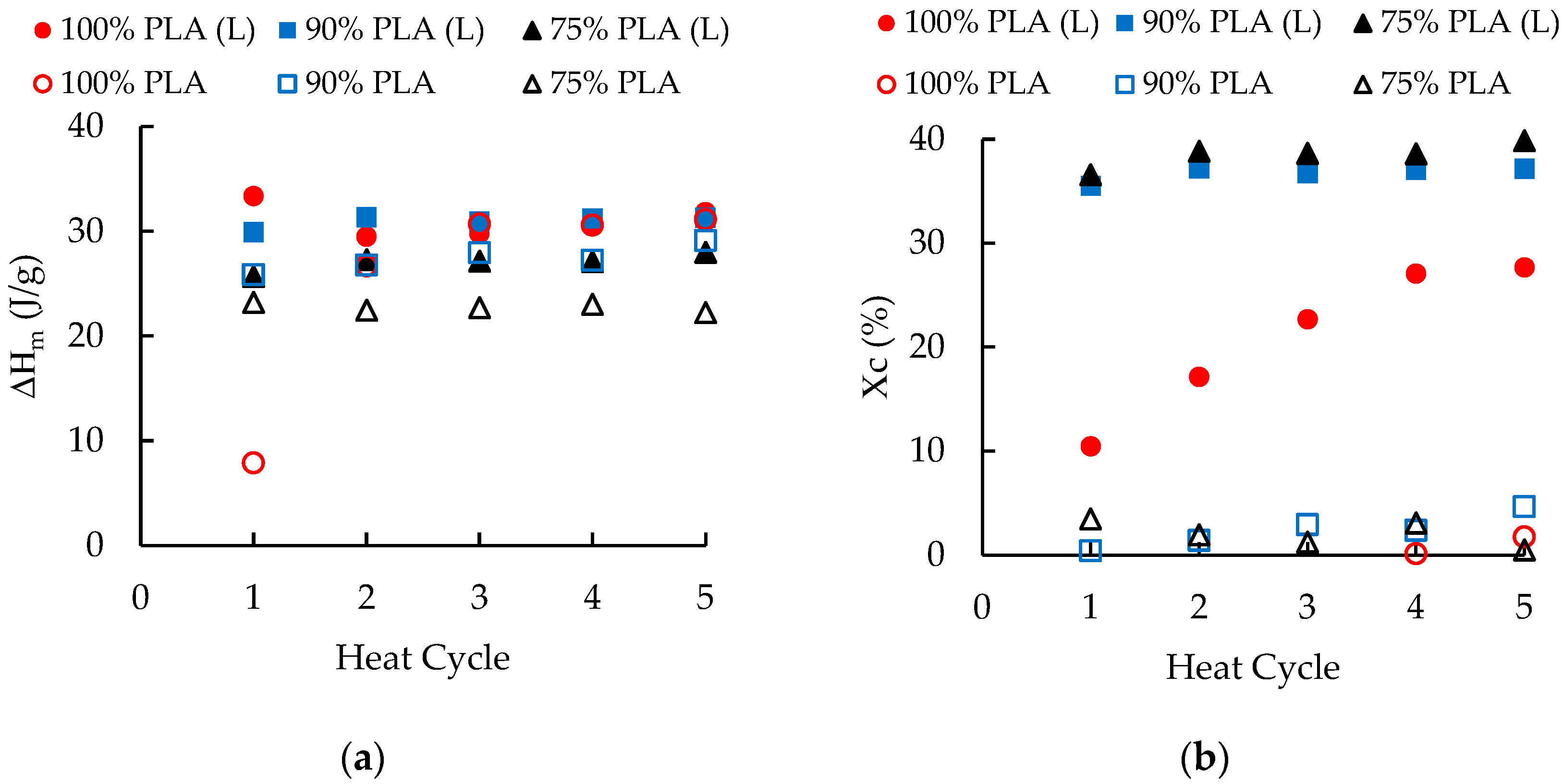
3.3. Differential Scanning Calorimetry
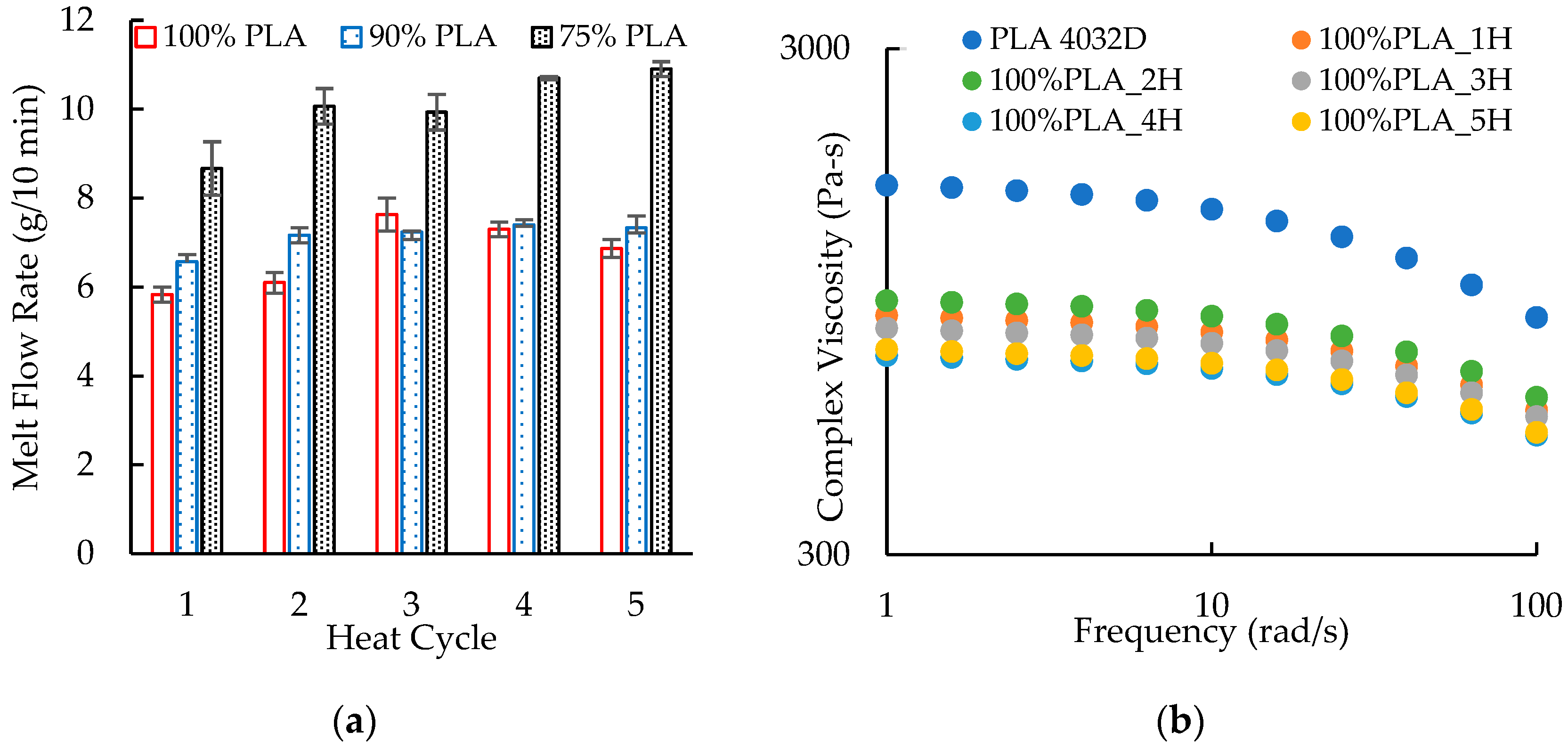
3.4. Melt Index
3.5. Parallel Plate Rheology
3.6. FTIR
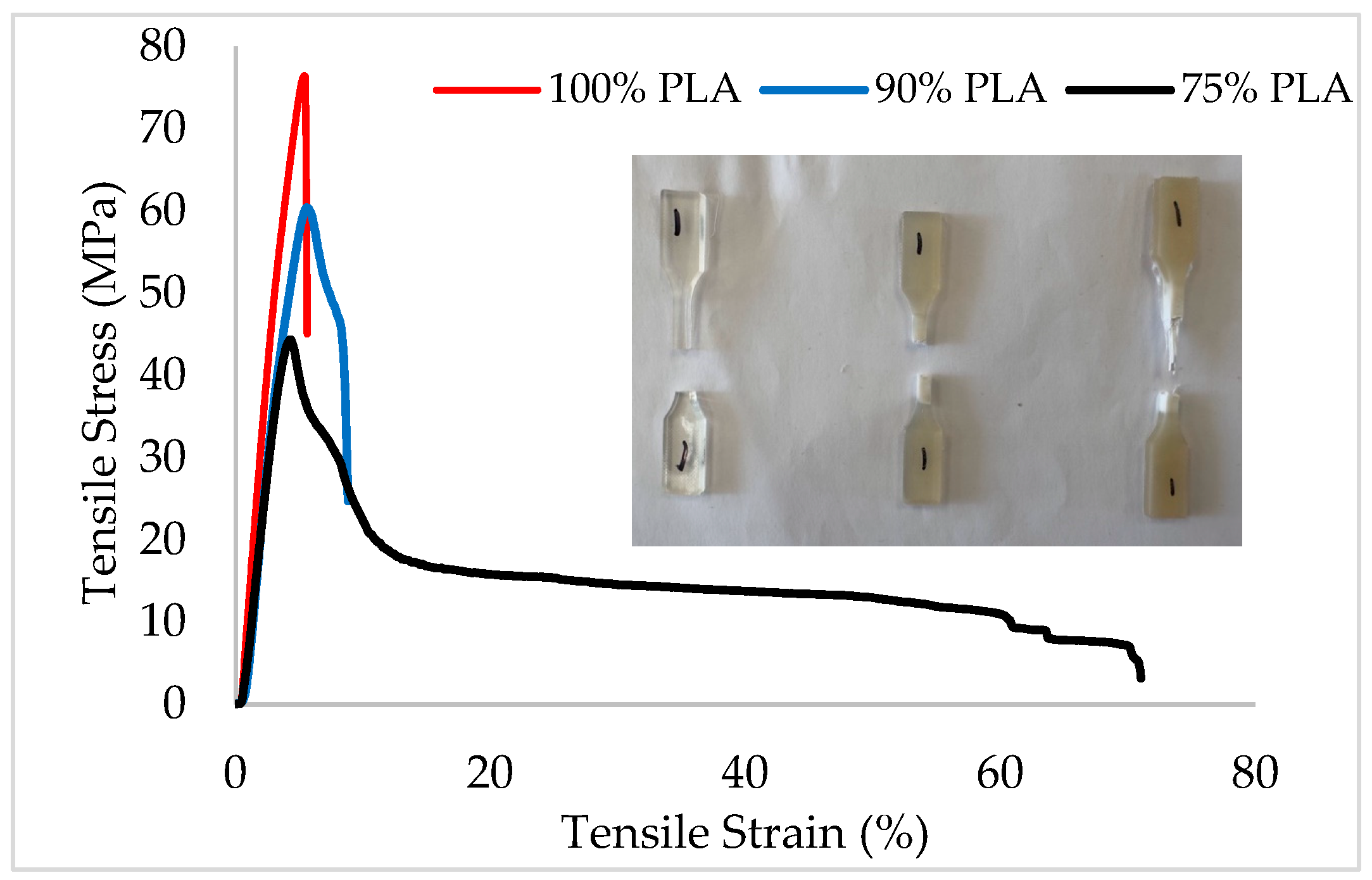
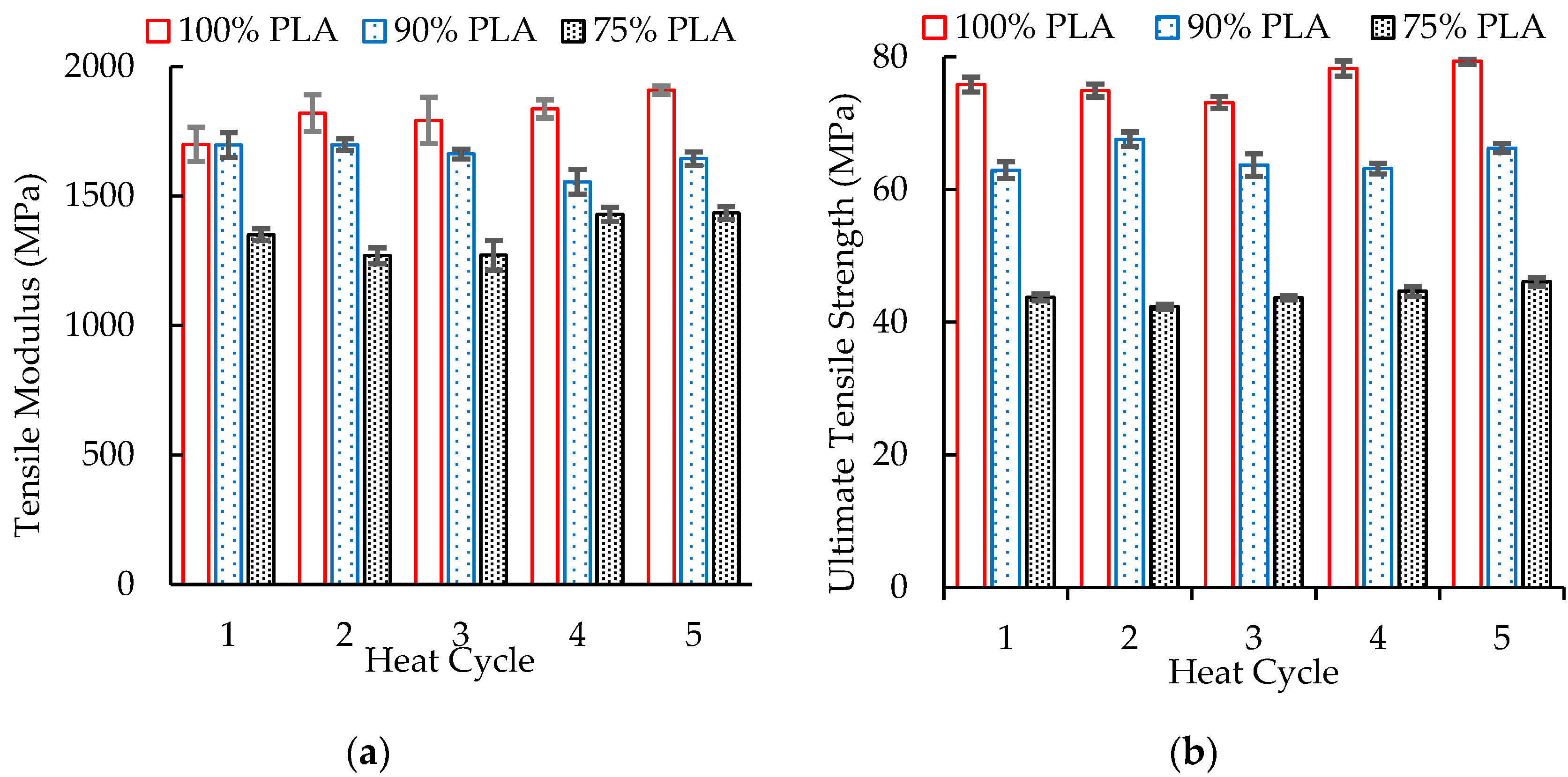
3.7. Mechanical Properties of Blends
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Law, K.L.; Narayan, R. Reducing Environmental Plastic Pollution by Designing Polymer Materials for Managed End-of-Life. Nat. Rev. Mater. 2021, 7, 104–116. [Google Scholar] [CrossRef]
- Thermoformed Plastics Market Size & Growth Report by 2030. Available online: https://marketresearchcommunity.com/thermoformed-plastics-market/?gad=1&gclid=EAIaIQobChMI1vn2taWa_wIVY4NbCh3c_QCOEAAYAiAAEgJrpPD_BwE (accessed on 28 March 2024).
- KC—Size Reduction|Plastics Technology. Available online: https://www.ptonline.com/kc/plastics-size-reduction (accessed on 28 March 2024).
- EXTRUSION: Managing Regrind|Plastics Technology. Available online: https://www.ptonline.com/articles/extrusion-managing-regrind (accessed on 28 March 2024).
- Jerez, A.; Partal, P.; Martínez, I.; Gallegos, C.; Guerrero, A. Rheology and Processing of Gluten Based Bioplastics. Biochem. Eng. J. 2005, 26, 131–138. [Google Scholar] [CrossRef]
- Jerez, A.; Partal, P.; Martínez, I.; Gallegos, C.; Guerrero, A. Protein-Based Bioplastics: Effect of Thermo-Mechanical Processing. Rheol. Acta 2007, 46, 711–720. [Google Scholar] [CrossRef]
- Nafchi, A.M.; Alias, A.K.; Mahmud, S.; Robal, M. Antimicrobial, Rheological, and Physicochemical Properties of Sago Starch Films Filled with Nanorod-Rich Zinc Oxide. J. Food Eng. 2012, 113, 511–519. [Google Scholar] [CrossRef]
- Xie, F.; Pollet, E.; Halley, P.J.; Avérous, L. Starch-Based Nano-Biocomposites. Prog. Polym. Sci. 2013, 38, 1590–1628. [Google Scholar] [CrossRef]
- Lackner, M. Biopolymers. In Handbook of Climate Change Mitigation and Adaptation; Springer: New York, NY, USA, 2016; Volume 4, pp. 3211–3230. [Google Scholar]
- Enriquez, E.; Mohanty, A.K.; Misra, M. Biobased Polymer Blends of Poly(Trimethylene Terephthalate) and High Density Polyethylene. Mater. Des. 2016, 90, 984–990. [Google Scholar] [CrossRef]
- Siracusa, V.; Blanco, I. Bio-Polyethylene (Bio-PE), Bio-Polypropylene (Bio-PP) and Bio-Poly(Ethylene Terephthalate) (Bio-PET): Recent Developments in Bio-Based Polymers Analogous to Petroleum-Derived Ones for Packaging and Engineering Applications. Polymers 2020, 12, 1641. [Google Scholar] [CrossRef] [PubMed]
- Samir, A.; Ashour, F.H.; Hakim, A.A.A.; Bassyouni, M. Recent Advances in Biodegradable Polymers for Sustainable Applications. NPJ Mater. Degrad. 2022, 6, 1–28. [Google Scholar] [CrossRef]
- Robertson, G.L. Food Packaging: Principles and Practice, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2016; pp. 1–688. [Google Scholar] [CrossRef]
- Alex Tullo. A Biodegradable Polymer Hits the Big Time. CEN Glob. Enterp. 2021, 99, 26–27. [Google Scholar] [CrossRef]
- Bo, L.; Guan, T.; Wu, G.; Ye, F.; Weng, Y. Biodegradation Behavior of Degradable Mulch with Poly (Butylene Adipate-Co-Terephthalate) (PBAT) and Poly (Butylene Succinate) (PBS) in Simulation Marine Environment. Polymers 2022, 14, 1515. [Google Scholar] [CrossRef] [PubMed]
- Thakur, M.; Majid, I.; Hussain, S.; Nanda, V. Poly(ε-Caprolactone): A Potential Polymer for Biodegradable Food Packaging Applications. Packag. Technol. Sci. 2021, 34, 449–461. [Google Scholar] [CrossRef]
- Peelman, N.; Ragaert, P.; De Meulenaer, B.; Adons, D.; Peeters, R.; Cardon, L.; Van Impe, F.; Devlieghere, F. Application of Bioplastics for Food Packaging. Trends Food Sci. Technol. 2013, 32, 128–141. [Google Scholar] [CrossRef]
- George, A.; Sanjay, M.R.; Srisuk, R.; Parameswaranpillai, J.; Siengchin, S. A Comprehensive Review on Chemical Properties and Applications of Biopolymers and Their Composites. Int. J. Biol. Macromol. 2020, 154, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Razza, F.; Briani, C.; Breton, T.; Marazza, D. Metrics for Quantifying the Circularity of Bioplastics: The Case of Bio-Based and Biodegradable Mulch Films. Resour. Conserv. Recycl. 2020, 159, 104753. [Google Scholar] [CrossRef]
- Ibrahim, N.I.; Shahar, F.S.; Hameed Sultan, M.T.; Md Shah, A.U.; Azrie Safri, S.N.; Mat Yazik, M.H. Overview of Bioplastic Introduction and Its Applications in Product Packaging. Coatings 2021, 11, 1423. [Google Scholar] [CrossRef]
- Madhavan Nampoothiri, K.; Nair, N.R.; John, R.P. An Overview of the Recent Developments in Polylactide (PLA) Research. Bioresour. Technol. 2010, 101, 8493–8501. [Google Scholar] [CrossRef]
- Tripathi, N.; Misra, M.; Mohanty, A.K. Durable Polylactic Acid (PLA)-Based Sustainable Engineered Blends and Biocomposites: Recent Developments, Challenges, and Opportunities. ACS Eng. Au 2021, 1, 7–38. [Google Scholar] [CrossRef]
- Balla, E.; Daniilidis, V.; Karlioti, G.; Kalamas, T.; Stefanidou, M.; Bikiaris, N.D.; Vlachopoulos, A.; Koumentakou, I.; Bikiaris, D.N. Poly(Lactic Acid): A Versatile Biobased Polymer for the Future with Multifunctional Properties—From Monomer Synthesis, Polymerization Techniques and Molecular Weight Increase to PLA Applications. Polymers 2021, 13, 1822. [Google Scholar] [CrossRef] [PubMed]
- Babu, R.P.; O’Connor, K.; Seeram, R. Current Progress on Bio-Based Polymers and Their Future Trends. Prog. Biomater. 2013, 2, 8. [Google Scholar] [CrossRef]
- Koller, M.; Mukherjee, A. A New Wave of Industrialization of PHA Biopolyesters. Bioengineering 2022, 9, 74. [Google Scholar] [CrossRef]
- Jo, M.; Jang, Y.; Lee, E.; Shin, S.; Kang, H.J. The Modification of Poly(3-Hydroxybutyrate-Co-4-Hydroxybutyrate) by Melt Blending. Polymers 2022, 14, 1725. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Diaz, C.; Yi, H.; Pao, S. Film Performance of Poly(Lactic Acid) Blends for Packaging Applications. J. Appl. Packag. Res. 2016, 8, 4. [Google Scholar] [CrossRef]
- Zhang, M.; Thomas, N.L. Blending Polylactic Acid with Polyhydroxybutyrate: The Effect on Thermal, Mechanical, and Biodegradation Properties. Adv. Polym. Technol. 2011, 30, 67–79. [Google Scholar] [CrossRef]
- Tri, P.N.; Domenek, S.; Guinault, A.; Sollogoub, C. Crystallization Behavior of Poly(Lactide)/Poly(β-Hydroxybutyrate)/Talc Composites. J. Appl. Polym. Sci. 2013, 129, 3355–3365. [Google Scholar] [CrossRef]
- Arrieta, M.P.; López, J.; Hernández, A.; Rayón, E. Ternary PLA–PHB–Limonene Blends Intended for Biodegradable Food Packaging Applications. Eur. Polym. J. 2014, 50, 255–270. [Google Scholar] [CrossRef]
- Burgos, N.; Armentano, I.; Fortunati, E.; Dominici, F.; Luzi, F.; Fiori, S.; Cristofaro, F.; Visai, L.; Jiménez, A.; Kenny, J.M. Functional Properties of Plasticized Bio-Based Poly(Lactic Acid)_Poly(Hydroxybutyrate) (PLA_PHB) Films for Active Food Packaging. Food Bioproc. Tech. 2017, 10, 770–780. [Google Scholar] [CrossRef]
- Pillin, I.; Montrelay, N.; Bourmaud, A.; Grohens, Y. Effect of Thermo-Mechanical Cycles on the Physico-Chemical Properties of Poly(Lactic Acid). Polym. Degrad. Stab. 2008, 93, 321–328. [Google Scholar] [CrossRef]
- Zenkiewicz, M.; Richert, J.; Rytlewski, P.; Moraczewski, K.; Stepczyńska, M.; Karasiewicz, T. Characterisation of Multi-Extruded Poly(Lactic Acid). Polym. Test. 2009, 28, 412–418. [Google Scholar] [CrossRef]
- Zembouai, I.; Bruzaud, S.; Kaci, M.; Benhamida, A.; Corre, Y.M.; Grohens, Y. Mechanical Recycling of Poly(3-Hydroxybutyrate-Co-3-Hydroxyvalerate)/Polylactide Based Blends. J. Polym. Environ. 2014, 22, 449–459. [Google Scholar] [CrossRef]
- Farias, N.C.; Major, I.; Devine, D.; Brennan Fournet, M.; Pezzoli, R.; Farshbaf Taghinezhad, S.; Hesabi, M. Multiple Recycling of a PLA/PHB Biopolymer Blend for Sustainable Packaging Applications: Rheology-Morphology, Thermal, and Mechanical Performance Analysis. Polym. Eng. Sci. 2022, 62, 1764–1774. [Google Scholar] [CrossRef]
- Krishnaswamy, R. Toughening Polylactic Acid with Polyhydroxyalkanoates. U.S. Patent No. 9,328,239, 17 May 2011. [Google Scholar]
- Ingeo Biopolymer 4032D Technical Data Sheet Extrusion Grade for Higher Heat Products, NatureWorks. Available online: https://www.natureworksllc.com/~/media/Files/NatureWorks/Technical-Documents/Technical-Data-Sheets/TechnicalDataSheet_4032D_general_pdf.pdf?la=en (accessed on 22 April 2024).
- ASTM D638-14; Standard Test Method for Tensile Properties of Plastics. ASTM International: West Conshohocken, PA, USA, 2022.
- ASTM D256-10 (2018); Standard Test Methods for Determining the Izod Pendulum Impact Resistance of Plastics. ASTM International: West Conshohocken, PA, USA, 2018.
- ASTM D1238-10; Standard Test Methods for Melt Flow Rates of Thermoplastics by Extrusion Plastometer. ASTM International: West Conshohocken, PA, USA, 2013.
- Yoo, H.M.; Jeong, S.Y.; Choi, S.W. Analysis of the Rheological Property and Crystallization Behavior of Polylactic Acid (IngeoTM Biopolymer 4032D) at Different Process Temperatures. E-Polymers 2021, 21, 702–709. [Google Scholar] [CrossRef]
- Mallet, B.; Lamnawar, K.; Maazouz, A. Improvement of Blown Film Extrusion of Poly(Lactic Acid): Structure–Processing–Properties Relationships. Polym. Eng. Sci. 2014, 54, 840–857. [Google Scholar] [CrossRef]
- Khoo, R.Z.; Ismail, H.; Chow, W.S. Thermal and Morphological Properties of Poly (Lactic Acid)/Nanocellulose Nanocomposites. Procedia Chem. 2016, 19, 788–794. [Google Scholar] [CrossRef]
- Othman, N.; Acosta-Ramírez, A.; Mehrkhodavandi, P.; Dorgan, J.R.; Hatzikiriakos, S.G. Solution and Melt Viscoelastic Properties of Controlled Microstructure Poly(Lactide). J. Rheol. 2011, 55, 987–1005. [Google Scholar] [CrossRef]
- Beltrán, F.R.; Lorenzo, V.; de la Orden, M.U.; Martínez-Urreaga, J. Effect of Different Mechanical Recycling Processes on the Hydrolytic Degradation of Poly(l-Lactic Acid). Polym. Degrad. Stab. 2016, 133, 339–348. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | 100% PLA | 90% PLA | 75% PLA |
|---|---|---|---|
| PLA (wt. %) | 100 | 90 | 75 |
| aPHA (wt. %) | 0 | 10 | 25 |
| Cooling Rate = 2 °C/min | Cooling Rate = 20 °C/min | ||||||
|---|---|---|---|---|---|---|---|
| Material | Heat Cycles | Tg, PLA (°C) | Tg, aPHA (°C) * | Tm, PLA (°C) | Tg, PLA (°C) | Tg, aPHA (°C) * | Tm, PLA (°C) |
| Neat PLA | 0 | 62 | --- | 168 | 64 | --- | 170 |
| Masterbatch | 0 | 64 | −15 | 170 | 63 | −11 | 170 |
| 100 wt. % PLA | 1–5 | 62 | --- | 169 | 63–64 | --- | 168–171 |
| 90 wt. % PLA | 1–5 | 65 | −19 | 170 | 62 | --- | 169–170 |
| 75 wt. % PLA | 1–5 | 66 | −19 | 170 | 61 | −14 | 169 |
| Cooling Rate = 2 °C/min | Cooling Rate = 20 °C/min | ||||||
|---|---|---|---|---|---|---|---|
| Material | Heat Cycles | Tc (°C) | dHc (J/g) | dHm (J/g) | Tc (°C) | dHc (J/g) | dHm (J/g) |
| Neat PLA | 0 | --- | --- | 17.99 | --- | --- | 1.96 |
| Masterbatch | 0 | 106 | 15.87 | 20.09 | --- | --- | 16.89 |
| 100 wt. % PLA | 1 | --- | --- | 33.36 | --- | --- | 7.88 |
| 100 wt. % PLA | 2–5 | 98–100 | 9.19–16.17 | 30.45 | --- | --- | 29.78 |
| 90 wt. % PLA | 1–5 | 102–103 | 23.64 | 30.95 | --- | --- | 27.39 |
| 75 wt. % PLA | 1–5 | 104–105 | 21.27 | 27.03 | --- | --- | 22.73 |
| Heat Cycle | 0 | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|---|
| η0 (Pa-s) | 1611 | 891 | 953 | 841 | 742 | 764 |
| Mw (g/mol) | 137,188 | 115,229 | 117,557 | 113,284 | 109,209 | 110,131 |
| Heat Cycle | 100% PLA | 90% PLA | 75% PLA |
|---|---|---|---|
| 1 | 5.66 ± 0.28 | 11.43 ± 7.79 | 121.8 ± 38.3 |
| 2 | 6.31 ± 0.96 | 10.14 ± 4.97 | 99.2 ± 26.6 |
| 3 | 7.01 ± 1.43 | 6.84 ± 1.74 | 103.9 ± 57.7 |
| 4 | 6.50 ± 0.26 | 11.32 ± 5.45 | 142.9 ± 47.2 |
| 5 | 6.57 ± 0.26 | 9.90 ± 5.20 | 158.3 ± 11.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chatrath, S.; Alotaibi, M.; Barry, C.F. Performance of Recycled Polylactic Acid/Amorphous Polyhydroxyalkanoate Blends. Polymers 2024, 16, 1230. https://doi.org/10.3390/polym16091230
Chatrath S, Alotaibi M, Barry CF. Performance of Recycled Polylactic Acid/Amorphous Polyhydroxyalkanoate Blends. Polymers. 2024; 16(9):1230. https://doi.org/10.3390/polym16091230
Chicago/Turabian StyleChatrath, Simran, Mansour Alotaibi, and Carol Forance Barry. 2024. "Performance of Recycled Polylactic Acid/Amorphous Polyhydroxyalkanoate Blends" Polymers 16, no. 9: 1230. https://doi.org/10.3390/polym16091230
APA StyleChatrath, S., Alotaibi, M., & Barry, C. F. (2024). Performance of Recycled Polylactic Acid/Amorphous Polyhydroxyalkanoate Blends. Polymers, 16(9), 1230. https://doi.org/10.3390/polym16091230