
A Comparative Study of Two Synthesis Methods for Poly(Acrylic Acid-Co-Acrylamide) Incorporating a Hyperbranched Star-Shaped Monomer

 , , ,
, , ,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis and Purification of Macroinitiator for ATRP and the Macromonomer Boltorn H30—PEGMA500—V (MM)
2.3. Synthesis and Purification of Acrylamide, Acrylic Acid, and Boltorn H30—PEGMA500—V Terpolymer and Poly(Acrylamide-Co-Acrylic Acid) Copolymer
2.3.1. Via Reverse Iodine Transfer Polymerization in Aqueous Medium (HPAM-H2O and HPAM-MM-H2O)
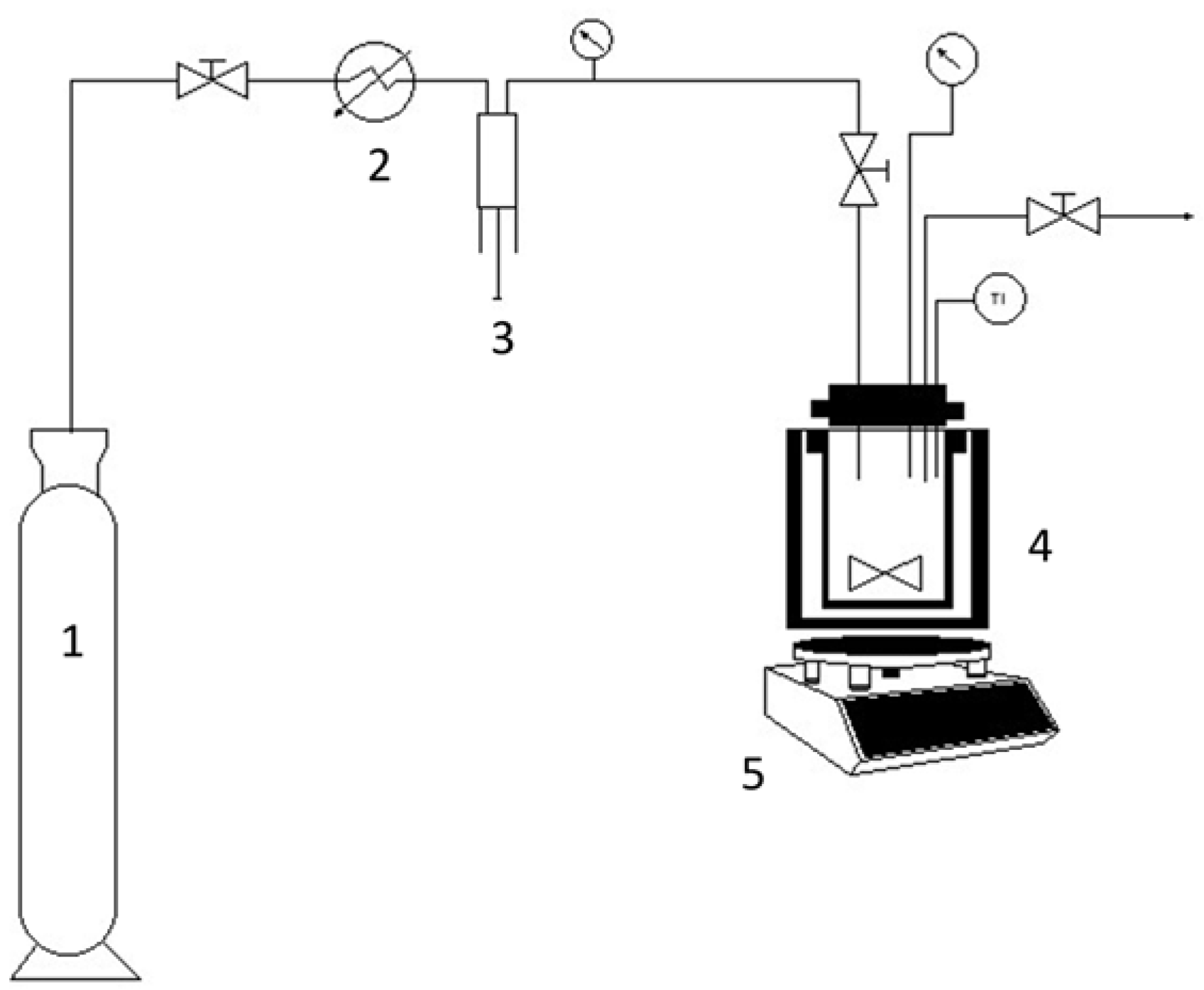
2.3.2. Via Reverse Iodine Transfer Polymerization in Supercritical Carbon Dioxide + Ethyl Acetate Solvent Mixture (HPAM-CO2 and HPAM-MM-CO2)
2.4. Purification of the Synthesized Copolymers
2.5. Characterization Techniques
2.5.1. Fourier Transform Infrared Spectroscopy (FTIR)
2.5.2. Proton and Carbon Nuclear Magnetic Resonance Spectroscopy (1H and 13C NMR)
2.5.3. Differential Scanning Calorimetry (DSC)
2.5.4. Scanning Electron Microscopy (SEM)
2.5.5. Size Exclusion Chromatography with Coupled Multi-Angle Light Scattering and Differential Refractometer Index Detectors (SEC-MALS-dRI)
2.5.6. Dynamic Light Scattering (DLS)
2.5.7. Apparent Viscosity and Rheological Measurements
3. Results and Discussion
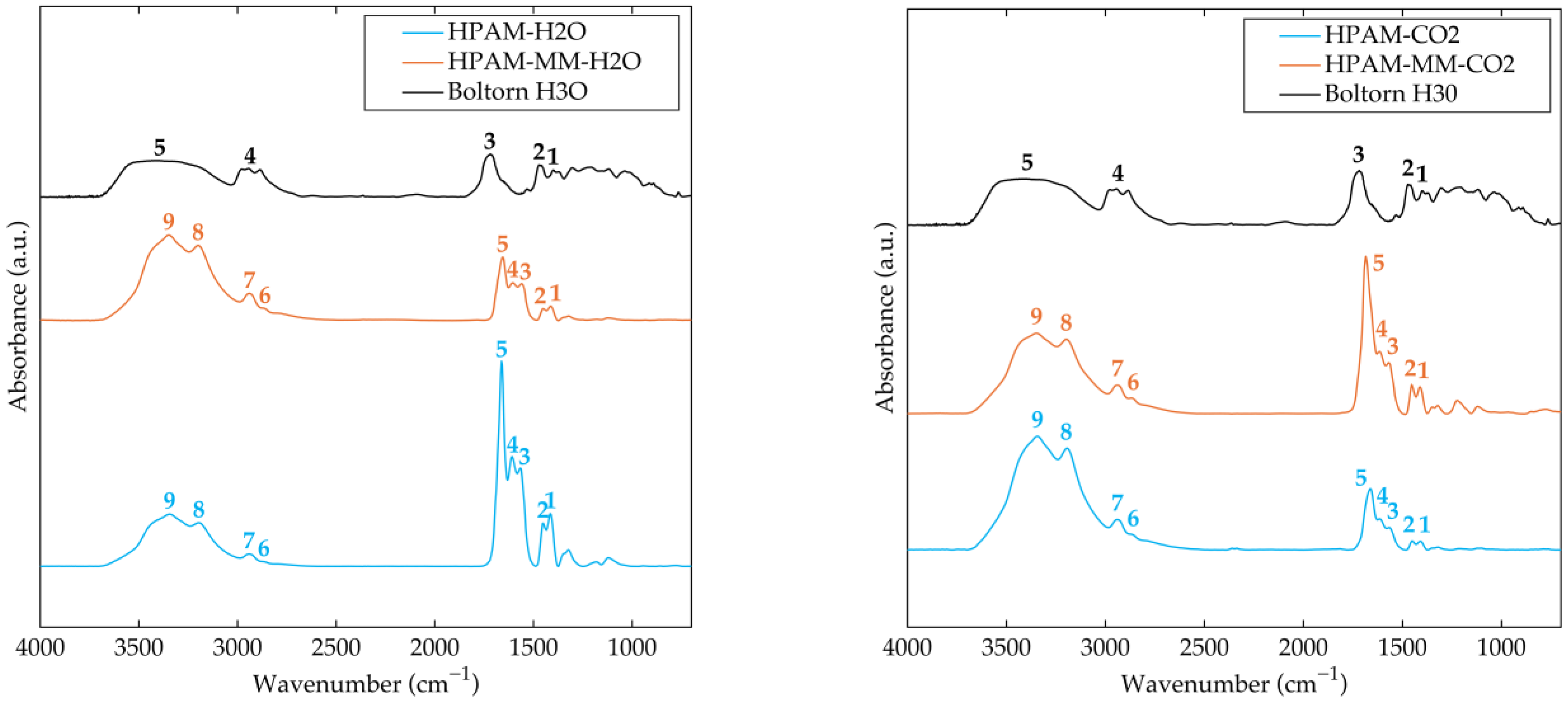
3.1. Synthesis and Spectral Analysis
3.2. Molar Mass and Molar Mass Distribution
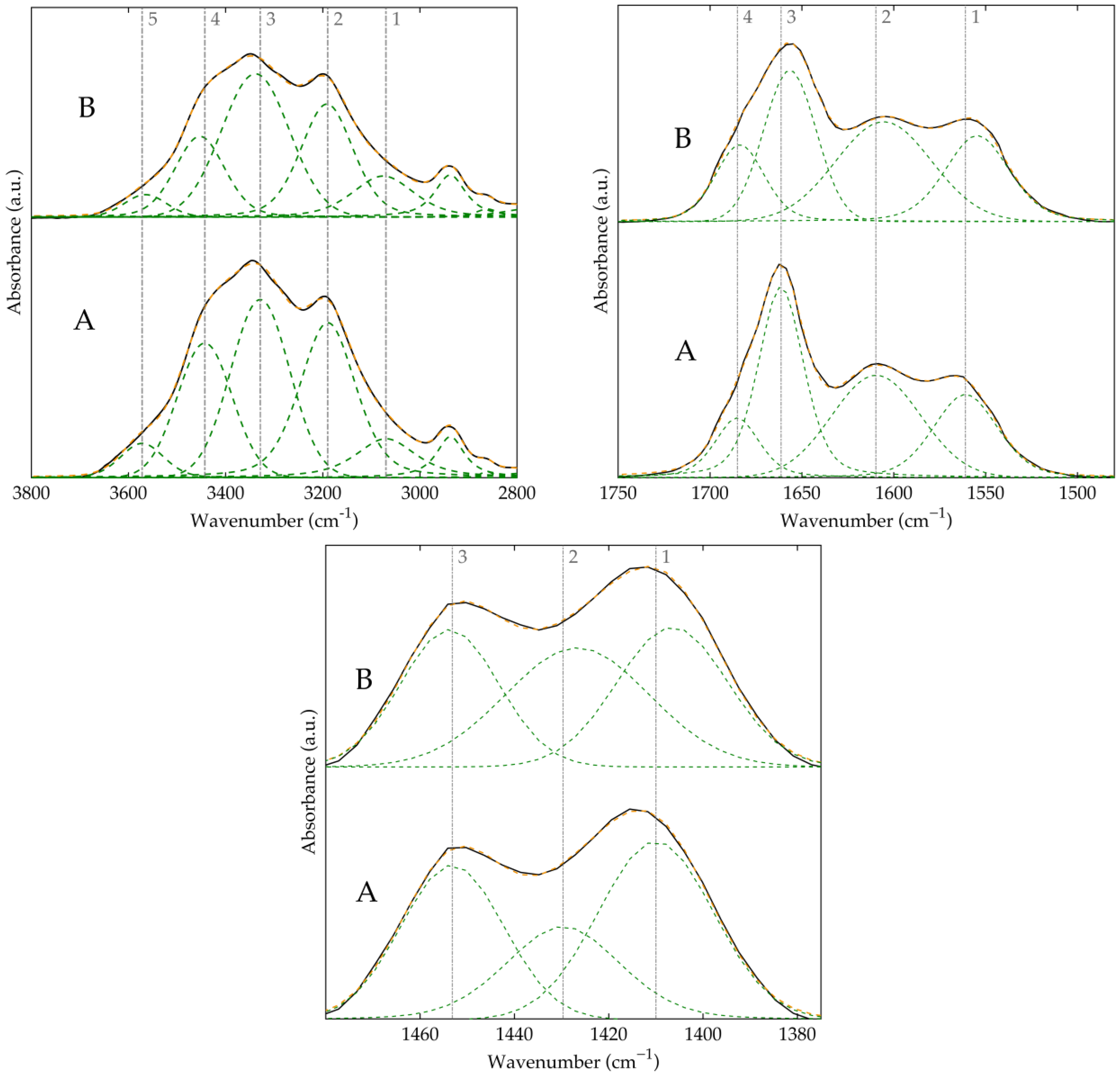
3.3. Structure and Conformational Analysis
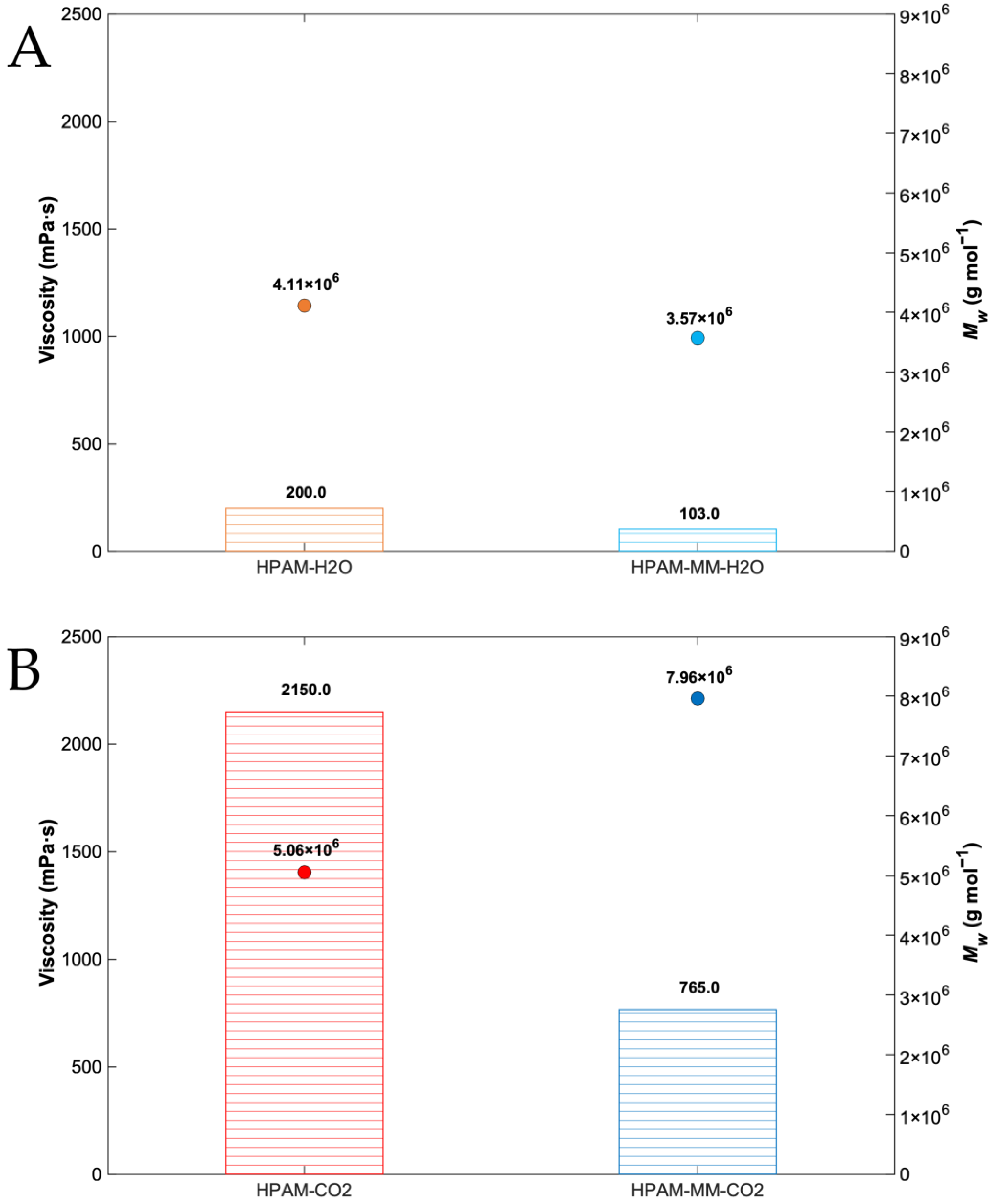
3.4. Rheological Properties
3.5. Thermal Analysis
- Region I (up to ~220 °C): This initial endothermic event is primarily associated with the release of surface and matrix-bound water. Near the upper end of this temperature range, decarboxylation reactions may also occur.
- Region II (up to ~350 °C): Two prominent endothermic peaks are observed in this region. These peaks correspond to the primary chemical modifications of the polymer—namely, cyclization reactions leading to imide and anhydride formation. The energy absorbed during these transitions reflects significant chemical modifications within the polymer.
- Region III (above ~350 °C): In this final stage, further degradation is evident, marked by the breakdown of imide and anhydride groups and chain scission, culminating in the disintegration of the polymer structure.
3.6. Morphological Analysis
3.7. Purification Process
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Druetta, P.; Raffa, P.; Picchioni, F. Chemical Enhanced Oil Recovery and the Role of Chemical Product Design. Appl. Energy 2019, 252, 113480. [Google Scholar] [CrossRef]
- Mandal, A.; Ojha, K. Enhanced Oil Recovery; CRC Press: Boca Raton, FL, USA, 2023; ISBN 9781003098850. [Google Scholar]
- Liang, K.; Han, P.; Chen, Q.; Su, X.; Feng, Y. Comparative Study on Enhancing Oil Recovery under High Temperature and High Salinity: Polysaccharides Versus Synthetic Polymer. ACS Omega 2019, 4, 10620–10628. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Billon, L.; Grassl, B.; Khoukh, A.; François, J. Hydrophobically Associating Polyacrylamides and Their Partially Hydrolyzed Derivatives Prepared by Post-Modification. 1. Synthesis and Characterization. Polymer 2002, 43, 2055–2064. [Google Scholar] [CrossRef]
- Wei, B.; Romero-Zerón, L.; Rodrigue, D. Evaluation of Two New Self-Assembly Polymeric Systems for Enhanced Heavy Oil Recovery. Ind. Eng. Chem. Res. 2014, 53, 16600–16611. [Google Scholar] [CrossRef]
- Crozzolo, J.; Formon, G.J.M.; Pomes-Hadda, M.; Aime, S.; Jouenne, S.; Nicolaÿ, R. Associative and Thermoresponsive Aqueous Polymer Formulations Based on Imine Chemistry. ACS Appl. Polym. Mater. 2022, 4, 4274–4285. [Google Scholar] [CrossRef]
- Scott, A.J.; Romero-Zerón, L.; Penlidis, A. Evaluation of Polymeric Materials for Chemical Enhanced Oil Recovery. Processes 2020, 8, 361. [Google Scholar] [CrossRef]
- Wever, D.A.Z.; Picchioni, F.; Broekhuis, A.A. Polymers for Enhanced Oil Recovery: A Paradigm for Structure–Property Relationship in Aqueous Solution. Prog. Polym. Sci. 2011, 36, 1558–1628. [Google Scholar] [CrossRef]
- Liu, T.; Gou, S.; Zhou, L.; Hao, J.; He, Y.; Liu, L.; Tang, L.; Fang, S. High-Viscoelastic Graft Modified Chitosan Hydrophobic Association Polymer for Enhanced Oil Recovery. J. Appl. Polym. Sci. 2021, 138, 50004. [Google Scholar] [CrossRef]
- Juárez Data, R.M.; Mattea, F.; Strumia, M.C.; Milanesio, J.M. Effect of Including a Hydrophobic Comonomer on the Rheology of an Acrylamide-acrylic Acid Based Copolymer. J. Appl. Polym. Sci. 2020, 137, 49532. [Google Scholar] [CrossRef]
- Viken, A.L.; Skauge, T.; Svendsen, P.E.; Time, P.A.; Spildo, K. Thermothickening and Salinity Tolerant Hydrophobically Modified Polyacrylamides for Polymer Flooding. Energy Fuels 2018, 32, 10421–10427. [Google Scholar] [CrossRef]
- Lai, N.; Qin, X.; Ye, Z.; Peng, Q.; Zhang, Y.; Ming, Z. Synthesis and Evaluation of a Water-Soluble Hyperbranched Polymer as Enhanced Oil Recovery Chemical. J. Chem. 2013, 2013, 824785. [Google Scholar] [CrossRef]
- Shi, L.; Liu, C.; Chen, M.; Hua, Z.; Ye, Z.; Zhang, J. Synthesis and Evaluation of a Hyperbranched Copolymer as Viscosity Reducer for Offshore Heavy Oil. J. Pet. Sci. Eng. 2021, 196, 108011. [Google Scholar] [CrossRef]
- Voit, B.I.; Lederer, A. Hyperbranched and Highly Branched Polymer Architectures—Synthetic Strategies and Major Characterization Aspects. Chem. Rev. 2009, 109, 5924–5973. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Ju, Z.; Zhang, C.; Yang, Y.; He, J. Dendritic Block and Dendritic Brush Copolymers through Anionic Macroinimer Approach. Macromolecules 2013, 46, 1437–1446. [Google Scholar] [CrossRef]
- Simon, P.F.W.; Müller, A.H.E. Kinetic Investigation of Self-Condensing Group Transfer Polymerization. Macromolecules 2004, 37, 7548–7558. [Google Scholar] [CrossRef]
- Simon, P.F.W.; Müller, A.H.E. Synthesis of Hyperbranched and Highly Branched Methacrylates by Self-Condensing Group Transfer Copolymerization. Macromolecules 2001, 34, 6206–6213. [Google Scholar] [CrossRef]
- Gao, C.; Yan, D. Hyperbranched Polymers: From Synthesis to Applications. Prog. Polym. Sci. 2004, 29, 183–275. [Google Scholar] [CrossRef]
- Ríos Valer, G.; Díaz, G.; Giussi, J.M.; Ceolín, M. Architecture-Behaviour-Properties Relationship in Star-Shaped MPA-PMMA and MPA-PS Hyper-Branched Copolymers. Adv. Mater. Lett. 2019, 10, 476–483. [Google Scholar] [CrossRef]
- Meleán Brito, R.S.; Padró, J.M.; Pinzón Barrantes, J.J.; Villa-Pérez, C.; Strumia, M.C.; Milanesio, J.M.; Mattea, F. The Role of Hyperbranched Polyesters in Acrylamide-based Polymers as Thickening Agents in Aqueous Solutions. J. Appl. Polym. Sci. 2024, 141, e56149. [Google Scholar] [CrossRef]
- Iborra, A.; Salvatierra, L.; Giussi, J.M.; Azzaroni, O. Synthesis of Lauryl Methacrylate and Poly(Ethylene Glycol) Methyl Ether Methacrylate Copolymers with Tunable Microstructure and Emulsifying Properties. Eur. Polym. J. 2019, 116, 117–125. [Google Scholar] [CrossRef]
- Meleán Brito, R.S.; Iborra, A.; Padró, J.M.; Vega, I.N.; Strumia, M.C.; Milanesio, J.M.; Mattea, F.; Giussi, J.M. Hyperbranched Star Monomer: A New Strategy to Improve HPAM in Harsh Environments. Ind. Eng. Chem. Res. 2025, 64, 948–958. [Google Scholar] [CrossRef]
- Romack, T.J.; Maury, E.E.; DeSimone, J.M. Precipitation Polymerization of Acrylic Acid in Supercritical Carbon Dioxide. Macromolecules 1995, 28, 912–915. [Google Scholar] [CrossRef]
- Cao, L.; Chen, L. Influence of Reaction Parameters on Synthesis of Temperature-Sensitive Materials in Supercritical Carbon Dioxide by Precipitation Polymerization. Polym. Bull. 2006, 57, 651–659. [Google Scholar] [CrossRef]
- Kendall, J.L.; Canelas, D.A.; Young, J.L.; DeSimone, J.M. Polymerizations in Supercritical Carbon Dioxide. Chem. Rev. 1999, 99, 543–564. [Google Scholar] [CrossRef]
- Versteeg, F.G.; Picchioni, F. Reversible Addition-Fragmentation Chain-Transfer Polymerization in Supercritical CO2: A Review. Macromol. Rapid Commun. 2024, 45, e2400514. [Google Scholar] [CrossRef]
- Tsai, W.-C.; Wang, Y. Progress of Supercritical Fluid Technology in Polymerization and Its Applications in Biomedical Engineering. Prog. Polym. Sci. 2019, 98, 101161. [Google Scholar] [CrossRef]
- Coelho, J.P.; Bernotaityte, K.; Miraldes, M.A.; Mendonça, A.F.; Stateva, R.P. Solubility of Ethanamide and 2-Propenamide in Supercritical Carbon Dioxide. Measurements and Correlation. J. Chem. Eng. Data 2009, 54, 2546–2549. [Google Scholar] [CrossRef]
- Claesson, H.; Malmström, E.; Johansson, M.; Hult, A. Synthesis and Characterisation of Star Branched Polyesters with Dendritic Cores and the Effect of Structural Variations on Zero Shear Rate Viscosity. Polymer 2002, 43, 3511–3518. [Google Scholar] [CrossRef]
- Boyer, C.; Lacroix-Desmazes, P.; Robin, J.-J.; Boutevin, B. Reverse Iodine Transfer Polymerization (RITP) of Methyl Methacrylate. Macromolecules 2006, 39, 4044–4053. [Google Scholar] [CrossRef]
- Bouilhac, C.; Chirat, M.; Joly-Duhamel, C.; Lacroix-Desmazes, P. Reverse Iodine Transfer Polymerization (RITP) of 1,1,2,2-Tetrahydroperfluorodecyl Acrylate in Supercritical Carbon Dioxide. Macromol. Chem. Phys. 2013, 214, 2259–2265. [Google Scholar] [CrossRef]
- Matson, J.B.; Steele, A.Q.; Mase, J.D.; Schulz, M.D. Polymer Characterization by Size-Exclusion Chromatography with Multi-Angle Light Scattering (SEC-MALS): A Tutorial Review. Polym. Chem. 2024, 15, 127–142. [Google Scholar] [CrossRef] [PubMed]
- de Lima, B.L.B.; Marques, N.d.N.; Villetti, M.A.; Balaban, R.d.C. HPAM-g-PEOPPO: Rheological Modifiers in Aqueous Media of High Temperature and High Ionic Strength. J. Appl. Polym. Sci. 2019, 136, 47453. [Google Scholar] [CrossRef]
- Simeonov, M.; Apostolov, A.A.; Georgieva, M.; Tzankov, D.; Vassileva, E. Poly(Acrylic Acid-Co-Acrylamide)/Polyacrylamide PIPNs/Magnetite Composite Hydrogels: Synthesis and Characterization. Gels 2023, 9, 365. [Google Scholar] [CrossRef] [PubMed]
- Craciun, G.; Ighigeanu, D.; Manaila, E.; Stelescu, M.D. Synthesis and Characterization of Poly(Acrylamide-Co-Acrylic Acid) Flocculant Obtained by Electron Beam Irradiation. Mater. Res. 2015, 18, 984–993. [Google Scholar] [CrossRef]
- Lee, Y.C.; Liew, C.W.; Buraidah, M.H.; Woo, H.J. Fourier Transform Infrared Studies of Gel Polymer Electrolyte Based on Poly(Acrylamide-Co-Acrylic Acid)—Ethylene Carbonate Incorporated with Water-Soluble Sodium Sulfide. Opt. Mater. 2023, 140, 113791. [Google Scholar] [CrossRef]
- Du, Y.; Chen, Q.; Shen, L.; Xing, Y.; Dai, J. Synthesis and Application of Long-Chain Alkyl Quaternary Ammonium-Functionalized Hyperbranched Polyester. J. Appl. Polym. Sci. 2011, 121, 2927–2935. [Google Scholar] [CrossRef]
- Kumari, S.; Mishra, A.K.; Chattopadhyay, D.K.; Raju, K.V.S.N. Synthesis and Characterization of Hyperbranched Polyesters and Polyurethane Coatings. J. Polym. Sci. A Polym. Chem. 2007, 45, 2673–2688. [Google Scholar] [CrossRef]
- Han, L.; Zhang, K.; Ishida, H.; Froimowicz, P. Study of the Effects of Intramolecular and Intermolecular Hydrogen-Bonding Systems on the Polymerization of Amide-Containing Benzoxazines. Macromol. Chem. Phys. 2017, 218, 1600562. [Google Scholar] [CrossRef]
- Arakawa, T.; Ejima, D.; Li, T.; Philo, J.S. The Critical Role of Mobile Phase Composition in Size Exclusion Chromatography of Protein Pharmaceuticals. J. Pharm. Sci. 2010, 99, 1674–1692. [Google Scholar] [CrossRef]
- Liu, T.; DeSimone, J.M.; Roberts, G.W. Kinetics of the Precipitation Polymerization of Acrylic Acid in Supercritical Carbon Dioxide: The Locus of Polymerization. Chem. Eng. Sci. 2006, 61, 3129–3139. [Google Scholar] [CrossRef]
- Liu, T.; Desimone, J.M.; Roberts, G.W. Continuous Precipitation Polymerization of Acrylic Acid in Supercritical Carbon Dioxide: The Polymerization Rate and the Polymer Molecular Weight. J. Polym. Sci. A Polym. Chem. 2005, 43, 2546–2555. [Google Scholar] [CrossRef]
- Cai, W.D.; Qiu, W.Y.; Ding, Z.C.; Wu, L.X.; Yan, J.K. Conformational and Rheological Properties of a Quaternary Ammonium Salt of Curdlan. Food Chem. 2019, 280, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Žagar, E.; Žigon, M. Aliphatic Hyperbranched Polyesters Based on 2,2-Bis(Methylol)Propionic Acid—Determination of Structure, Solution and Bulk Properties. Prog. Polym. Sci. 2011, 36, 53–88. [Google Scholar] [CrossRef]
- Maurer, J.J.; Harvey, G.D. Thermal Degradation Characteristics of Poly(Acrylamide-Co-Acrylic Acid) and Poly(Acrylamide-Co-Sodium Acrylate) Copolymers. Thermochim. Acta 1987, 121, 295–306. [Google Scholar] [CrossRef]
- Caulfield, M.J.; Qiao, G.G.; Solomon, D.H. Some Aspects of the Properties and Degradation of Polyacrylamides. Chem. Rev. 2002, 102, 3067–3083. [Google Scholar] [CrossRef]
- Voronova, M.I.; Surov, O.V.; Afineevskii, A.V.; Zakharov, A.G. Properties of Polyacrylamide Composites Reinforced by Cellulose Nanocrystals. Heliyon 2020, 6, e05529. [Google Scholar] [CrossRef]
- Lv, Y.; Zhang, S.; Zhang, Y.; Yin, H.; Feng, Y. Hydrophobically Associating Polyacrylamide “Water-in-Water” Emulsion Prepared by Aqueous Dispersion Polymerization: Synthesis, Characterization and Rheological Behavior. Molecules 2023, 28, 2698. [Google Scholar] [CrossRef]
- Van Dyke, J.D.; Kasperski, K.L. Thermogravimetric Study of Polyacrylamide with Evolved Gas Analysis. J. Polym. Sci. Part A Polym. Chem. 1993, 31, 1807–1823. [Google Scholar] [CrossRef]
- Moharram, M.A.; Allam, M.A. Study of the Interaction of Poly(Acrylic Acid) and Poly(Acrylic Acid-Poly Acrylamide) Complex with Bone Powders and Hydroxyapatite by Using TGA and DSC. J. Appl. Polym. Sci. 2007, 105, 3220–3227. [Google Scholar] [CrossRef]
- Moharram, M.A.; Khafagi, M.G. Thermal Behavior of Poly(Acrylic Acid)-Poly(Vinyl Pyrrolidone) and Poly(Acrylic Acid)-Metal-Roly(Vinyl Pyrrolidone) Complexes. J. Appl. Polym. Sci. 2006, 102, 4049–4057. [Google Scholar] [CrossRef]
- Zhang, X.; Han, M.; Xu, L.; AlSofi, A.M. Long-Term Stability Prediction of Polyacrylamide-Type Polymers at Harsh Conditions via Thermogravimetric Analysis. Chem. Phys. Lett. 2022, 795, 139538. [Google Scholar] [CrossRef]
- Kuldasheva, S.; Abdikamalova, A.; Eshmetov, I.; Abdurahimov, D.; Sharipova, A.; Nortojiyeva, G. Investigation of Changes in the Viscosity Properties of Acrylamide (Co)Polymer and Their Hydrolyzed Forms Depending on the Conditions of Their Preparation. Polym. Bull. 2024, 81, 4065–4091. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Copolymer | Boltorn H30—PEGMA500—V (g) | Boltorn H30—PEGMA500—V Content Relative to the Total Weight of Monomers in the Feed (wt%) | AAm (g) | AAm (mol) | AAm (%mol) | AAc (g) | AAc (mol) | AAc (%mol) | Initiator (%mol) (×10−2) | Temperature (°C) | Initial Pressure (bar) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HPAM-H2O | - | 10.6795 | 0.15 | 72.92 | 4.0198 | 0.05 | 27.08 | 3.08 | 65 | - | |
| HPAM-MM-H2O | 0.1865 | 0.85 | 10.6895 | 0.15 | 73.32 | 3.9415 | 0.05 | 26.66 | 3.01 | 65 | - |
| HPAM-CO2 | - | 10.7187 | 0.15 | 74.99 | 3.6242 | 0.05 | 25.01 | 3.23 | 90 | 320 | |
| HPAM-MM-CO2 | 0.1723 | 0.85 | 10.7174 | 0.15 | 74.92 | 3.6345 | 0.05 | 25.06 | 3.23 | 90 | 320 |
| Copolymer | Global Residual Monomers Fraction (mol%) a | Global Monomer Conversion (mol%) a | AAc Precipitation (mol%) b | AAc Dialysis (mol%) b | Boltorn H30—PEGMA500–V Incorporation (Precipitation) (mol%) | Boltorn H30—PEGMA500–V Incorporation (Dialysis) (mol%) |
|---|---|---|---|---|---|---|
| HPAM-H2O | 1.5 | 98.5 | 23.3 | 24.4 | - | - |
| HPAM-MM-H2O | 0.9 | 99.1 | 23.6 | 23.3 | 0.0220 ± 0.0005 | 0.0614 ± 0.0005 |
| HPAM-CO2 | 2.7 | 97.3 | 24.3 | 41.4 | - | - |
| HPAM-MM-CO2 | 0.5 | 99.5 | 20.0 | 33.4 | 0.0278 ± 0.0005 | 0.0477 ± 0.0005 |
| Sample | Precipitation | Dialysis | ||||
|---|---|---|---|---|---|---|
| Mw (g mol−1) (×106) | Mn (g mol−1) (×106) | Ð | Mw (g mol−1) (×106) | Mn (g mol−1) (×106) | Ð | |
| HPAM-H2O | 2.54 ± 0.03 | 1.47 ± 0.03 | 1.72 | 3.57± 0.04 | 2.60 ± 0.04 | 1.37 |
| HPAM-MM-H2O | 3.52 ± 0.04 | 2.78 ± 0.05 | 1.27 | 4.11 ± 0.04 | 3.20 ± 0.05 | 1.28 |
| HPAM-CO2 | 0.96 ± 0.01 | 0.39 ± 0.01 | 2.47 | 7.96 ± 0.06 | 7.24 ± 0.07 | 1.10 |
| HPAM-MM-CO2 | 0.86 ± 0.01 | 0.32 ± 0.01 | 2.68 | 5.06 ± 0.04 | 3.52 ± 0.06 | 1.44 |
| MM | - | - | - | 0.14 ± 0.01 | 0.06 ± 0.01 | 2.44 |
| Dialyzed Copolymer | Rg (nm) | Rh(fil) (nm) | ρ (Rg/Rh(fil)) | Conformation |
|---|---|---|---|---|
| MM | 5.2 ± 0.1 | - | - | - |
| HPAM-H2O | 141.9 ± 0.7 | 98 ± 3 | 1.45 | Random coil |
| HPAM-MM-H2O | 148.8 ± 0.7 | 178 ± 2 | 0.84 | Compact sphere |
| HPAM-CO2 | 47.8 ± 0.3 | 64 ± 1 | 0.75 | Compact sphere |
| HPAM-MM-CO2 | 60.0 ± 0.4 | 143 ± 4 | 0.42 | Highly compact structures |
| Copolymer | Rh(agg) (nm) | Rh(son) (nm) |
|---|---|---|
| HPAM-H2O | 2243 ± 98 | 271 ± 24 |
| HPAM-MM-H2O | 2493 ± 65 | 2304 ± 57 |
| HPAM-CO2 | 1670 ± 92 | 304 ± 20 |
| HPAM-MM-CO2 | 2468 ± 64 | 2046 ± 82 |
| HPAM-H2O | HPAM-MM-H2O | HPAM-CO2 | HPAM-MM-CO2 | |
|---|---|---|---|---|
| Initial Temperature (°C) | 200.0 | 208.0 | 192.8 | 190.0 |
| Maximum Temperature (°C) | 245.6 | 241.8 | 248.4 | 253.7 |
| Final Temperature (°C) | 266.0 | 291.7 | 283.5 | 289.4 |
| Peak Enthalpy (J·g−1) | 76.3 | 118.1 | 107.6 | 44.0 |
| Initial Temperature (°C) | 282.9 | 291.7 | 285.5 | 289.4 |
| Maximum Temperature (°C) | 311.3 | 319.1 | 314.8 | 360.0 |
| Final Temperature (°C) | 342.5 | 359.3 | 353.2 | 316.5 |
| Peak Enthalpy (J·g−1) | 108.9 | 92.9 | 113.7 | 58.4 |
| Initial Temperature (°C) | 357.0 | 359.3 | 355.0 | - |
| Maximum Temperature (°C) | 379.7 | 392.3 | 388.2 | - |
| Final Temperature (°C) | 389.8 | 399.0 | 392.4 | - |
| Peak Enthalpy (J·g−1) | 23.6 | 13.9 | 9.11 | - |
| Copolymer | Lost Mass (%) | Purified Mass (%) |
|---|---|---|
| HPAM-H2O | 62.7 | 37.3 |
| HPAM-MM-H2O | 87.6 | 12.4 |
| HPAM-CO2 | 73.5 | 26.5 |
| HPAM-MM-CO2 | 91.9 | 8.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brito, R.S.M.; Iborra, A.; Padró, J.M.; Villa-Pérez, C.; Strumia, M.C.; Mattea, F.; Giussi, J.M.; Milanesio, J.M. A Comparative Study of Two Synthesis Methods for Poly(Acrylic Acid-Co-Acrylamide) Incorporating a Hyperbranched Star-Shaped Monomer. Polymers 2025, 17, 964. https://doi.org/10.3390/polym17070964
Brito RSM, Iborra A, Padró JM, Villa-Pérez C, Strumia MC, Mattea F, Giussi JM, Milanesio JM. A Comparative Study of Two Synthesis Methods for Poly(Acrylic Acid-Co-Acrylamide) Incorporating a Hyperbranched Star-Shaped Monomer. Polymers. 2025; 17(7):964. https://doi.org/10.3390/polym17070964
Chicago/Turabian StyleBrito, Ramses S. Meleán, Agustín Iborra, Juan M. Padró, Cristian Villa-Pérez, Miriam C. Strumia, Facundo Mattea, Juan M. Giussi, and Juan M. Milanesio. 2025. "A Comparative Study of Two Synthesis Methods for Poly(Acrylic Acid-Co-Acrylamide) Incorporating a Hyperbranched Star-Shaped Monomer" Polymers 17, no. 7: 964. https://doi.org/10.3390/polym17070964
APA StyleBrito, R. S. M., Iborra, A., Padró, J. M., Villa-Pérez, C., Strumia, M. C., Mattea, F., Giussi, J. M., & Milanesio, J. M. (2025). A Comparative Study of Two Synthesis Methods for Poly(Acrylic Acid-Co-Acrylamide) Incorporating a Hyperbranched Star-Shaped Monomer. Polymers, 17(7), 964. https://doi.org/10.3390/polym17070964