
Effect of Proteins on the Network Formation and Degradation of Peroxide Cross-Linked Natural Rubber Elucidated by Time-Domain NMR

 , ,
, ,  , ,
, ,  and
and 
Abstract

1. Introduction
2. Experimentation
2.1. Sample Preparation
2.2. Characterization
2.2.1. 1H DQ-NMR Experiment
2.2.2. Hahn Echo Experiments Measured on Swollen Samples
3. Results and Discussion
3.1. Characterization of Rubber Samples
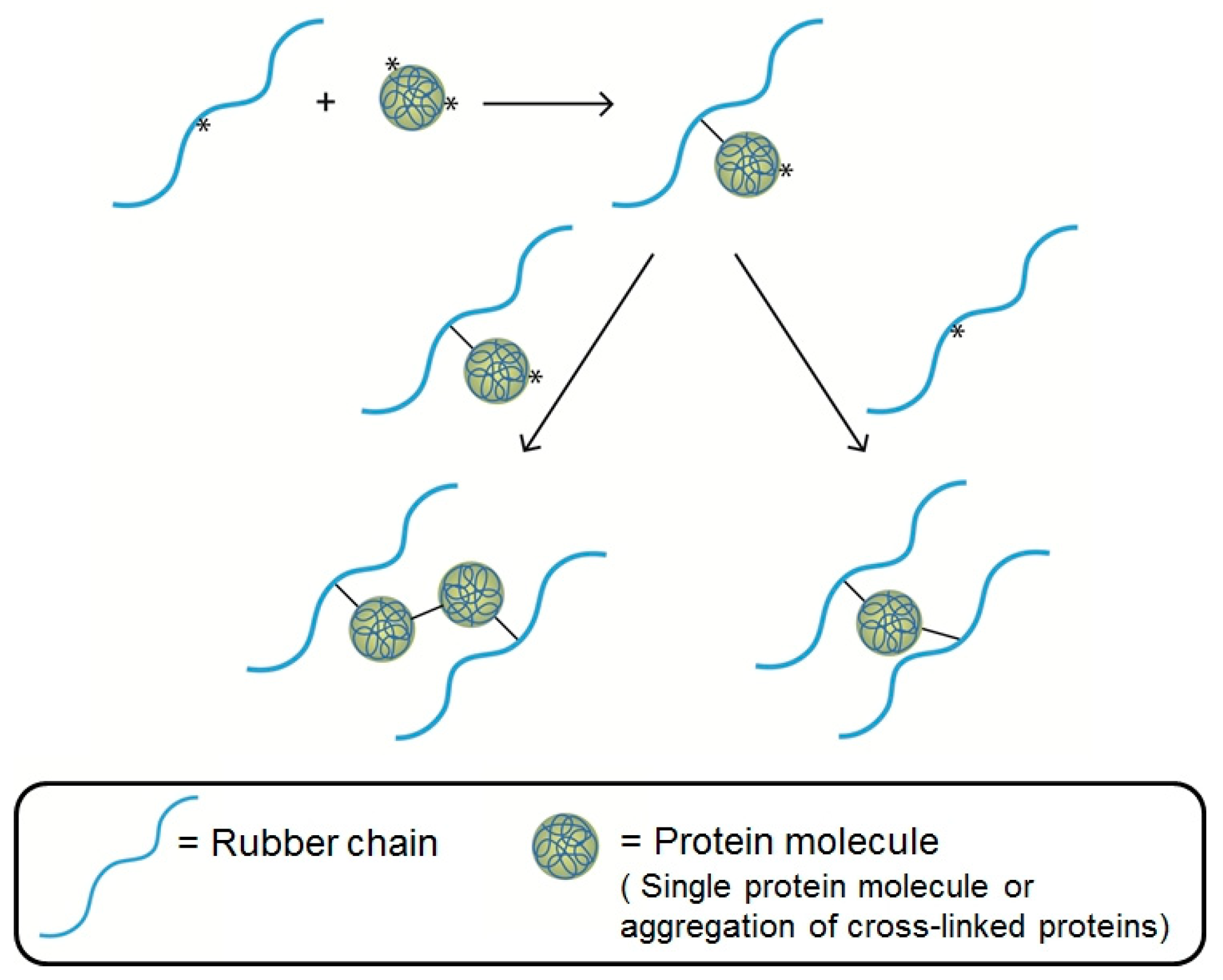
3.2. Pseudo-End-Linked Network
3.3. Evolution of Cross-Link Density During the Vulcanization Process
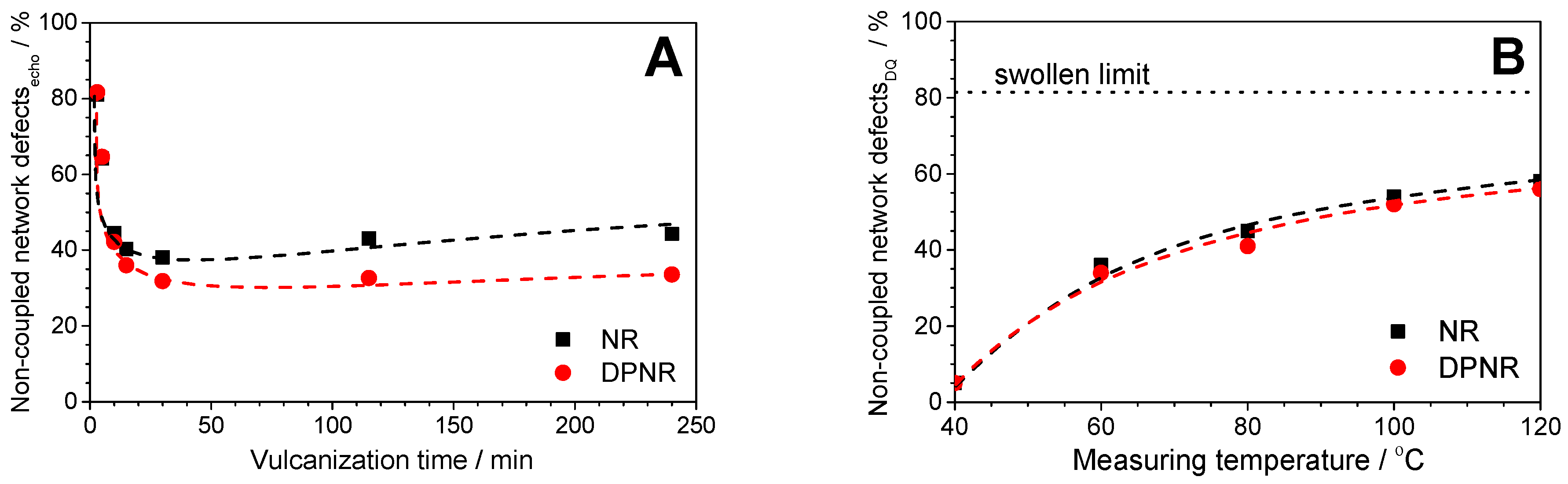
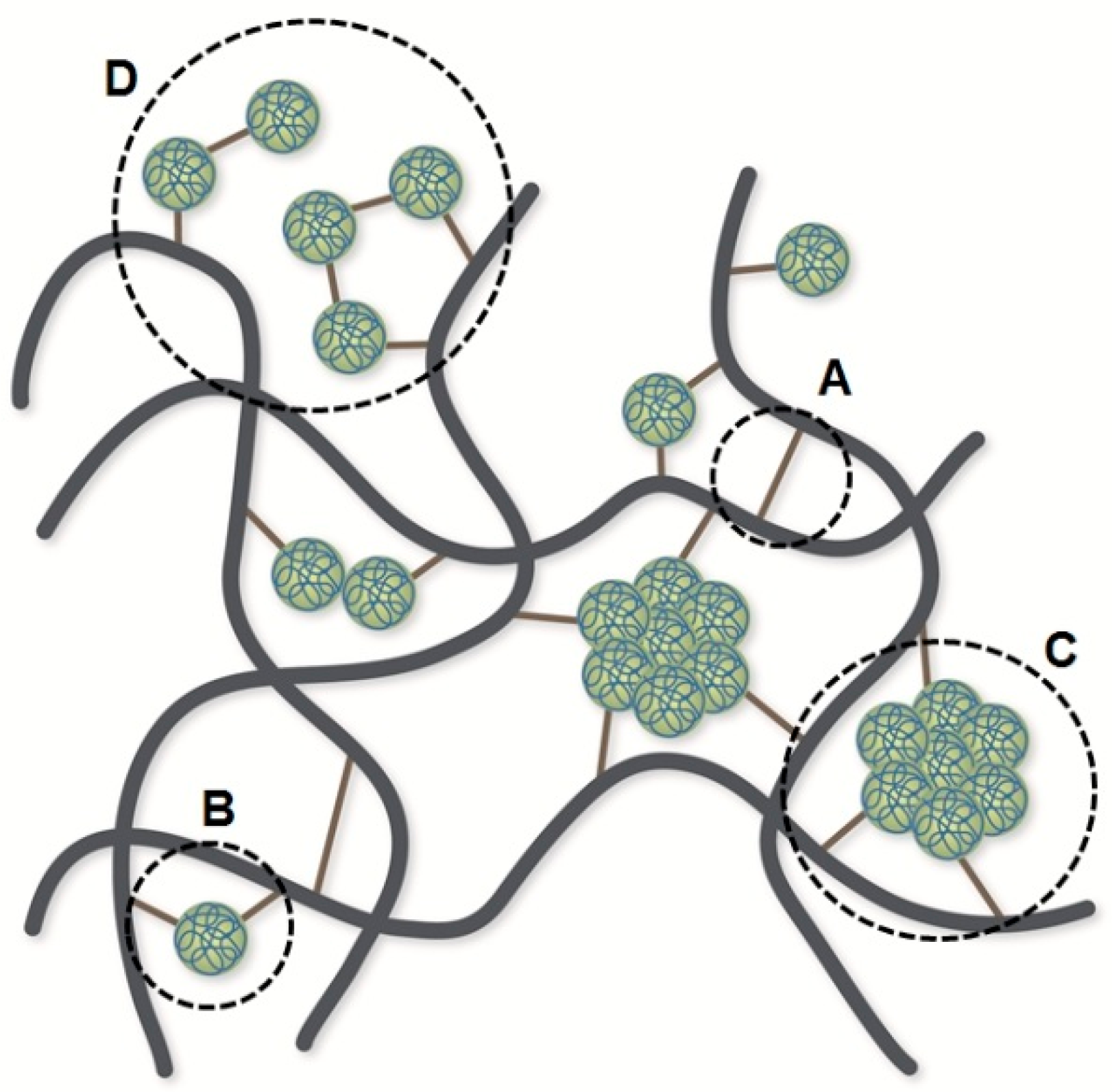
3.4. Evolution of Non-Elastic Network Defects During the Vulcanization Process
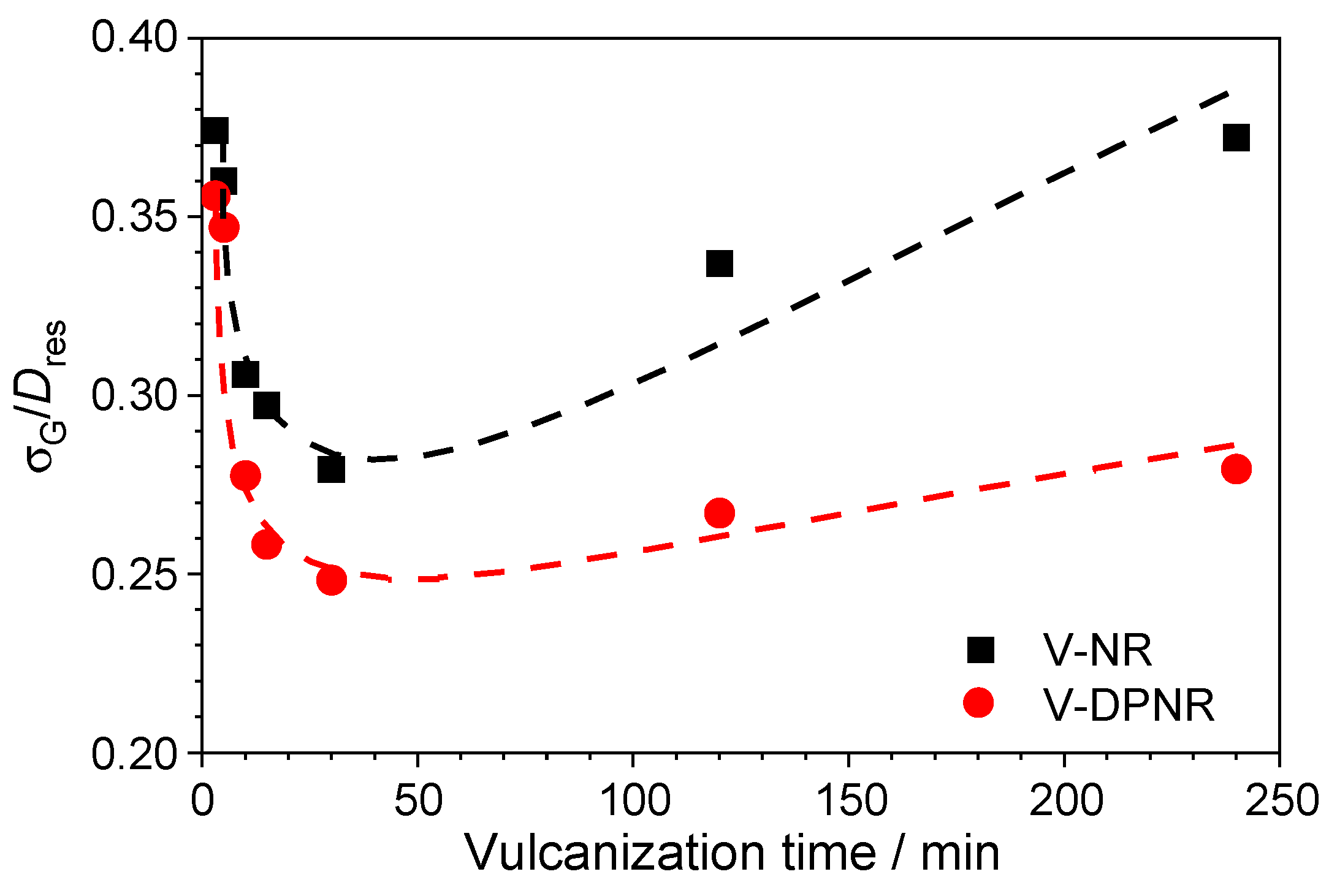
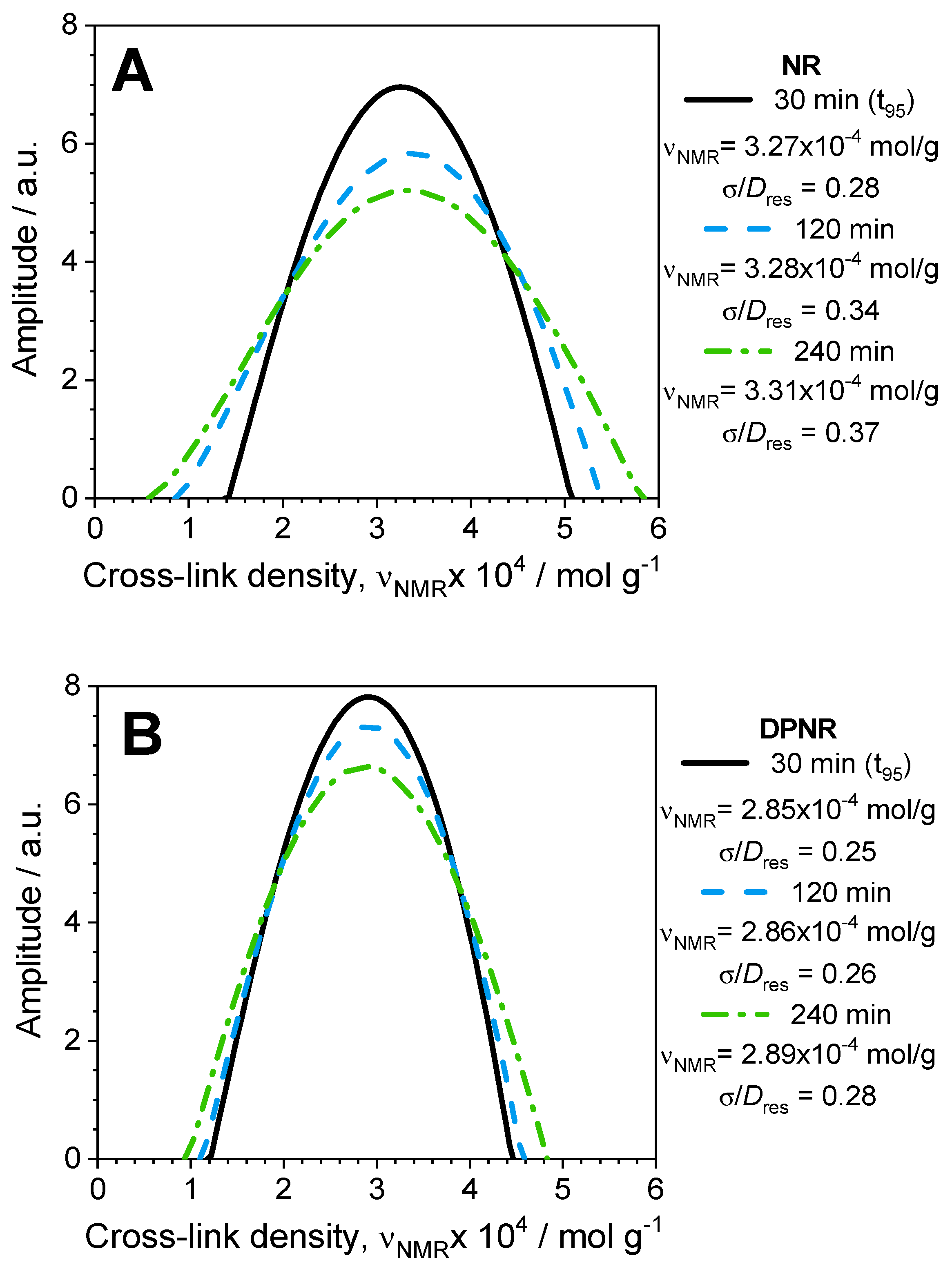
3.5. Evolution of Spatial Distribution of Cross-Links During the Vulcanization Process
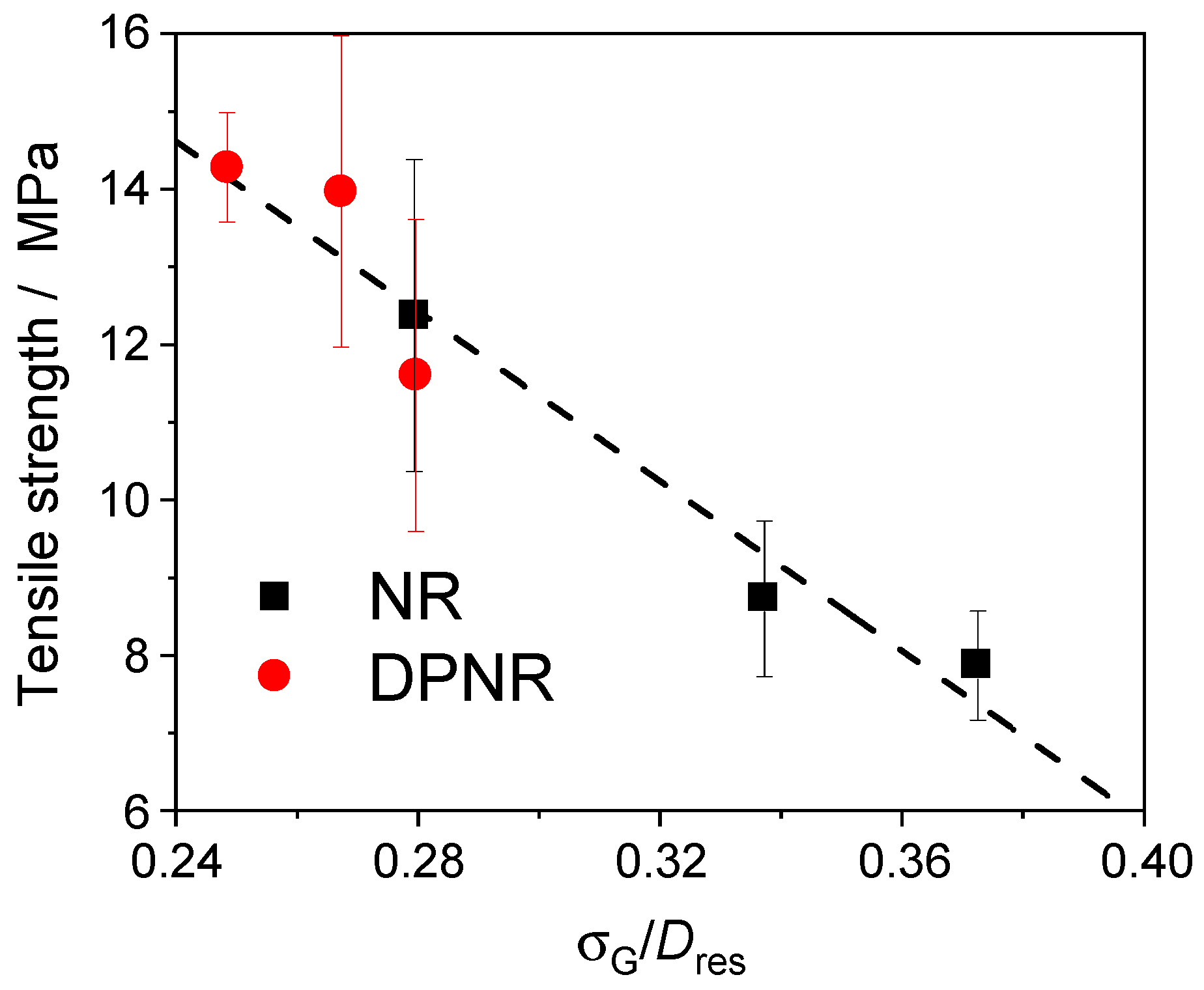
3.6. Effect of Network Heterogeneities on the Mechanical Properties
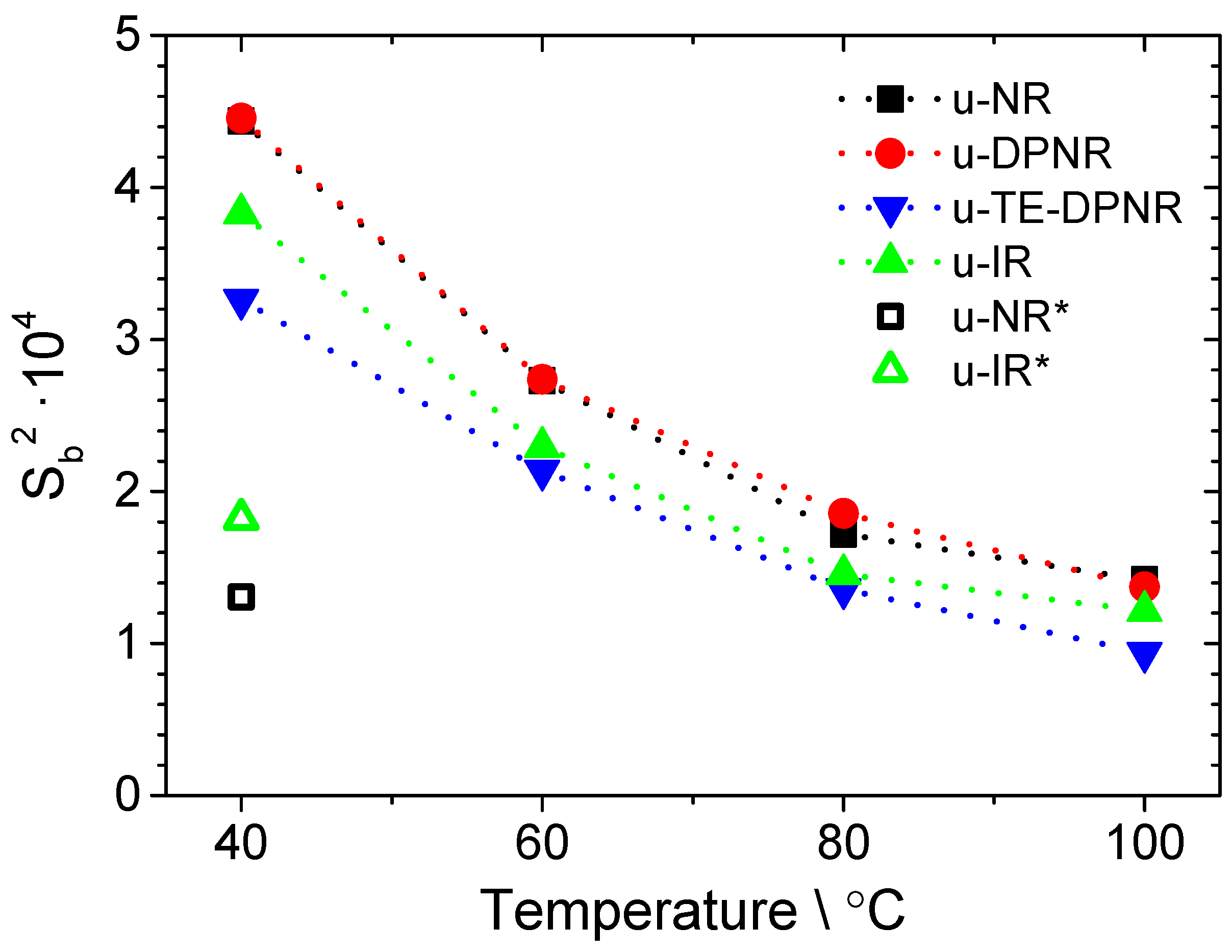
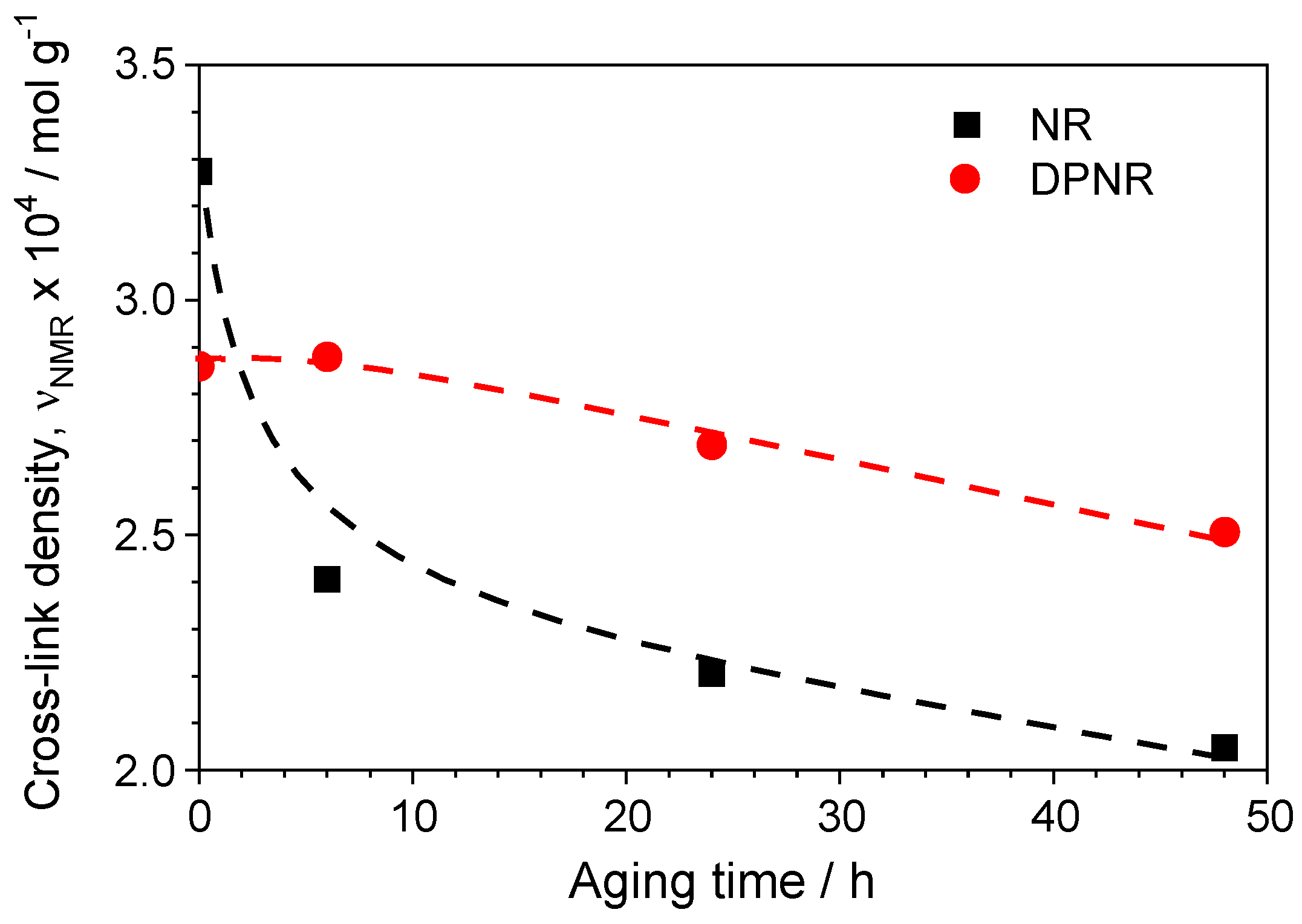
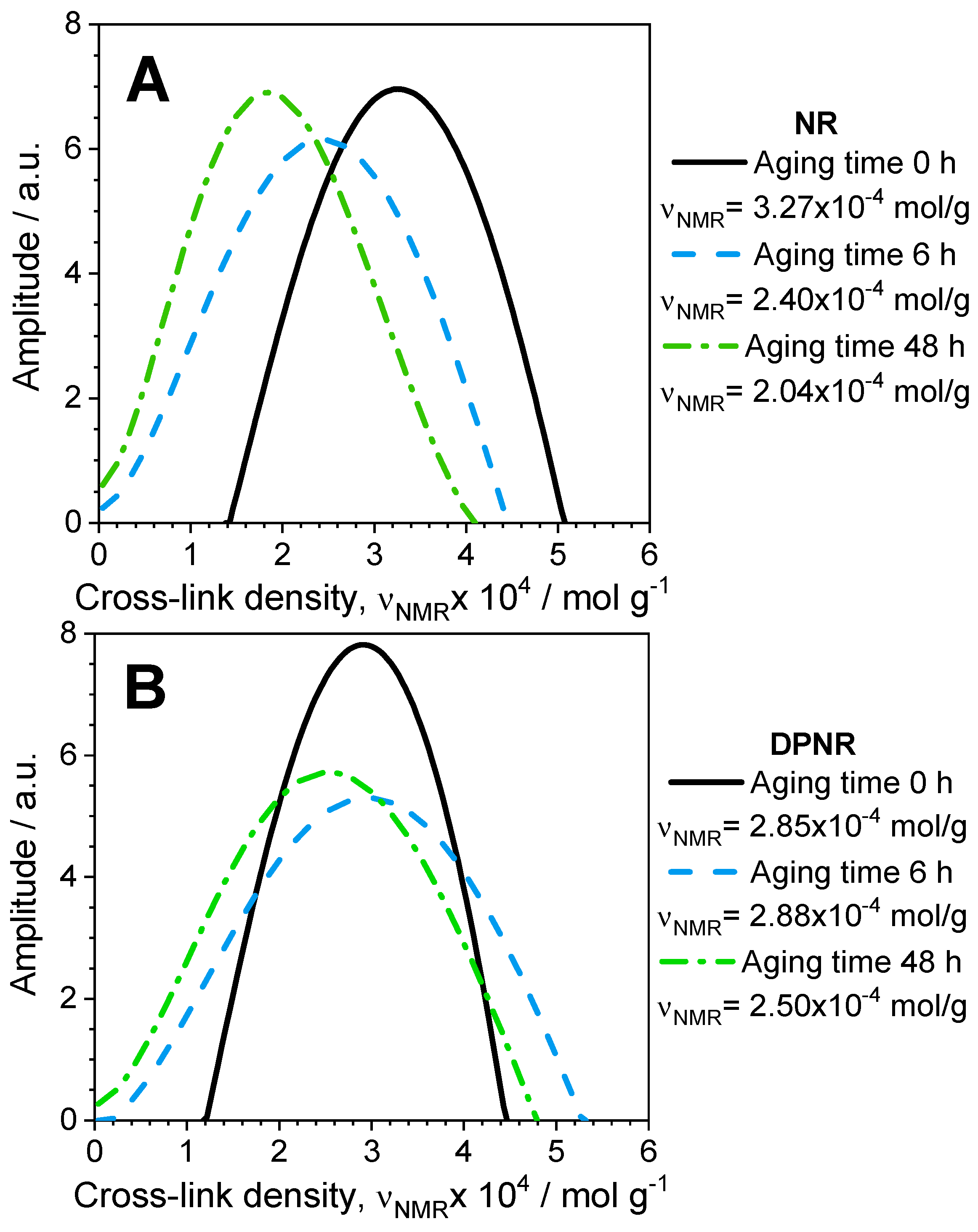
3.7. Thermal–Oxidative Degradation of Peroxide Cross-Linked NR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saleh, T.A. Polymer Hybrid Materials and Nanocomposites: Fundamentals and Applications; William Andrew: Jeddah, Saudi Arabia, 2021. [Google Scholar]
- Blume, A.; van Elburg, F.; Grunert, F.; Talma, A. “Re-Think” Sulfur Curing. Molecules 2024, 29, 5198. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Romain, C.; Williams, C. Sustainable polymers from renewable resources. Nature 2016, 540, 354–362. [Google Scholar] [CrossRef]
- Mohanty, A.K.; Wu, F.; Mincheva, R.; Hakkarainen, M.; Raquez, J.M.; Mielewski, D.F.; Narayan, R.; Netravali, A.N.; Misra, M. Sustainable polymers. Nat. Rev. Methods Primers 2022, 2, 46. [Google Scholar] [CrossRef]
- Allen, P.W.; Bloomfield, G.F. Natural rubber hydrocarbon. In Chemistry and Physics of Rubber-like Substance; Bateman, L., Ed.; MacLaren and Sons: London, UK, 1963; Chapter 1; pp. 1–17. [Google Scholar]
- Eng, A.H.; Tanaka, Y.; Gan, S.N. FTIR studies on amino groups in purified Hevea rubber. J. Nat. Rubber Res. 1992, 7, 152–155. [Google Scholar]
- Tangpakdee, J.; Tanaka, Y. Characterization of sol and gel in Hevea natural rubber. Rubber Chem. Technol. 1997, 70, 707–713. [Google Scholar] [CrossRef]
- Tarachiwin, L.; Sakdapipanich, J.T.; Ute, K.; Kitayama, T.; Bamba, T.; Fukusaki, E.; Kobayashi, A.; Tanaka, Y. Structural characterization of α-terminal group of natural rubber 1. Decomposition of branch-points by lipase and phosphatase treatments. Biomacromolecules 2005, 6, 1851–1857. [Google Scholar] [CrossRef] [PubMed]
- Tarachiwin, L.; Sakdapipanich, J.T.; Ute k Kitayama, T.; Tanaka, Y. Structural characterization of α-terminal group of natural rubber 2. Decomposition of branch-points by phospholipase and chemical treatments. Biomacromolecules 2005, 6, 1858–1863. [Google Scholar] [CrossRef]
- Tarachiwin, L.; Sakdapipanich, J.; Tanaka, Y. Structure and origin of long-chain branching and gel in natural rubber. Kautsch. Gummi Kunstst. 2005, 58, 115–122. [Google Scholar]
- Amnuaypornsri, S.; Sakdapipanich, J.; Toki, S.; Hsiao, B.; Ichikawa, N.; Tanaka, Y. Strain-induced crystallization of natural rubber: Effect of proteins and phospholipids. Rubber Chem. Technol. 2008, 81, 753–766. [Google Scholar] [CrossRef]
- Food and Drug Administration. Allergic Reaction to Latex-Containing Medical Devices; FDA Medical Alert 1991; Food and Drug Administration: Silver Spring, MD, USA, 1991; MDA91-1. [Google Scholar]
- Ichikawa, N.; Aik-Hwee, E.; Tanaka, Y. Properties of deproteinised natural rubber. In Natural Rubber: Current Development in Product Manufacture and Application, Proceedings of the International Rubber Technology Conference 1993, Kuala Lumpur, Malaysia, 14–16 June 1993; Kadir, A.A.S.A., Ed.; Rubber Research Institute of Malaysia: Kuala Lumpur, Malaysia, 1993; pp. 101–110. [Google Scholar]
- Nakade, S.; Kuga, A.; Hayashi, M.; Tanaka, Y. Highly purified natural rubber. IV. Preparation and characteristics of gloves and condoms. J. Nat. Rubb. Res. 1997, 12, 33–42. [Google Scholar]
- Coran, A.Y. Vulcanization: Conventional and dynamic. Rubber Chem. Technol. 1995, 68, 351–375. [Google Scholar] [CrossRef]
- Aprem, A.S.; Joseph, K.; Thomas, S. Recent developments in crosslinking of elastomers. Rubber Chem. Technol. 2005, 78, 458–488. [Google Scholar] [CrossRef]
- Dluzneski, P.R. Peroxide vulcanization of elastomers. Rubber Chem. Technol. 2001, 74, 451–492. [Google Scholar] [CrossRef]
- Coleman, M.M.; Shelton, J.R.; Koenig, J.L. Sulfur vulcanization of hydrocarbon diene elastomers. Ind. Eng. Chem. Prod. Res. Dev. 1974, 13, 154–165. [Google Scholar] [CrossRef]
- Krejsa, M.R.; Koenig, J.L. A review of sulfur crosslinking fundamentals for accelerated and unaccelerated vulcanization. Rubber Chem. Technol. 1993, 66, 376–410. [Google Scholar] [CrossRef]
- Akiba, M.; Hashim, A.S. Vulcanization and crosslinking in elastomers. Prog. Polym. Sci. 1997, 22, 475–521. [Google Scholar] [CrossRef]
- Heideman, G.; Datta, R.N.; Noordermeer, J.W.M. Activators in accelerated sulfur vulcanization. Rubber Chem. Technol. 2004, 77, 512–541. [Google Scholar] [CrossRef]
- Suchiva, K.; Kowitteerawut, T.; Srichantamit, L. Structure properties of purified natural rubber. J. Appl. Polym. Sci. 2000, 78, 1495–1504. [Google Scholar] [CrossRef]
- Conde-Salazar, L.; del-Río, E.; Guimaraens, D.; González Domingo, A. Type IV allergy to rubber additives: A 10-year study of 686 cases. J. Am. Acad. Dermatol. 1993, 29, 176–180. [Google Scholar] [CrossRef]
- Tan, Z.; Jaeger, R.; Vancso, G.J. Crosslinking studies of poly(dimethylsiloxane) networks: A comparison of inverse gas chromatography, swelling experiments and mechanical analysis. Polymer 1994, 35, 3230–3236. [Google Scholar] [CrossRef]
- Arndt, K.F.; Schreck, J. Netzwerkcharakterisierung durch dampfdruckosmotische Messungen. Acta Polym. 1985, 36, 56–57. [Google Scholar] [CrossRef]
- Patel, S.K.; Malone, S.; Cohen, C.; Gillmor, J.R.; Colby, R.H. Elastic modulus and equilibrium swelling of poly(dimethylsiloxane) networks. Macromolecules 1992, 25, 5241–5251. [Google Scholar] [CrossRef]
- Valentín, J.L.; Carretero-González, J.; Mora-Barrantes, I.; Chassé, W.; Saalwächter, K. Uncertainties in the determination of cross-link density by equilibrium swelling experiments in natural rubber. Macromolecules 2008, 41, 4717–4729. [Google Scholar] [CrossRef]
- Imanishi, Y.; Adachi, K.; Kotaka, T.J. Further investigation of the dielectric normal mode process in undiluted cis-polyisoprene with narrow distribution of molecular weight. Chem. Phys. 1988, 89, 7585–7592. [Google Scholar]
- Zaper, A.M.; Koenig, J.L. Solid State Carbon-13 NMR Studies of Vulcanized Elastomers. II, Sulfur Vulcanization of Natural Rubber. Rubber Chem. Technol. 1987, 60, 252–277. [Google Scholar] [CrossRef]
- Orza, R.A.; Magusin, P.C.M.M.; Litvinov, V.M.; van Duin, M.; Michels, M.A.J. Mechanism for peroxide cross-linking of EPDM rubber from MAS 13C NMR Spectroscopy. Macromolecules 2009, 42, 8914–8924. [Google Scholar] [CrossRef]
- Pyckout-Hintzen, W.; Springer, T.; Forster, F.; Gronski, W. Small-angle neutron scattering investigation of a multilinked polybutadiene network crosslinked in solution. Macromolecules 1991, 24, 1269–1274. [Google Scholar] [CrossRef]
- Ikeda, Y.; Higashitani, N.; Hijikata, K.; Kokubo, Y.; Morita, Y.; Shibayama, M.; Osaka, N.; Suzuki, T.; Endo, H.; Kohjiya, S. Vulcanization: New focus on a traditional technology by small-angle neutron scattering. Macromolecules 2009, 42, 2741–2748. [Google Scholar] [CrossRef]
- Litvinov, V.M.; De, P.P. (Eds.) Spectroscopy of Rubbers and Rubbery Materials; Rapra Technology Ltd.: Shawbury, UK, 2002. [Google Scholar]
- Cohen-Addad, J.P. Effect of the anisotropic chain motion in molten polymers: The solidlike contribution of the nonzero average dipolar coupling to NMR signals theoretical description. J. Chem. Phys. 1973, 60, 2440–2453. [Google Scholar] [CrossRef]
- Brereton, M.G. NMR transverse relaxation function calculated for constrained polymer chains: Application to entanglements and networks. Macromolecules 1990, 23, 1119–1131. [Google Scholar] [CrossRef]
- Graf, R.; Heuer, A.; Spiess, H.W. Chain-order effects in polymer melts probed by 1H double-quantum NMR spectrocopy. Phys. Rev. Lett. 1998, 80, 5738–5741. [Google Scholar] [CrossRef]
- Saalwächter, K. Proton multiple-quantum NMR for the study of chain dynamics and structural constraints in polymeric soft materials. Prog. Nucl. Magn. Reason. Spectrosc. 2007, 51, 1–35. [Google Scholar] [CrossRef]
- Vieyres, A.; Pérez-Aparicio, R.; Albouy, P.-A.; Sanseau, O.; Saalwächter, K.; Long, D.R.; Sotta, P. Sulfur-Cured Natural Rubber Elastomer Networks: Correlating Cross-Link Density, Chain Orientation, and Mechanical Response by Combined Techniques. Macromolecules 2013, 46, 889–899. [Google Scholar] [CrossRef]
- Basterra-Beroiz, B.; Rommel, R.; Kayser, F.; Westermann, S.; Valentín, J.L.; Heinrich, G. New Insights into Rubber Network Structure by a Combination of Experimental Techniques. Rubber Chem. Technol. 2017, 90, 347–366. [Google Scholar] [CrossRef]
- Litvinov, V.M. EPDM/PP thermoplastic vulcanizates as studied by proton NMR relaxation: Phase composition, molecular mobility, network structure in the rubbery phase, and network heterogeneity. Macromolecules 2006, 39, 8727–8741. [Google Scholar] [CrossRef]
- Demco, D.E.; Hafner, S.; Fülber, C.; Graf, R.; Spiess, H.W. Two-dimensional proton magnetization-exchange NMR spectroscopy in cross-linked elastomers. J. Chem. Phys. 1996, 105, 11285–11296. [Google Scholar] [CrossRef]
- Callaghan, P.T.; Samulski, E.T. Molecular ordering and the direct measurement of weak proton-proton dipolar interactions in a rubber network. Macromolecules 1997, 30, 113–122. [Google Scholar] [CrossRef]
- Valentín, J.; Posadas, P.; Fernández-Torres, A.; Malmierca, M.A.; González, L.; Chassé, W.; Saalwächter, K. Inhomogeneities and chain dynamics in diene rubbers vulcanized with different cure systems. Macromolecules 2010, 43, 4210–4222. [Google Scholar] [CrossRef]
- Basterra-Beroiz, B.; Rommel, R.; Kayser, F.; Valentín, J.L.; Westermann, S.; Heinrich, G. Revisiting Segmental Order: A Simplified Approach for Sulfur-Cured Rubbers Considering Junction Fluctuations and Entanglements. Macromolecules 2018, 51, 2076–2088. [Google Scholar] [CrossRef]
- Saalwächter, K.; Herrero, B.; López-Manchado, M.A. Chain Order and Cross-Link Density of Elastomers As Investigated by Proton Multiple-Quantum NMR. Macromolecules 2005, 38, 9650–9660. [Google Scholar] [CrossRef]
- Saleesung, T.; Reichert, D.; Saalwächter, K.; Sirisinha, C. Correlation of crosslink densities using solid state NMR and conventional techniques in peroxide-crosslinked EPDM rubber. Polymer 2015, 56, 309–317. [Google Scholar] [CrossRef]
- Saalwächter, K.; Ziegler, P.; Spyckerelle, O.; Haidar, B.; Vidal, A.; Sommer, J.U. 1H multiple-quantum nuclear magnetic resonance investigations of molecular order distributions in poly(dimethylsiloxane) networks: Evidence for a linear mixing law in bimodal systems. J. Chem. Phys. 2003, 119, 3468–3482. [Google Scholar] [CrossRef]
- Chassé, W.; Valentín, J.L.; Genesky, G.D.; Cohen, C.; Saalwächter, K. Precise dipolar coupling constant distribution analysis in proton multiple-quantum NMR of elastomers. J. Chem. Phys. 2011, 134, 044907. [Google Scholar] [CrossRef] [PubMed]
- Chassé, W.; Lang, M.; Sommer, J.-U.; Saalwächter, K. Cross-link density estimation of PDMS networks with precise consideration of networks defects. Macromolecules 2012, 45, 899–912. [Google Scholar] [CrossRef]
- Acosta, R.H.; Vega, D.A.; Villar, M.A.; Monti, G.A.; Vallés, E.M. Double quantum NMR applied to polymer networks with low concentration of pendant chains. Macromolecules 2006, 39, 4788–4792. [Google Scholar] [CrossRef]
- Acosta, R.H.; Monti, G.A.; Villar, M.A.; Vallés, E.M.; Vega, D.A. Transiently trapped entanglements in model polymer networks. Macromolecules 2009, 42, 4674–4680. [Google Scholar] [CrossRef]
- Sommer, J.-U.; Chassé, W.; Valentín, J.L.; Saalwächter, K. Effect of excluded volume on segmental orientation correlations in polymer chains. Phys. Rev. E 2008, 78, 051803. [Google Scholar] [CrossRef]
- Iqbal, J.; Bhatia, B.; Nayyar, N.K. Transition metal-promoted free-radical reactions in organic synthesis: The formation of carbon-carbon bonds. Chem. Rev. 1994, 94, 519–564. [Google Scholar] [CrossRef]
- Tanaka, Y. Structural characterization of natural polyisoprenes: Solve the mystery of natural rubber based on structural study. Rubber Chem. Technol. 2001, 74, 355–375. [Google Scholar] [CrossRef]
- Toki, S.; Che, J.; Rong, L.; Hsiao, B.S.; Amnuaypornsri, S.; Nimpaiboon, A.; Sakdapipanich, J. Entanglements and networks to strain-induced crystallization and stress−strain relations in natural rubber and synthetic polyisoprene at various temperatures. Macromolecules 2013, 46, 5238–5248. [Google Scholar] [CrossRef]
- Valentin, J.L.; Lopez, D.; Hernandez, R.; Mijangos, C.; Saalwachter, K. Structure of Poly(vinyl alcohol) Cryo-Hydrogels as Studied by Proton Low-Field NMR Spectroscopy. Macromolecules 2009, 42, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Che, J.; Toki, S.; Valentin, J.L.; Brasero, J.; Nimpaiboon, A.; Rong, L.; Hsiao, B.S. Chain Dynamics and Strain-Induced Crystallization of Pre- and Postvulcanized Natural Rubber Latex Using Proton Multiple Quantum NMR and Uniaxial Deformation by in Situ Synchrotron X-ray Diffraction. Macromolecules 2012, 45, 6491–6503. [Google Scholar] [CrossRef]
- Malmierca, M.A.; Mora-Barrantes, I.; Posadas, P.; Rodríguez, A.; Ibarra, L.; Gonzalez-Jiménez, A.; Nogales, A.; Saalwachter, K.; Valentín, J.L. Characterization of Network Structure and Chain Dynamics of Elastomeric Ionomers by Means of 1H Low-Field NMR. Macromolecules 2014, 47, 5655–5667. [Google Scholar] [CrossRef]
- Vaca Chávez, F.; Saalwächter, K. Time-domain NMR observation of entangled polymer dynamics: Analytical theory of signal functions. Macromolecules 2011, 44, 1560–1569. [Google Scholar] [CrossRef]
- Vaca Chávez, F.; Saalwächter, K. Time-domain NMR observation of entangled polymer dynamics: Universal behavior of flexible homopolymers and applicability of the tube model. Macromolecules 2011, 44, 1549–1559. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Claredon Press: Oxford, UK, 1986. [Google Scholar]
- Amnuaypornsri, S.; Sakdapipanich, J.; Tanaka, Y. Green strength of natural rubber: The origin of the stress–strain behavior of natural rubber. J. Appl. Polym. Sci. 2009, 111, 2127–2133. [Google Scholar] [CrossRef]
- Suzuki, T.; Osaka, N.; Endo, H.; Shibayama, M.; Ikeda, Y.; Asai, H.; Higashitani, N.; Kokubo, Y.; Kohjiya, S. Nonuniformity in cross-linked natural rubber as revealed by contrast-variation small-angle neutron scattering. Macromolecules 2010, 43, 1556–1563. [Google Scholar] [CrossRef]
- Whelan, A.; Lee, K.S. (Eds.) Development in Rubber Technology; Applied Science: London, UK, 1979; Volume 1. [Google Scholar]
- García-Quesada, J.C.; Gilbert, M. Peroxide crosslinking of unplasticized poly(vinyl chloride). J. Appl. Polym. Sci. 2000, 77, 2657–2666. [Google Scholar] [CrossRef]
- Busci, A.; Szocs, F. Kinetics of radical generation in PVC with dibenzoyl peroxide utilizing high-pressure technique. Macromol. Chem. Phys. 2000, 201, 435–438. [Google Scholar]
- Keller, R.C. Peroxide curing of ethylene-propylene elastomers. Rubber Chem. Technol. 1988, 61, 238–254. [Google Scholar] [CrossRef]
- Murgic, Z.H.; Jelencic, J.; Murgic, L. The mechanism of triallylcyanurate as a coagent in EPDM peroxide vulcanization. Polym. Eng. Sci. 1998, 38, 689–692. [Google Scholar] [CrossRef]
- Class, J. A review of the fundamentals of crosslinking with peroxides. Rubber World 1999, 220, 35–39. [Google Scholar]
- Oh, S.J.; Koenig, J.L. Studies of peroxide curing of cis-1,4-Polyisoprene/Diallyl phthalate blends by spectroscopic techniques. Rubber Chem. Technol. 1999, 72, 334–342. [Google Scholar] [CrossRef]
- Oh, S.J.; Koenig, J.L. Studies of peroxide curing of polybutadiene/zinc diacrylate blends by fast FT-IR imaging. Rubber Chem. Technol. 2000, 73, 74–79. [Google Scholar] [CrossRef]
- Schaich, K.M.; Karel, M. Free radical reactions of peroxidizing lipids with amino acids and proteins: An ESR study. Lipids 1976, 11, 392–400. [Google Scholar] [CrossRef]
- Gardner, H.W. Lipid hydroperoxide reactivity with proteins and amino acids: A review. J. Agric. Food Chem. 1979, 27, 220–229. [Google Scholar] [CrossRef]
- Saeed, S.; Fawthrop, S.A.; Howell, N.K. Electron spin resonance (ESR) study on free radical transfer in fish lipid–protein interaction. J. Sci. Food Agric. 1999, 79, 1809–1816. [Google Scholar] [CrossRef]
- Endstra, W.C. Application of coagents for peroxide crosslinking. Kautsch. Gummi Kunstst. 1990, 43, 790–793. [Google Scholar]
- Simpson, J.A.; Narita, S.; Gieseg, S.; Gebicki SGebick, J.M.; Dean, R.T. Long-lived reactive species on free-radical-damaged proteins. Biochem. J. 1992, 282, 621–624. [Google Scholar] [CrossRef]
- Gebicki, S.; Gebicki, J.M. Formation of peroxides in amino acids and proteins exposed to oxygen free radicals. Biochem. J. 1993, 289, 743–749. [Google Scholar] [CrossRef]
- Dean, R.T.; Fu, S.; Stocker, R.; Davies, M.J. Biochemistry and pathology of radical-mediated protein oxidation. Biochem. J. 1997, 324, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, C.L.; Davies, M.J. Generation and propagation of radical reactions on proteins. Biochim. Biophys. Acta 2001, 1504, 196–219. [Google Scholar] [CrossRef]
- Curro, J.G.; Pincus, P. A theoretical basis for viscoelastic relaxation of elastomers the long-time limit. Macromolecules 1983, 16, 559–562. [Google Scholar] [CrossRef]
- Roth, L.E.; Vega, D.A.; Valles, E.M.; Villar, M.A. Viscoelastic properties of networks with low concentration of pendant chains. Polymer 2004, 45, 5923–5931. [Google Scholar] [CrossRef]
- Batra, A.; Cohen, C.; Archer, L. Stress relaxation of end-linked Polydimethylsiloxane elastomers with long pendent chains. Macromolecules 2005, 38, 7174–7180. [Google Scholar] [CrossRef]
- Moore, C.G.; Scanlan, J.J. Determination of degree of crosslinking in natural rubber vulcanizates. Part VI. Evidence for chain scission during the crosslinking of natural rubber with organic peroxides. Polym. Sci. 1960, 43, 23–33. [Google Scholar] [CrossRef]
- Mark, H.F. Elastomers-Past, Present, and Future. Rubber Chem. Technol. 1988, 61, G73–G96. [Google Scholar] [CrossRef]
- González, L.; Rodríguez, A.; Marcos, A.; Chamorro, C. Crosslink reaction mechanisms of diene rubber with dicumyl peroxide. Rubber Chem. Technol. 1996, 69, 203–214. [Google Scholar] [CrossRef]
- Valentín, J.L.; Fernández-Torres, A.; Posadas, P.; Marcos-Fernandez, A.; Rodríguez, A.; González, L. Measurements of freezing-point depression to evaluate rubber network structure. Crosslinking of natural rubber with dicumyl peroxide. J. Polym. Sci. Part B Polym. Phys. 2007, 45, 544–556. [Google Scholar] [CrossRef]
- Karino, T.; Ikeda, Y.; Yasuda, Y.; Kohjiya, S.; Shibayama, M. Nonuniformity in natural rubber as revealed by small-angle neutron scattering, small-angle x-ray scattering, and atomic force microscopy. Biomacromolecules 2007, 8, 693–699. [Google Scholar] [CrossRef]
- Pailhories, G. Reducing proteins in latex gloves: The industrial approach. Clin. Rev. Allergy 1993, 11, 391–402. [Google Scholar] [CrossRef]
- Ng, K.P.; Yip, E.; Mok, K.L. Production of natural rubber latex gloves with low extractable protein content: Some practical recommendations. J. Nat. Rubber Res. 1994, 9, 87–95. [Google Scholar]
- Ghazaly, H.M. Factory production of examination gloves from low protein latex. J. Nat. Rubber Res. 1994, 9, 96–108. [Google Scholar]
- Dalrymple, S.J.; Audley, B.G. Allergenic proteins in dipped products: Factors influencing extractable protein levels. Rubber Dev. 1992, 45, 51–60. [Google Scholar]
- Leynadier, F.; Xuan, T.T.; Dry, J. Allergenicity suppression in natural latex surgical gloves. Allergy 1991, 46, 619–625. [Google Scholar] [CrossRef]
- Klüppel, M.; Heinrich, G. Network Structure and Mechanical Properties of Sulfur Cured Rubbers. Macromolecules 1994, 27, 3595–3603. [Google Scholar] [CrossRef]
- Wu, B.; Chassé, W.; Peters, R.; Brooijmans, T.; Dias, A.A.; Heise, A.; Duxbury, C.J.; Kentgens, A.P.M.; Brougham, D.F.; Litvinov, V.M. Network Structure in Acrylate Systems: Effect of Junction Topology on Cross-Link Density and Macroscopic Gel Properties. Macromolecules 2016, 49, 6531–6540. [Google Scholar] [CrossRef]
- Lang, M. Relation between Cross-Link Fluctuations and Elasticity in Entangled Polymer Networks. Macromolecules 2017, 50, 2547–2555. [Google Scholar] [CrossRef]
- Vilgis, T.A.; Heinrich, G. Statics and dynamics of heterogeneous polymer networks. Macromol. Theory Simul. 1971, 3, 271–293. [Google Scholar] [CrossRef]
- Nadarajah, M.; Tirimanne, A.S.L.; Coomarasamy, A.; Kasinathan, S. Some naturally occurring antioxidants in Hevea brasiliensis latex. J. Rubb. Res. Inst. Sri Lanka 1971, 48, 202–211. [Google Scholar]
- Tuampeomsab, S.; Sakdapipanich, J.; Tanaka, Y. Influence of some non-rubber components on aging behavior of purified natural rubber. Rubber Chem. Technol. 2007, 80, 159–169. [Google Scholar] [CrossRef]
- Hasma, H. Role of some non-rubber constituents on thermal oxidative aging of natural rubber. J. Nat. Rubb. Res. 1990, 5, 1–18. [Google Scholar]
- Tuampeomsab, S.; Sakdapipanich, J. Role of naturally occurring lipids and proteins on thermal aging behaviour of purified natural rubber. Kautsch. Gummi Kunstst. 2007, 12, 678–684. [Google Scholar]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 3rd ed.; Clarendon Press: Oxford, UK, 1999. [Google Scholar]
- Gebicki, J.M. Protein hydroperoxides as new reactive oxygen species. Redox Rep. 1997, 3, 99–110. [Google Scholar] [CrossRef]
- Tanaka, Y.; Tarachiwin, L. Recent advances in structural characterization of natural rubber. Rubber Chem. Technol. 2009, 82, 283–314. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Protein Content (% w/w) | Ester Content (mmol/kg-Rubber) | Metal Content (% w/w) | ||
|---|---|---|---|---|---|
| Cu2+ | Fe2+ | Mn2+ | |||
| NR | 2.62 | 14.28 | ND | 0.0016 | ND |
| DPNR | 0.06 | 14.23 | ND | 0.0014 | ND |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nimpaiboon, A.; González-Jiménez, A.; Pérez-Aparicio, R.; Martín-Salamanca, F.; Zepeda-Rodríguez, Z.; López-Valentín, J.; Sakdapipanich, J. Effect of Proteins on the Network Formation and Degradation of Peroxide Cross-Linked Natural Rubber Elucidated by Time-Domain NMR. Polymers 2025, 17, 1063. https://doi.org/10.3390/polym17081063
Nimpaiboon A, González-Jiménez A, Pérez-Aparicio R, Martín-Salamanca F, Zepeda-Rodríguez Z, López-Valentín J, Sakdapipanich J. Effect of Proteins on the Network Formation and Degradation of Peroxide Cross-Linked Natural Rubber Elucidated by Time-Domain NMR. Polymers. 2025; 17(8):1063. https://doi.org/10.3390/polym17081063
Chicago/Turabian StyleNimpaiboon, Adun, Antonio González-Jiménez, Roberto Pérez-Aparicio, Fernando Martín-Salamanca, Zenen Zepeda-Rodríguez, Juan López-Valentín, and Jitladda Sakdapipanich. 2025. "Effect of Proteins on the Network Formation and Degradation of Peroxide Cross-Linked Natural Rubber Elucidated by Time-Domain NMR" Polymers 17, no. 8: 1063. https://doi.org/10.3390/polym17081063
APA StyleNimpaiboon, A., González-Jiménez, A., Pérez-Aparicio, R., Martín-Salamanca, F., Zepeda-Rodríguez, Z., López-Valentín, J., & Sakdapipanich, J. (2025). Effect of Proteins on the Network Formation and Degradation of Peroxide Cross-Linked Natural Rubber Elucidated by Time-Domain NMR. Polymers, 17(8), 1063. https://doi.org/10.3390/polym17081063