Synthesis of Ethylene Glycol Dimethacrylate-Methyl Methacrylate Copolymers, Determination of their Reactivity Ratios, and a Study of Dopant and Temperature Effects on their Conductivities

Abstract
:1. Introduction
2. Experimental
2.1. Materials
2.2. Preparation of Polymers
2.3. Preparation of Copolymers
3. Theory
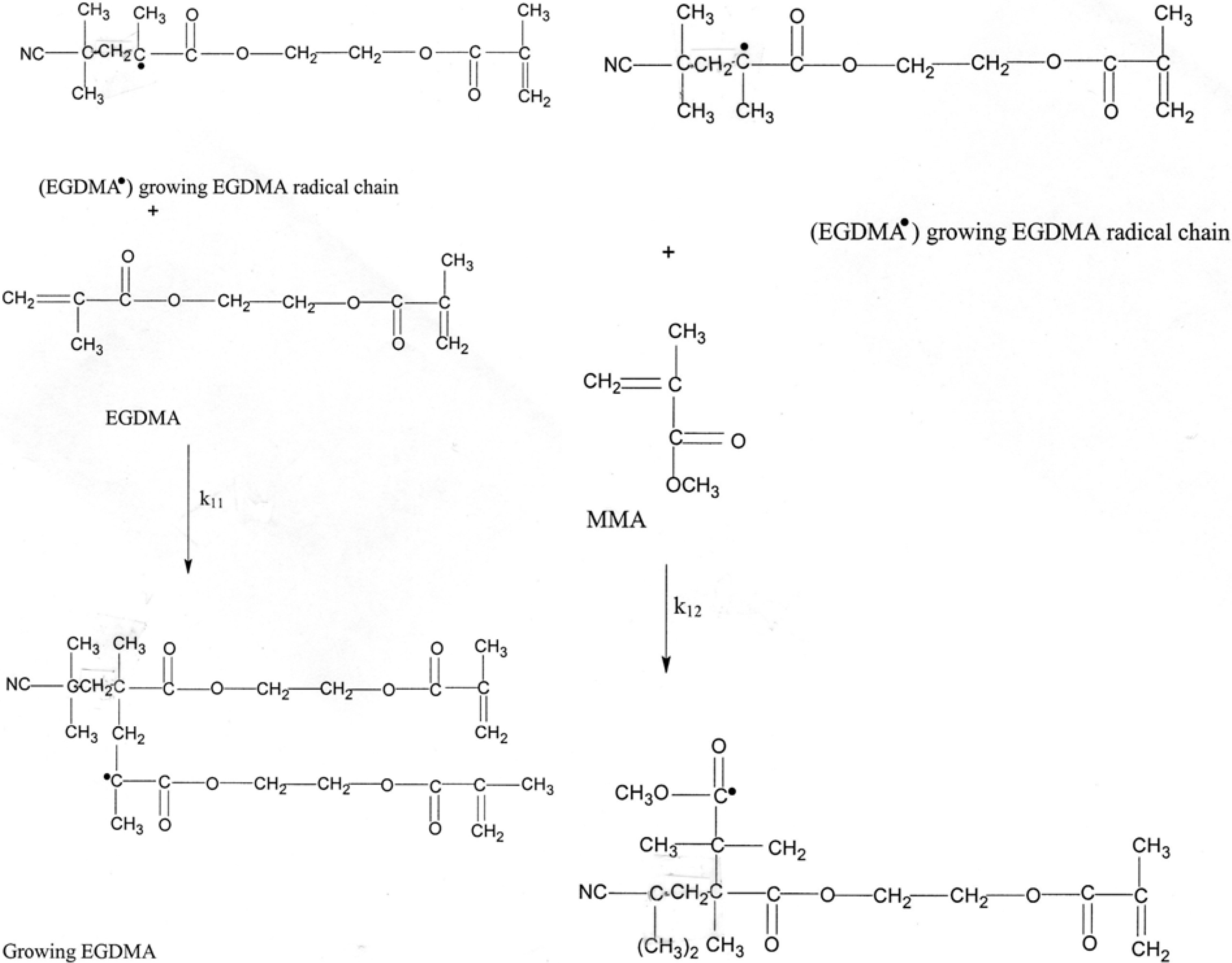
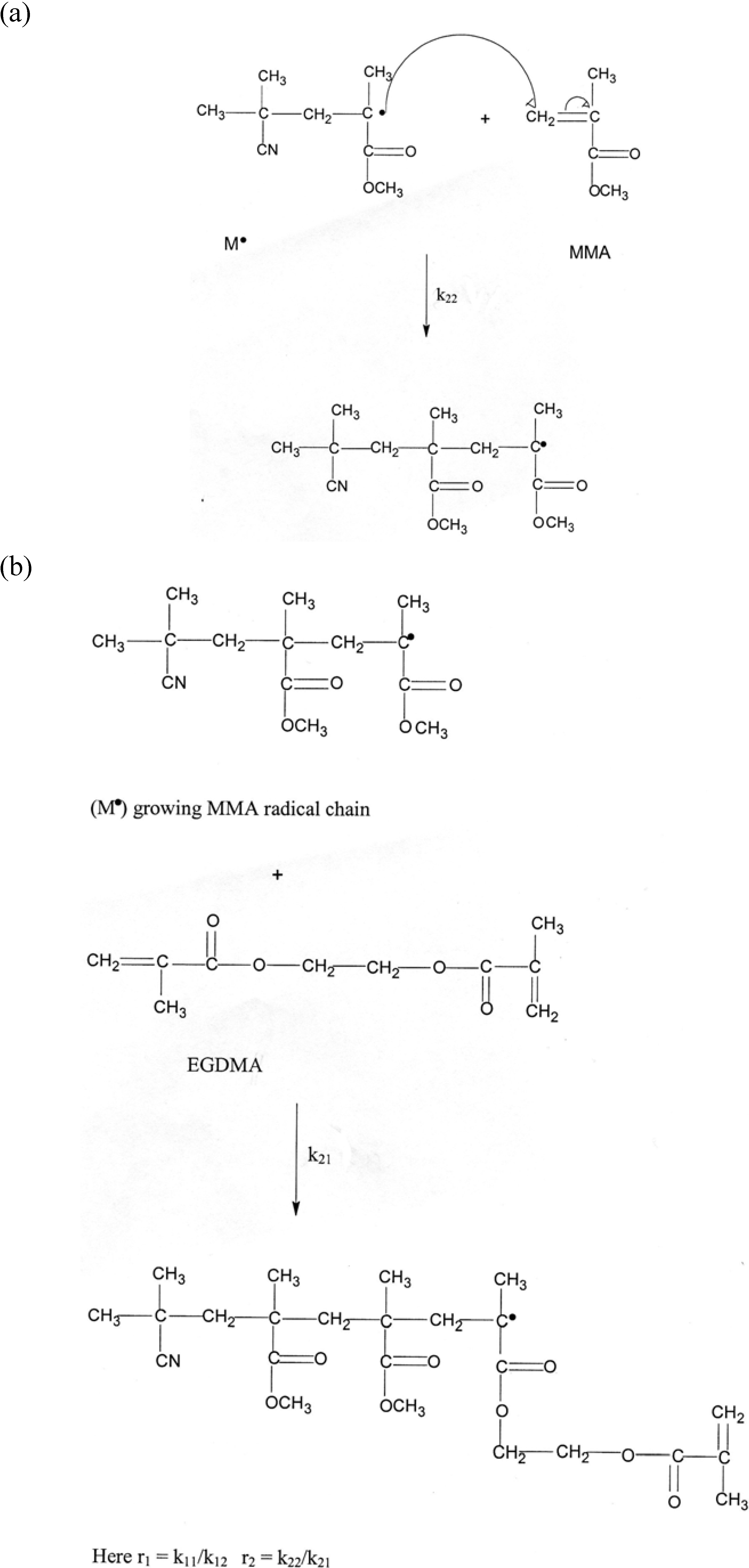
3.1. Mechanisms





EGDMA● + EGDMA ➔ EGDMA● (growing radical)
EGDMA● + MMA ➔ MMA●
MMA● + EGDMA ➔ EGDMA●
MMA● + MMA ➔ MMA● (growing radical)
3.2. Rate of Creation of Radicals
4. Results and Discussion
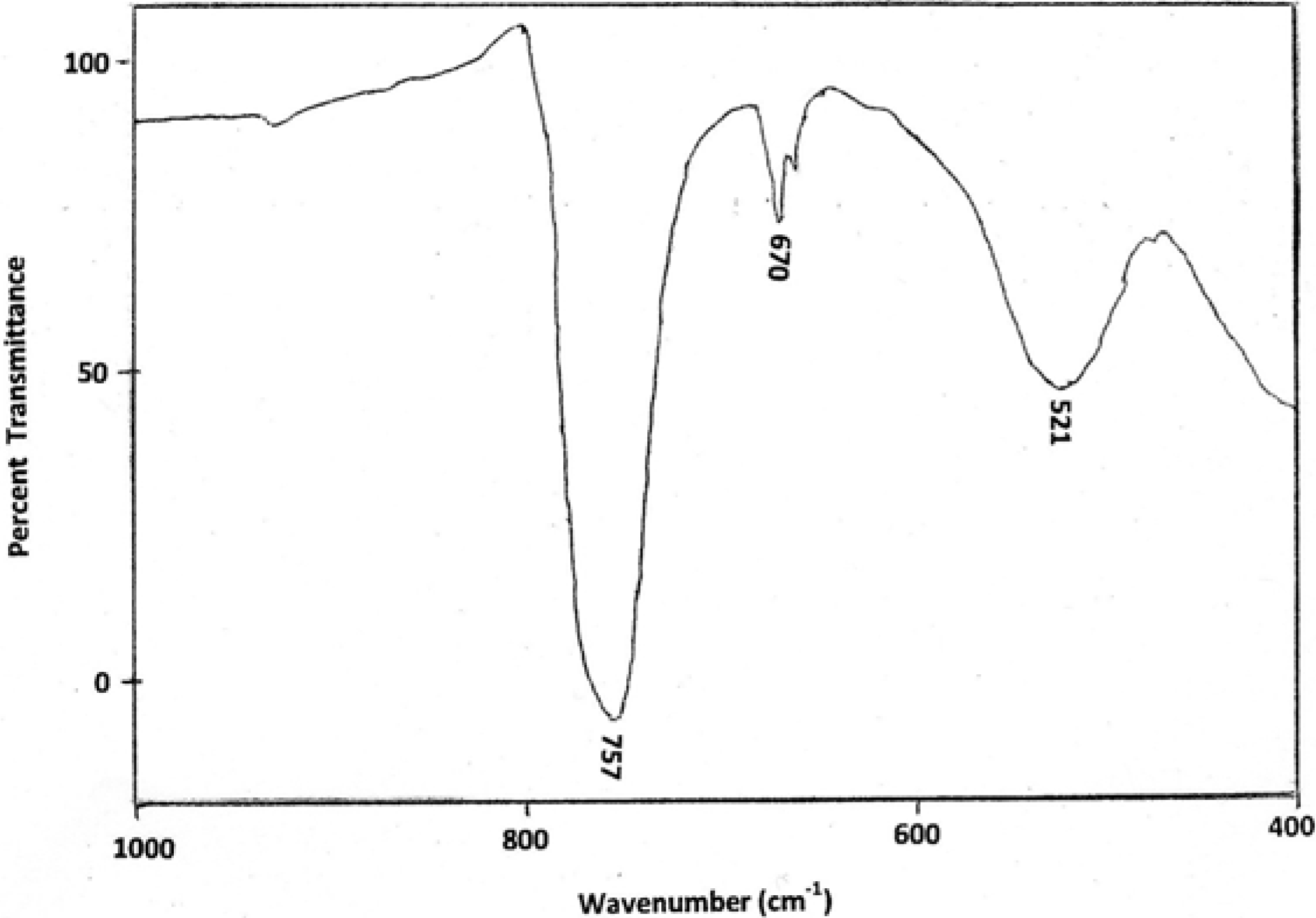
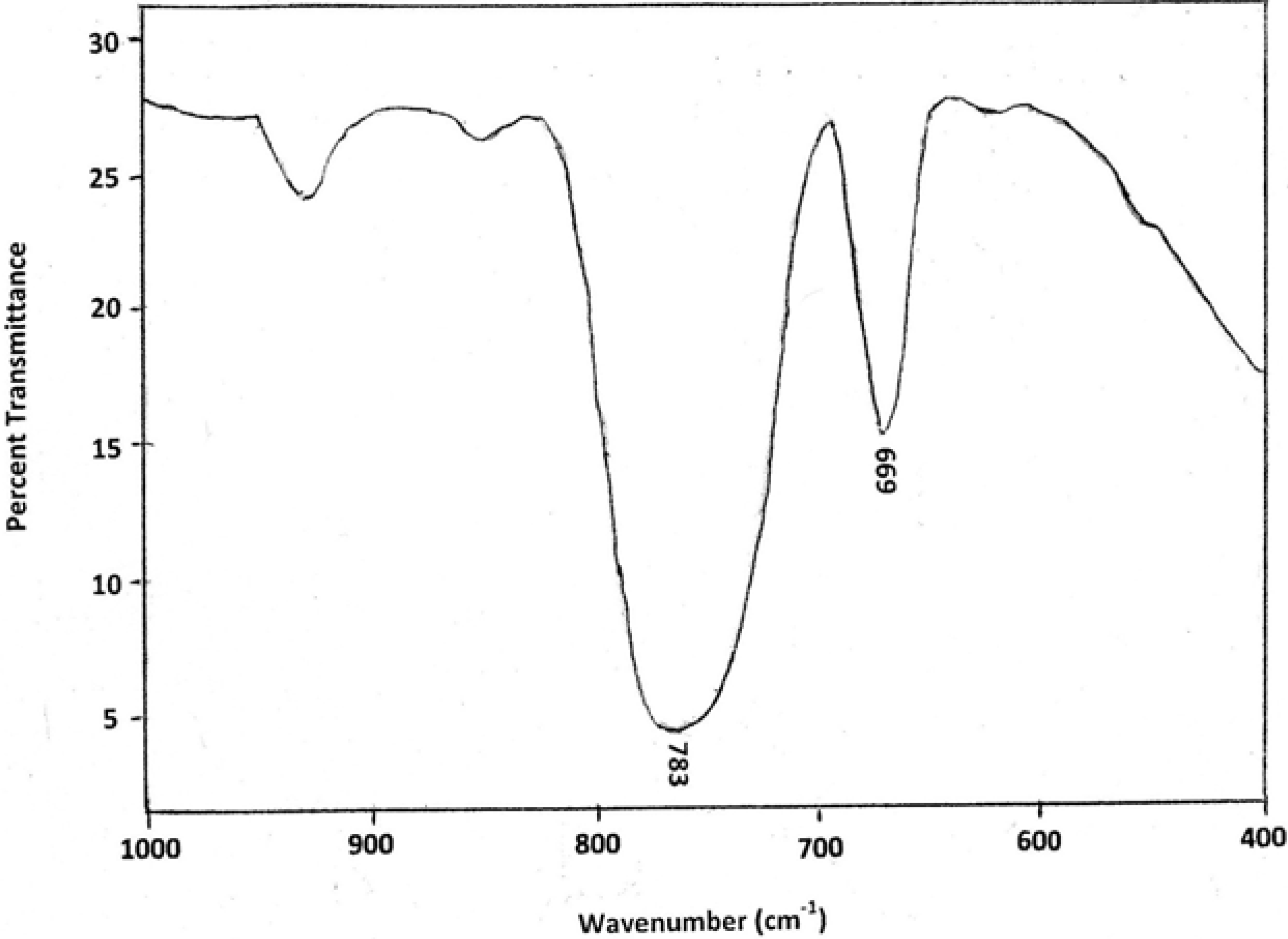
4.1. IR Analysis of EGDMA/MMA Copolymers





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Copolymer | MMA (mL) | EGDMA (mL) | THF (mL) | Volume ratio of EGDMA to MMA in feed |
|---|---|---|---|---|
| 1 | 0.75 | 4.25 | 10 | 5.66 |
| 2 | 1.00 | 4.00 | 10 | 4.00 |
| 3 | 1.25 | 3.75 | 10 | 3.00 |
| 4 | 1.50 | 3.50 | 10 | 2.33 |
| 5 | 1.75 | 3.25 | 10 | 1.86 |
| Copolymer | (A)Copolymer Taken g/mL in chl *103 | Absorbance at 520–540 cm−1 | (B)(MMA)B1 *103 in chl. | (A−B) (EGDMA)A-B2 *103 in chl. | [MMA] mol/L in co-polymer | [EDGMA] mol/L in copolymer | (F2) mol % of MMA in copolymer |
|---|---|---|---|---|---|---|---|
| 1 | 7.0 | 0.27 | 1.35 | 5.66 | 0.01348 | 0.02855 | 32.09 |
| 2 | 4.2 | 0.32 | 1.80 | 2.40 | 0.01797 | 0.01210 | 59.76 |
| 3 | 5.4 | 0.37 | 2.20 | 5.03 | 0.02190 | 0.02537 | 46.41 |
| 4 | 7.7 | 0.48 | 3.10 | 4.61 | 0.03096 | 0.02325 | 57.12 |
| 5 | 9.9/ | 0.85 | 6.30 | 3.60 | 0.06292 | 0.01816 | 77.60 |
| Copolymer | [EGDMA]/[MMA] volume ratio in feed from Table 1 | [MMA] in feed, mol/L, in chloroform | [EGDMA] in feed, mol/L, in chloroform | (f2) mol % MMA in feed |
|---|---|---|---|---|
| 1 | 5.66 | 0.467 | 1.502 | 23.72 |
| 2 | 4.00 | 0.623 | 1.414 | 30.20 |
| 3 | 3.00 | 0.779 | 1.325 | 37.02 |
| 4 | 2.33 | 0.935 | 1.237 | 43.05 |
| 5 | 1.86 | 1.091 | 1.149 | 48.71 |
| Copolymer No. | Mole ratio in feed H = [EGDMA][MMA] from Table 3 | Mole ratio in copolymer H = [EGDMA]/[MMA] | H(1-h)/h | H2/h |
|---|---|---|---|---|
| 1 | 3.216 | 2.118 | −1.697 | 4.883 |
| 2 | 2.269 | 1.330 | −0.563 | 3.870 |
| 3 | 1.701 | 1.155 | −0.228 | 2.505 |
| 4 | 1.323 | 0.751 | 0.439 | 2.331 |

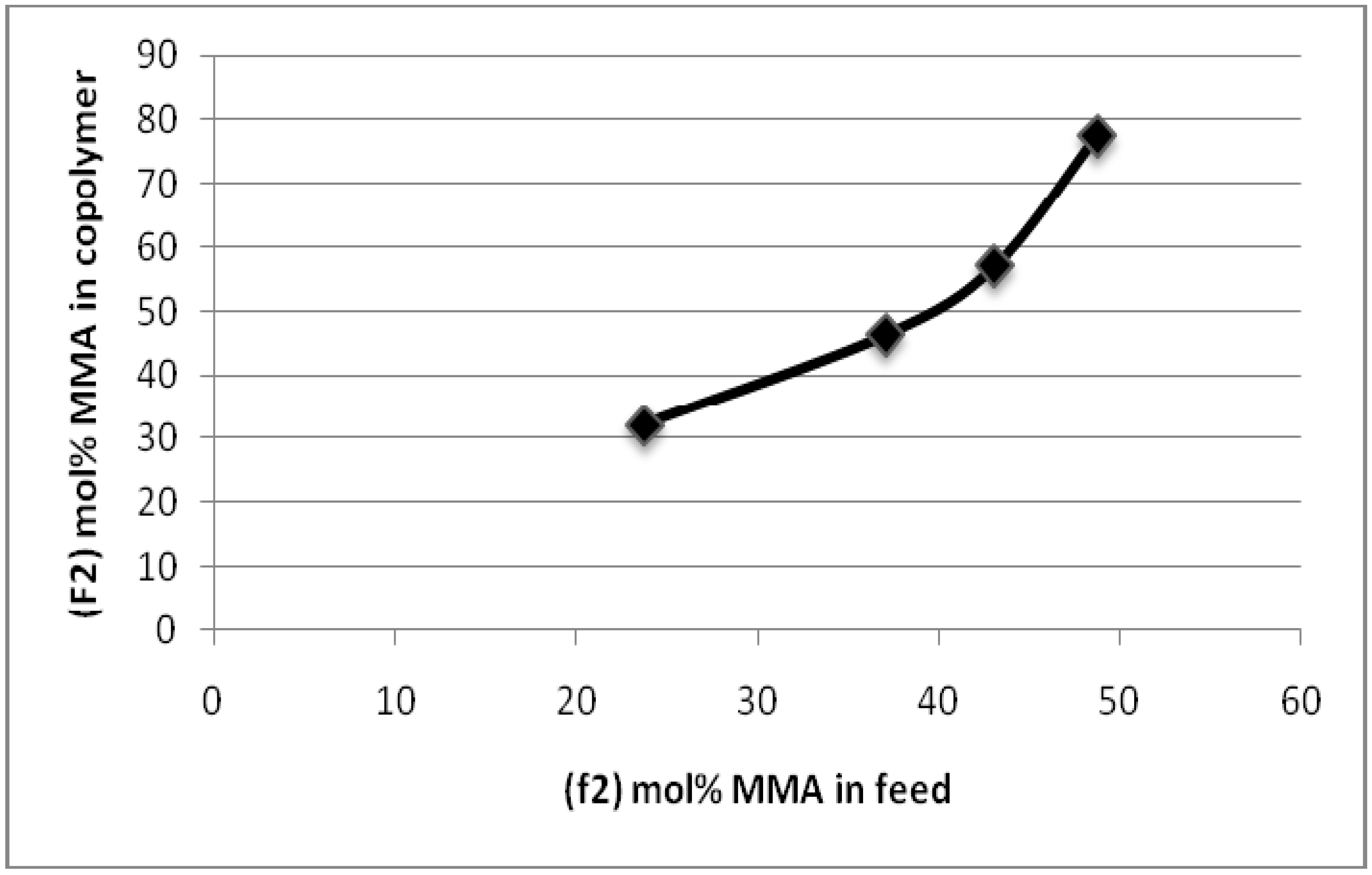
| f2 (mol% MMA in feed) | F2 (mol% MMA in Copolymer) |
|---|---|
| 23.72 | 32.09 |
| 37.02 | 46.41 |
| 43.05 | 57.12 |
| 48.71 | 77.60 |

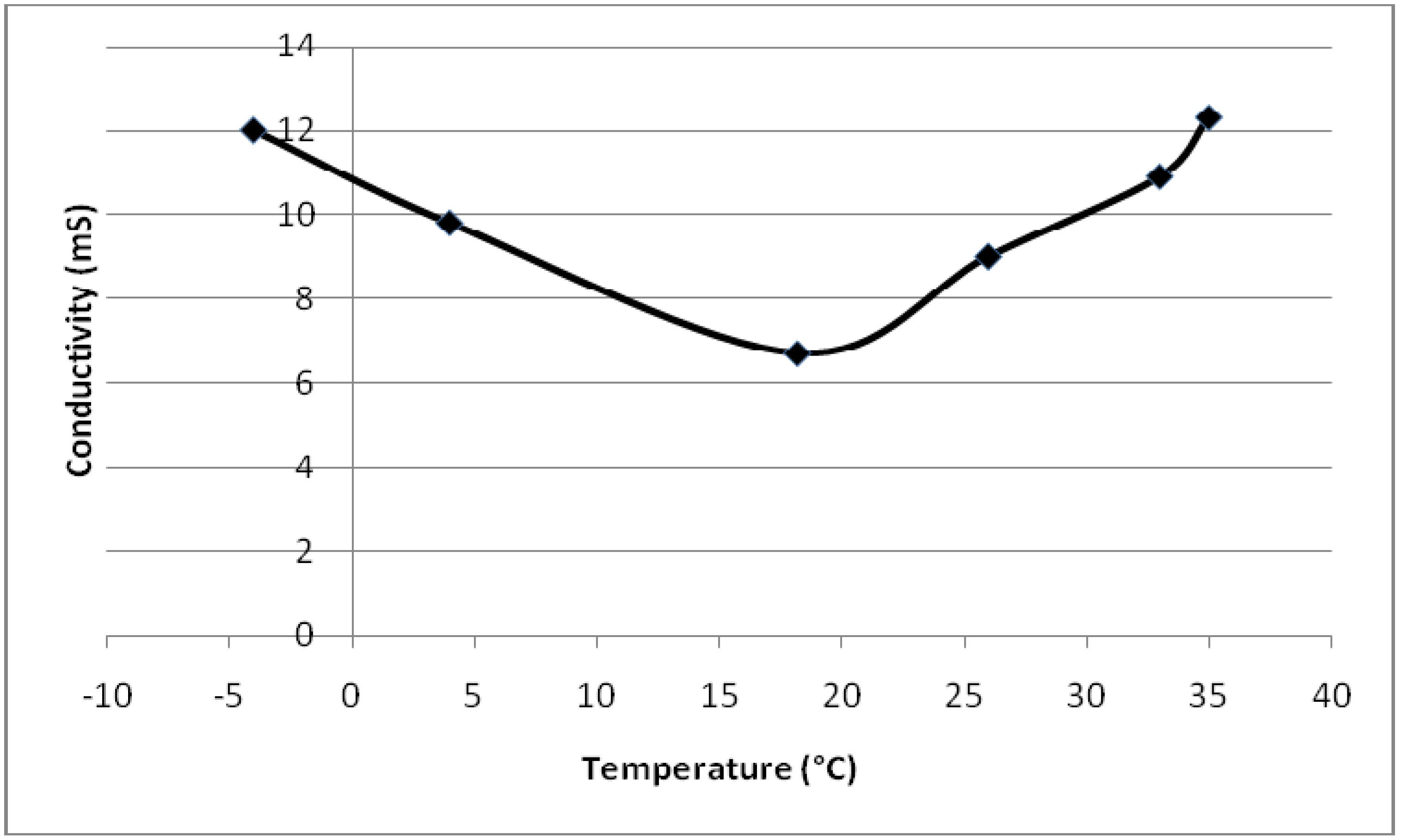
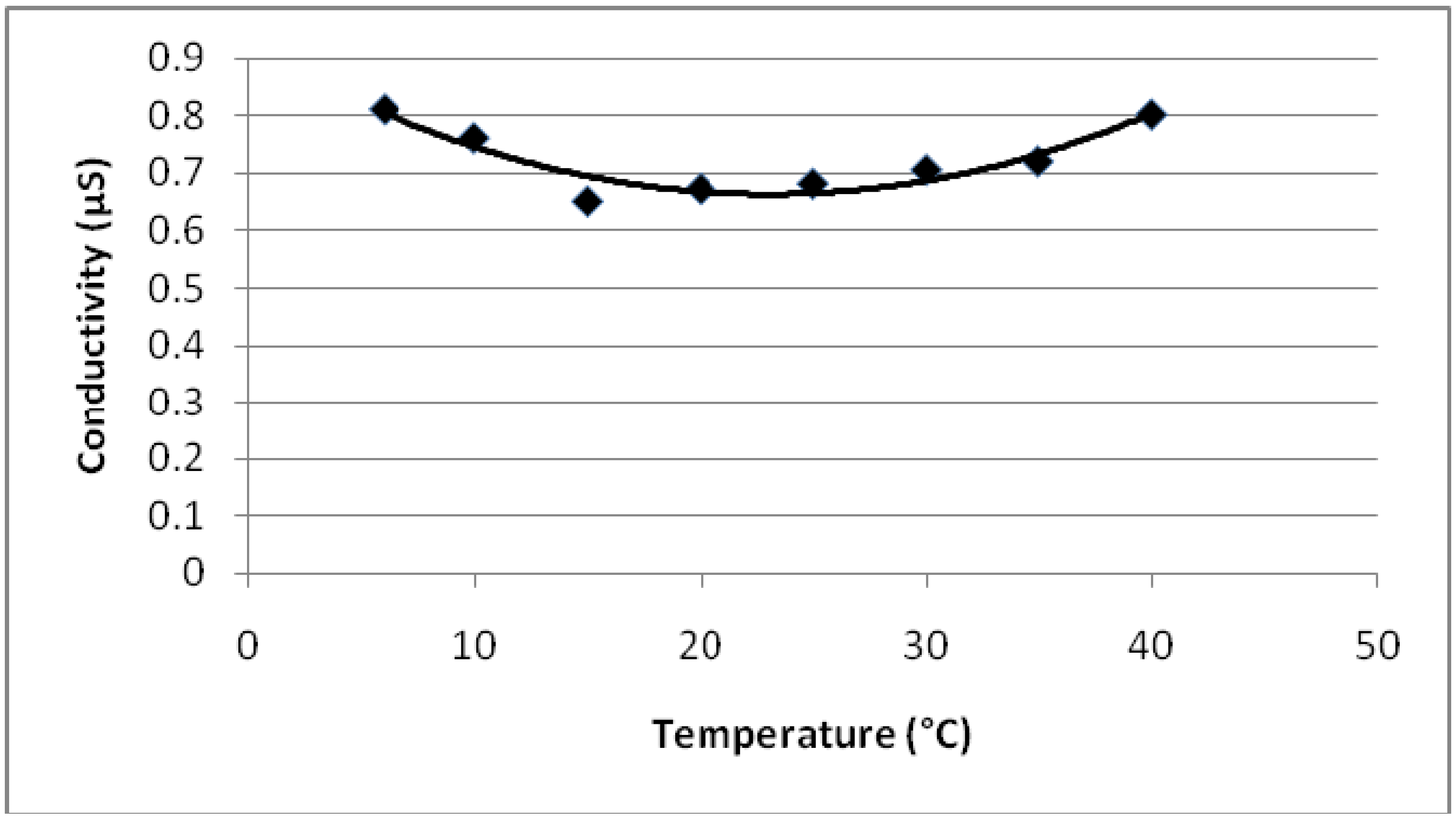
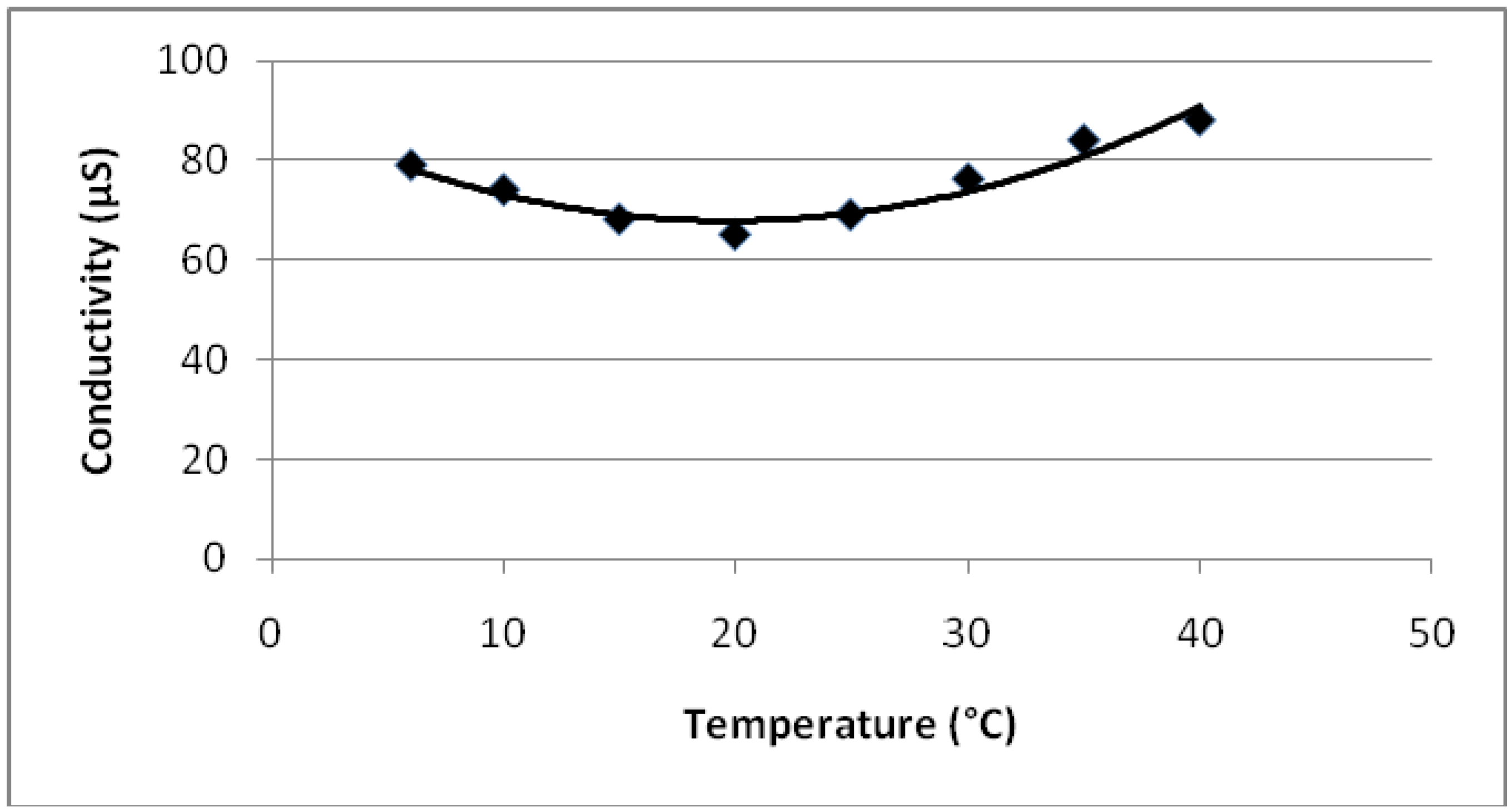
4.2. Conductivity Change of Monomers and Copolymers with Temperature and Dopant

| Temperature (°C) | Conductivity (µS) |
|---|---|
| 35.0 | 12.3 |
| 33.0 | 10.9 |
| 26.0 | 9.0 |
| 18.2 | 6.7 |
| 4.0 | 9.8 |
| −4.0 | 12.0 |






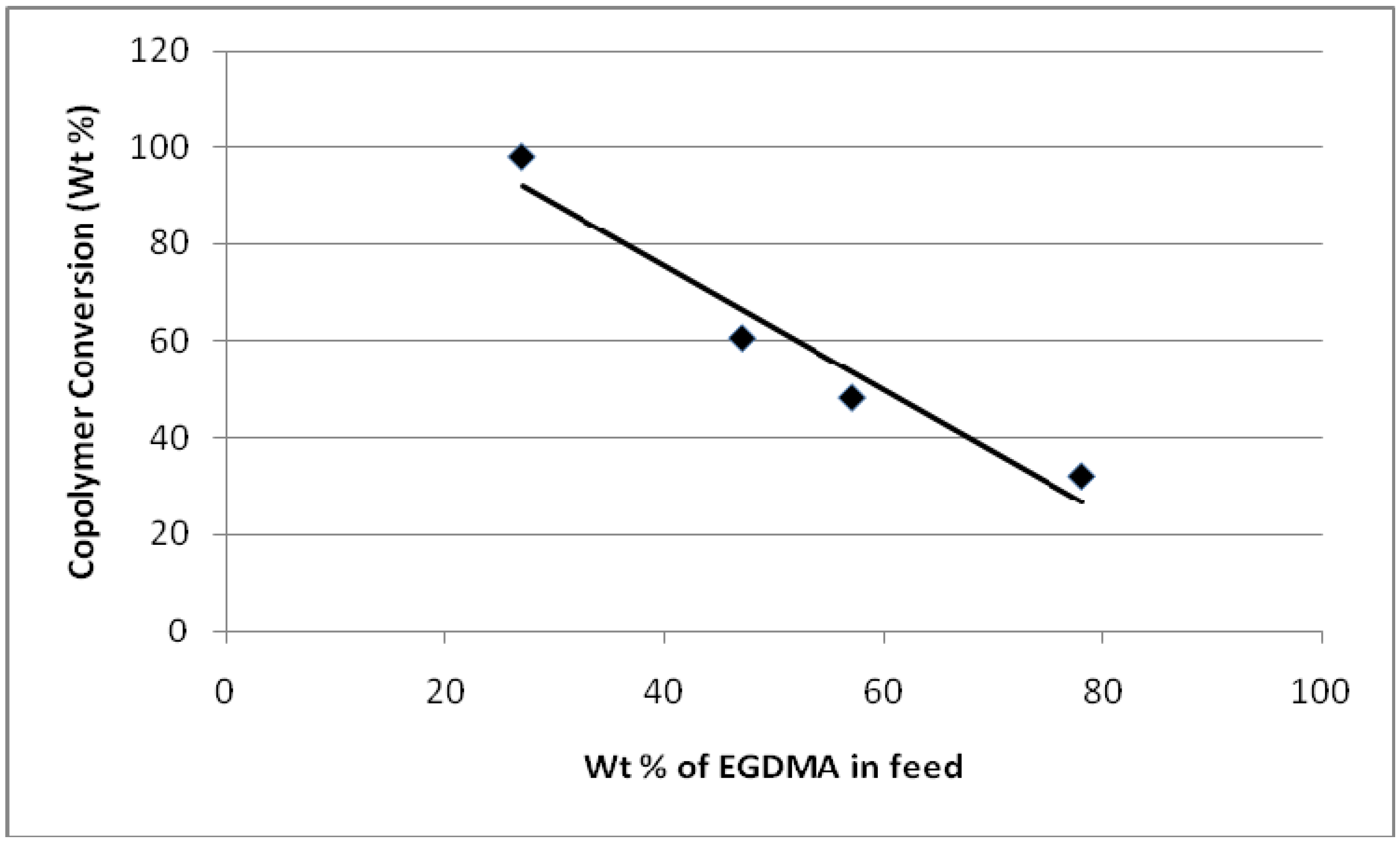
4.3. Copolymer Conversion


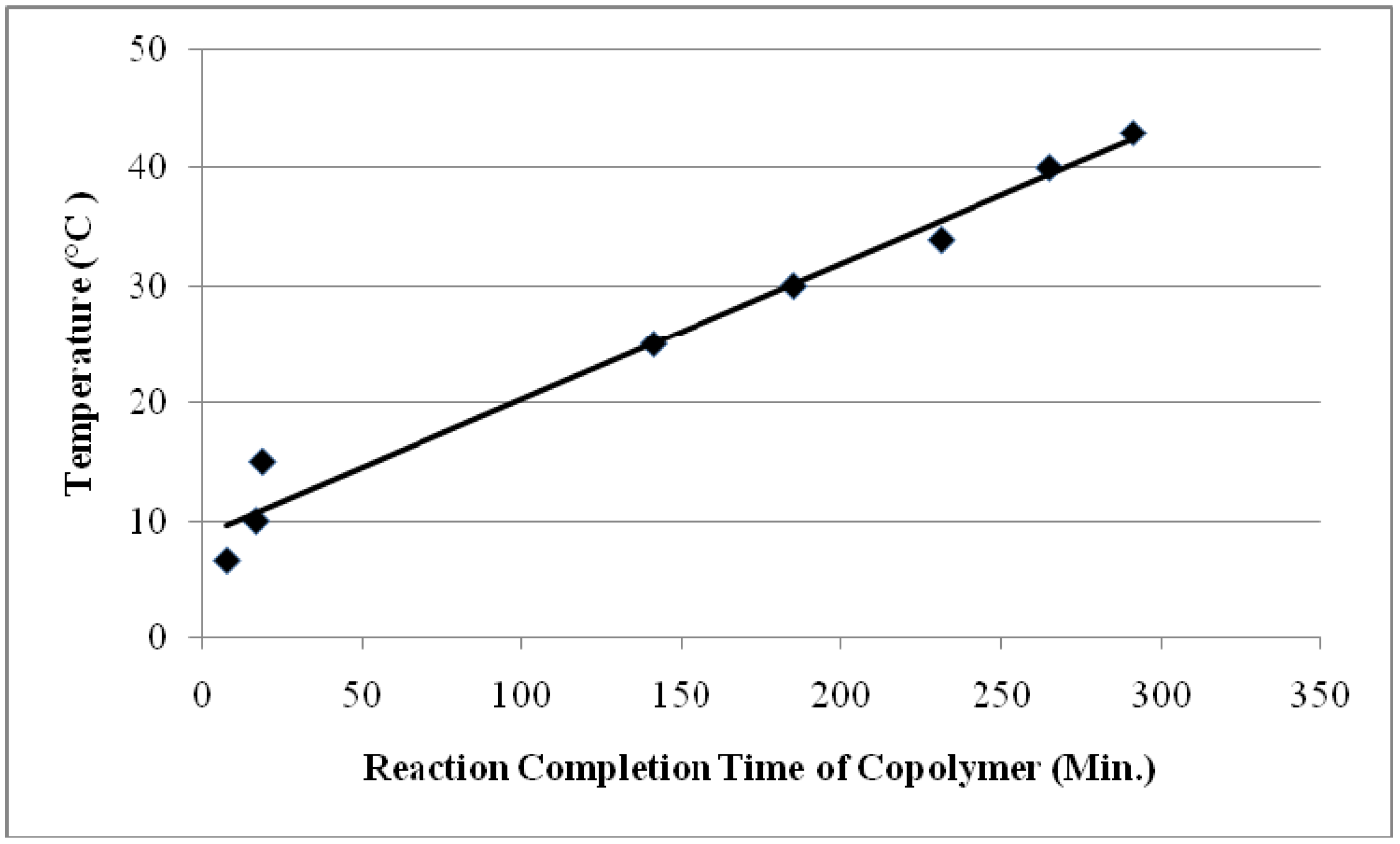
4.4. Activation Energy Determination for the Reaction of EGDMA/MMA Copolymer with Lithium Perchlorate

| Temperature (°C) | Reaction completion time of copolymer with dopant LiClO4 (Min.) |
|---|---|
| 6.5 | 8 |
| 10.0 | 17 |
| 15.0 | 19 |
| 25.0 | 141 |
| 30.0 | 185 |
| 33.9 | 231 |
| 40.0 | 265 |
| 43.0 | 291 |
5. Conclusions
- Using IR spectroscopy of PEGDMA and PMMA homopolymers and their copolymers, the reactivity ratio of EGDMA (r2) and MMA (r1) were calculated as 0.6993 and 1.8635, respectively.
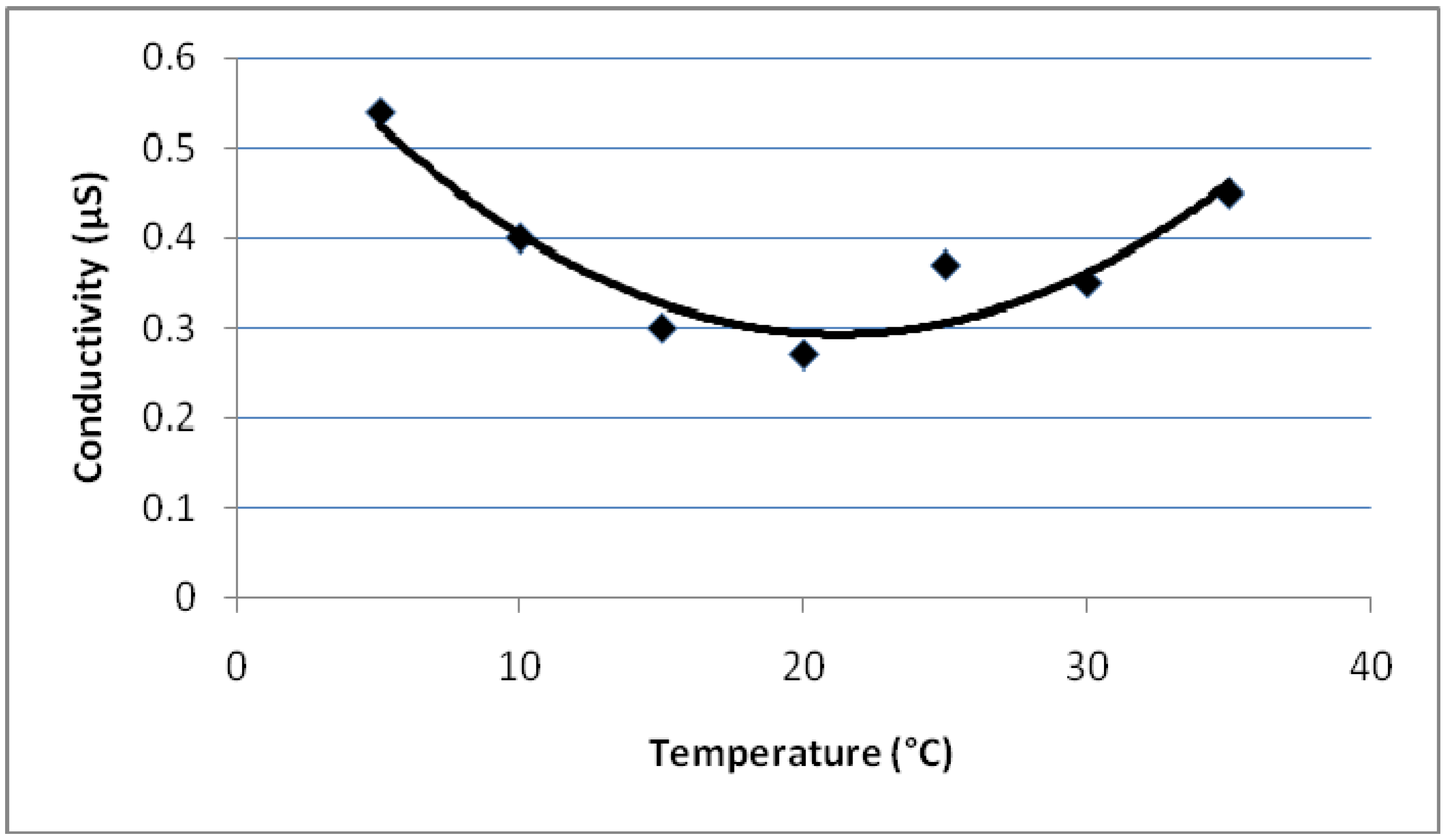
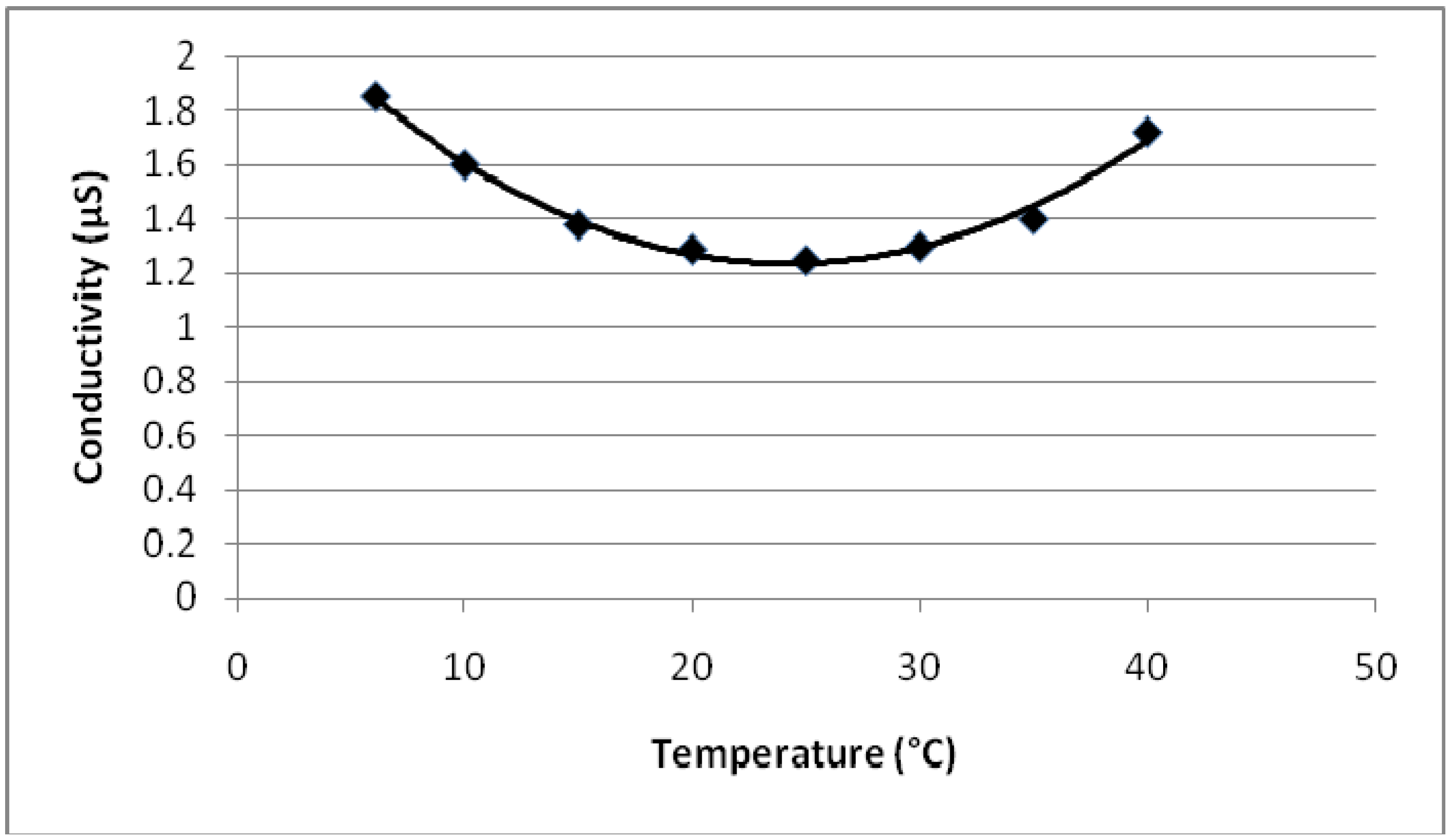
- The conductivities of PEGDMA (mS region) is much greater than the conductivities of PMMA (µS region).
- The conductivities of PEGDMA and PMMA both increase with added dopant, LiClO4.
- As the MMA concentration in the polymer increases, the conductivity value decreases (mS to µS).
- As the MMA percentage in the monomer feed increases, the percent conversion (polymerization) decreases.
- Conductivity vs. temperature graph for PEGDMA shows a minimum at 18 °C. PMMA and PEGDMA/PMMA copolymers also show the same kind of behavior, with minimum values observed at 22–24 °C. This indicates that these polymers first act as a conductor, then after a minimum temperature become semiconductors and can be potentially used to control the current in electrical devices via temperature change (thermal switch).
- The measurement of conductivity change with time provides an excellent and new way to follow the kinetics of the copolymer-dopant reactions. By measuring the reaction completion times at different temperatures of the PEGDMA/MMA copolymer, activation energy of interaction with perchlorate ion was determined to be 31.52 kJ/mol. For PEGDMA alone this value was found to be 54.7 kJ/mol. As the temperature increases, the reaction completion time also increases.
- When EGDMA is copolymerized with MMA to increase its mechanical properties, copolymers still show the same trend of conductivity change with temperature even though the conductivity of the copolymer decreases slightly with increasing percentage of MMA. Therefore, interest in using these copolymers in industry as thermal switches still exists.
References
- Hush, N.S. An Overview of the First Half-Centruy of Molecular Electronics. Ann. N.Y. Acad. Sci. 2003, 1006, 1–20. [Google Scholar] [CrossRef] [PubMed]
- McGiness, J.; Corry, P.; Proctor, P. Amorphous semiconductor switching in melanins. Science 1974, 183, 853–855. [Google Scholar] [CrossRef] [PubMed]
- Bhadra, S.D.; Nikhil, K.S.; Lee, J.H. Progress in preparation, processing and applications of polyaniline. Prog. Polym. Sci. 2009, 34, 783–810. [Google Scholar] [CrossRef]
- Peres, R.C.D.; DePaoli, M.A.; Panero, S.; Scrosati, B. A new electrode for poly(pyrrole)-based rechargeable battery. J. Power Sources 1992, 40, 299–305. [Google Scholar] [CrossRef]
- Diaz, A.F.; Castillo, J.I.; Logan, T.A.; Lee, W.J. Electrochemistry of conducting polypyrrole films. J. Electroanal. Chem. 1981, 129, 115–132. [Google Scholar] [CrossRef]
- Pellagrino, J.; Radebaugh, R.; Mattes, B.R. Gas sorption in polyaniline. 1. Emeradline base. Macromolecules 1996, 29, 4985. [Google Scholar]
- Selampinar, F.; Akbulut, U.; Ozden, M.Y.; Toppare, L. Immobilization of invertase in conducting polymer matrices. Biomaterials 1997, 92, 1163. [Google Scholar] [CrossRef]
- Kizilyar, N.; Akbulut, U.; Toppare, L.; Ozden, M. M.; Yagci, Y. Immoblization of invertase in conducting polypyrrole/polytetrahydrofuran graft polymer matrices. Synthet. Metal. 1999, 104, 45. [Google Scholar] [CrossRef]
- Ramelow, U.S.; Ma, J.; Darbeau, R. Electrical conductivities of polypyrrole reacted with dopant solutions. Mater. Res. Innov. 2001, 5, 40. [Google Scholar] [CrossRef]
- Ramelow, U.S.; Darbeau, R.; Ma, J.; Glenn, G.; Garrison, J. Effects of concentration and speciation of iron and solvent nucleophilicity/basicity on the electrical conductivty of polypyrrole: Introduction of a novel guage of polypyrrole conductivity. Mater. Res. Innov. 2004, 8, 29. [Google Scholar]
- Wang, H.L.; Toppare, L.; Ferandez, J.E. Conducting polymer blends: Polythiophene and polypyrrole blends with polystyrene and poly(bisphenol A carbonate). Macromolecules 1990, 23, 1053–1059. [Google Scholar]
- Yurtsever, M.; Toppare, L. Theoretical investigation of block copolymerization of tetrahydrofuran and pyrrole. Polymer 1990, 40, 5459. [Google Scholar] [CrossRef]
- DePaoli, M.; Panero, S.; Prosperi, P.; Scrosanti, B. Study of electrochromism of polypyrrole/dodecyl sulfate in aqueous soluton. Electrochim. Acta 1990, 35, 1145. [Google Scholar] [CrossRef]
- Alkan, S.; Toppare, L.; Hepuzer, Y.; Yagci, Y. Block copolymers of thiophene-capped poly(methyl methacrylate) with pyrrole. J. Polym. Sci. Part A Polym. Chem. 1999, 37, 4218–4225. [Google Scholar] [CrossRef]
- Alkan, S.; Toppare, L.; Bakir, U.; Yagci, Y. Immobilization of urease in conducting thiophene-capped polymethylmethacrylate/pyrrole matrices. Synthet. Metal. 2001, 123, 95–99. [Google Scholar] [CrossRef]
- Sanchez, M.; Tompkins, J.; Rizk, W. Thermal Curing and Strength of PMMA Bone Cement. In Proceedings of ASME Summer Heat Transfer Conference, Las Vegas, NV, USA, July 2003.
- Ramelow, U.; Braganza, S.; Darbeau, R.; Ramelow, G. Relaxation time, activation energy, and reaction pathway determination of polyethylene glycol/dimethacrylate—dopant (I2) interaction with the nuclear magnetic resonance technique. J. Appl. Polym. Sci. 2006, 100, 5087–5101. [Google Scholar] [CrossRef]
- Ramelow, U.S.; Qui, G. Monomer reactivity ratios in UV-initiated free radical copolymerization reactions. J. Appl. Polym. Sci. 1995, 57, 911–920. [Google Scholar] [CrossRef]
- Stevens, M.P. Polymer Chemistry, An Introduction, 2nd ed.; Oxford University Press: Oxford, UK, 1990; p. 224. [Google Scholar]
- Ebemele, R.O. Polymer Science and Technology; CRC Press: Boca Raton, FL, USA, 2000; pp. 215–216. [Google Scholar]
- Billmeyer, F.W. Textbook of Polymer Science, 2nd ed.; John Wiley and Sons: New York, NY, USA, 1971; p. 330. [Google Scholar]
- Ramelow, U.S.; Braganza, S.N.; Ramelow, G.J. Electrical conductivities of polyethylene glycol dimethacrylate reacted with iodine and lithium perchlorate dopants, and activation energy determination of polymer-dopant interaction. J. Appl. Polym. Sci. 2009, 112, 1916–1926. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ramelow, U.S.; Pingili, S. Synthesis of Ethylene Glycol Dimethacrylate-Methyl Methacrylate Copolymers, Determination of their Reactivity Ratios, and a Study of Dopant and Temperature Effects on their Conductivities. Polymers 2010, 2, 265-285. https://doi.org/10.3390/polym2030265
Ramelow US, Pingili S. Synthesis of Ethylene Glycol Dimethacrylate-Methyl Methacrylate Copolymers, Determination of their Reactivity Ratios, and a Study of Dopant and Temperature Effects on their Conductivities. Polymers. 2010; 2(3):265-285. https://doi.org/10.3390/polym2030265
Chicago/Turabian StyleRamelow, Ulku S., and Sreedhar Pingili. 2010. "Synthesis of Ethylene Glycol Dimethacrylate-Methyl Methacrylate Copolymers, Determination of their Reactivity Ratios, and a Study of Dopant and Temperature Effects on their Conductivities" Polymers 2, no. 3: 265-285. https://doi.org/10.3390/polym2030265
APA StyleRamelow, U. S., & Pingili, S. (2010). Synthesis of Ethylene Glycol Dimethacrylate-Methyl Methacrylate Copolymers, Determination of their Reactivity Ratios, and a Study of Dopant and Temperature Effects on their Conductivities. Polymers, 2(3), 265-285. https://doi.org/10.3390/polym2030265