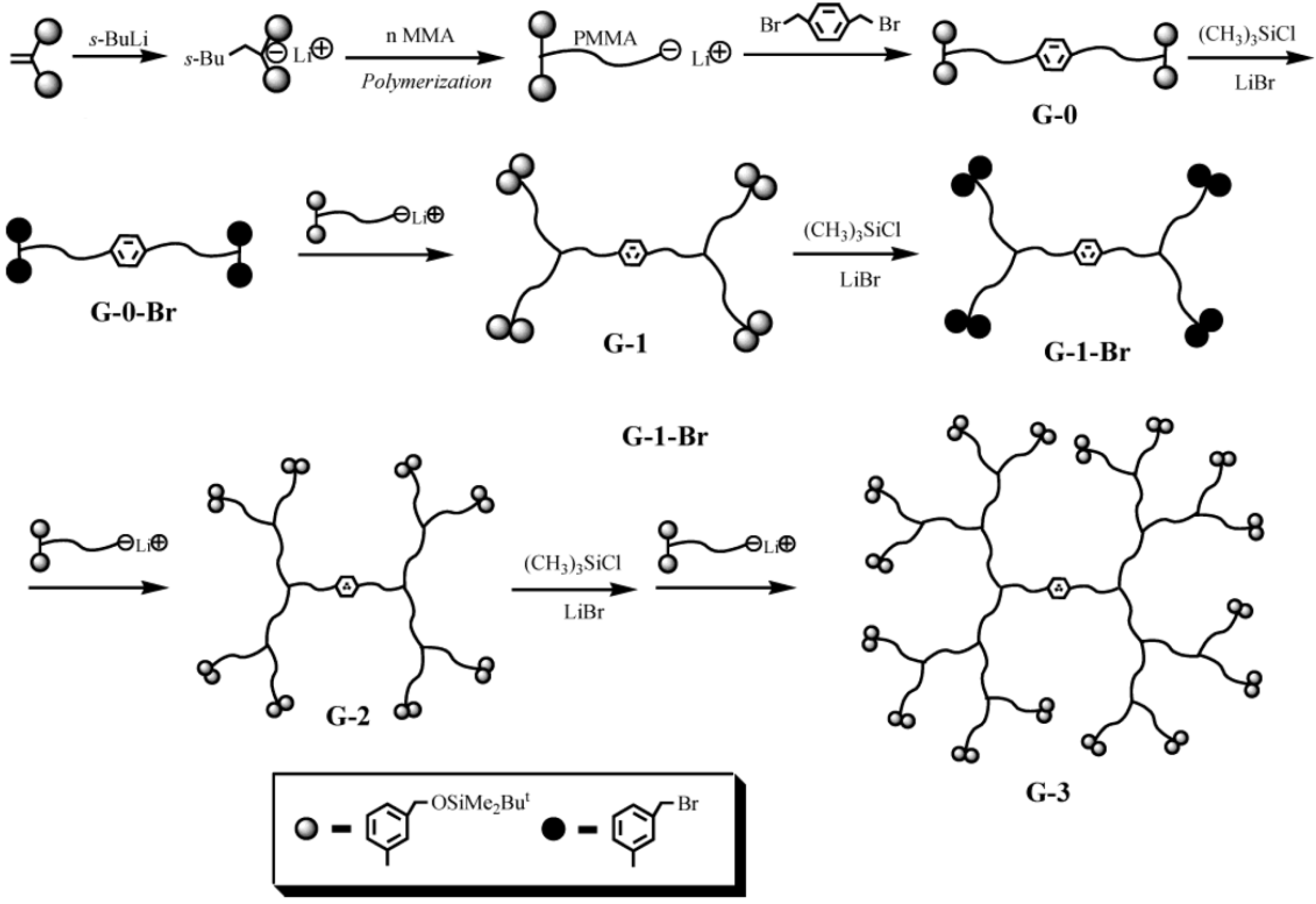
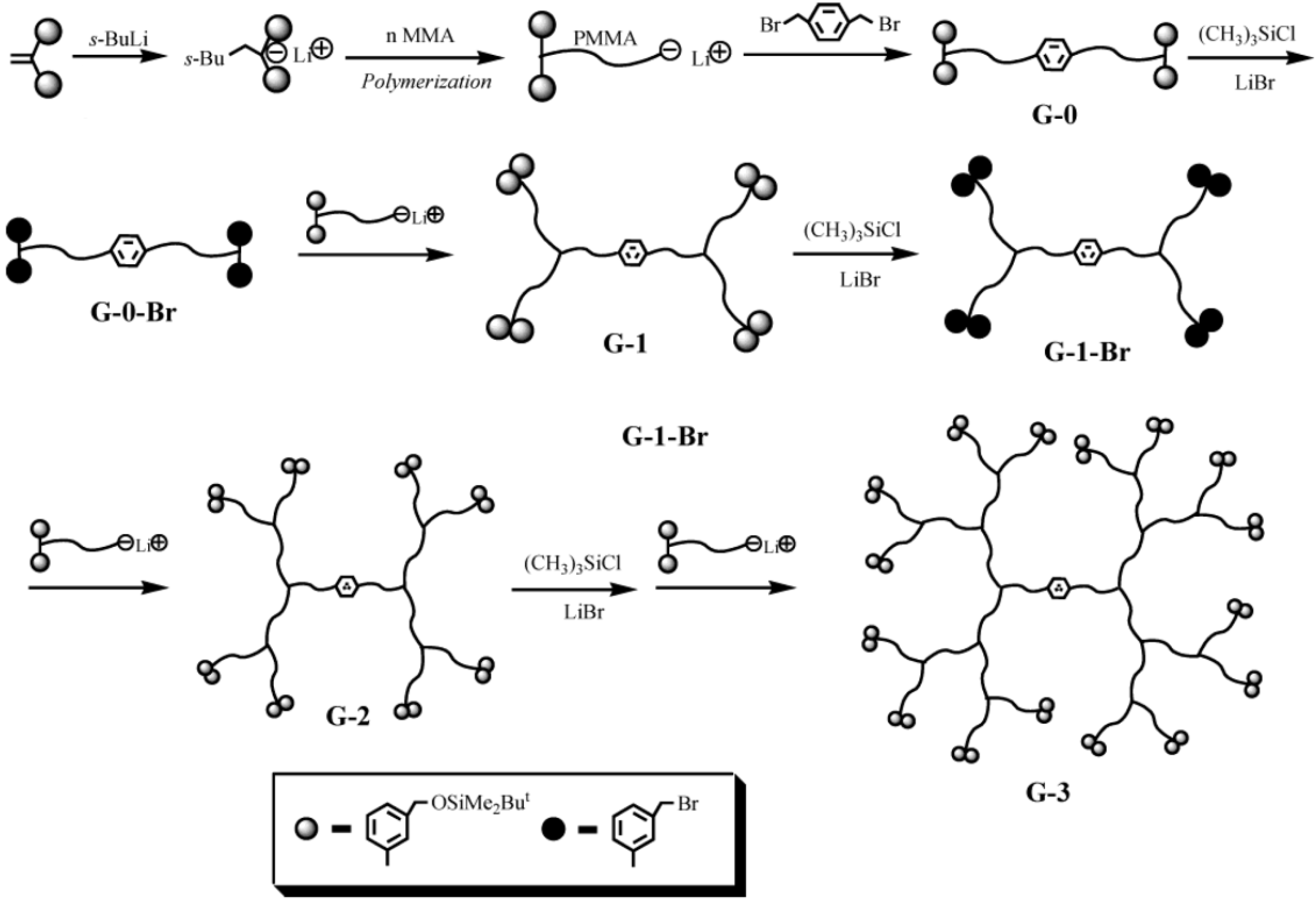
2.1. Arborescent Polystyrene
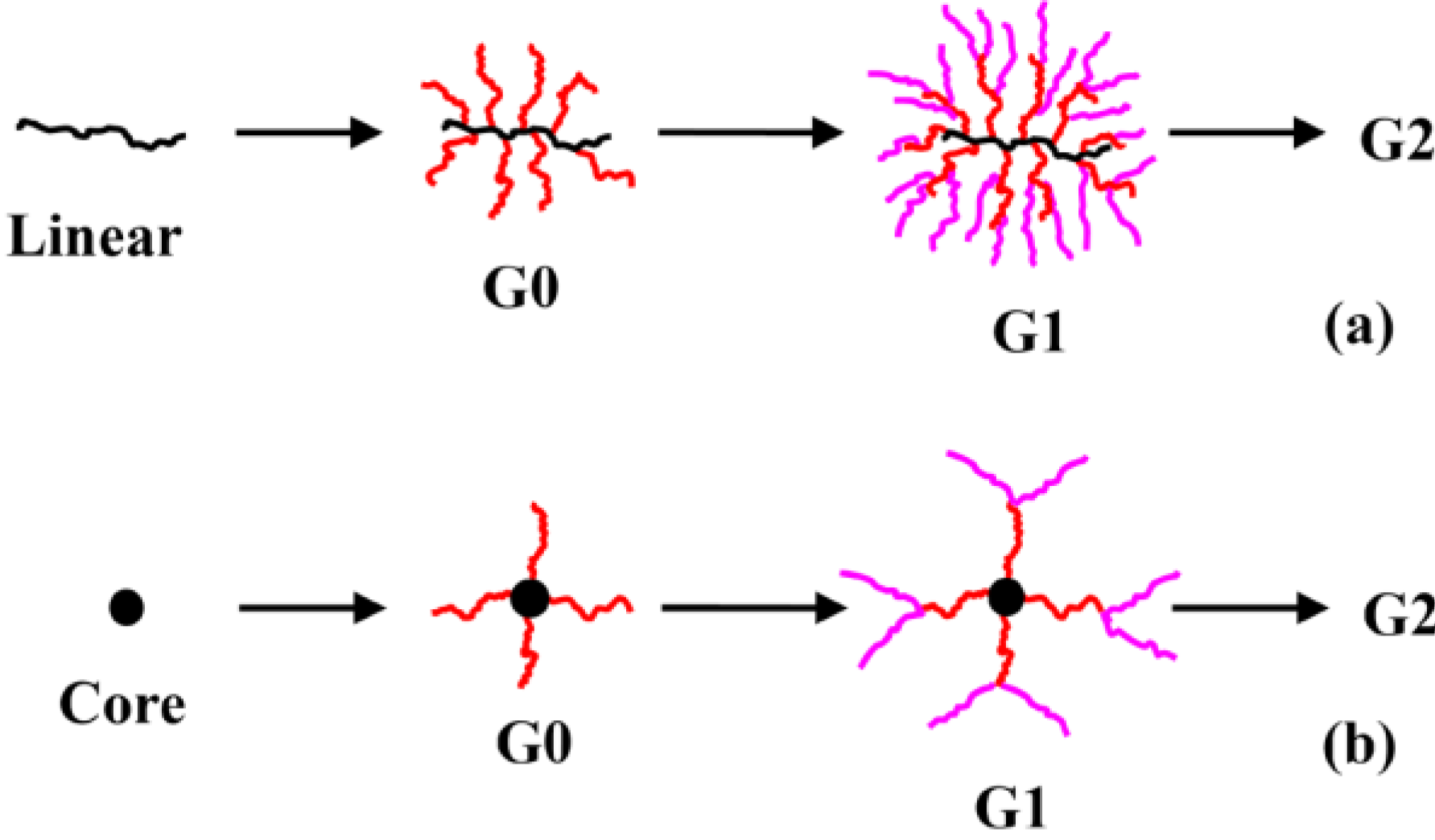
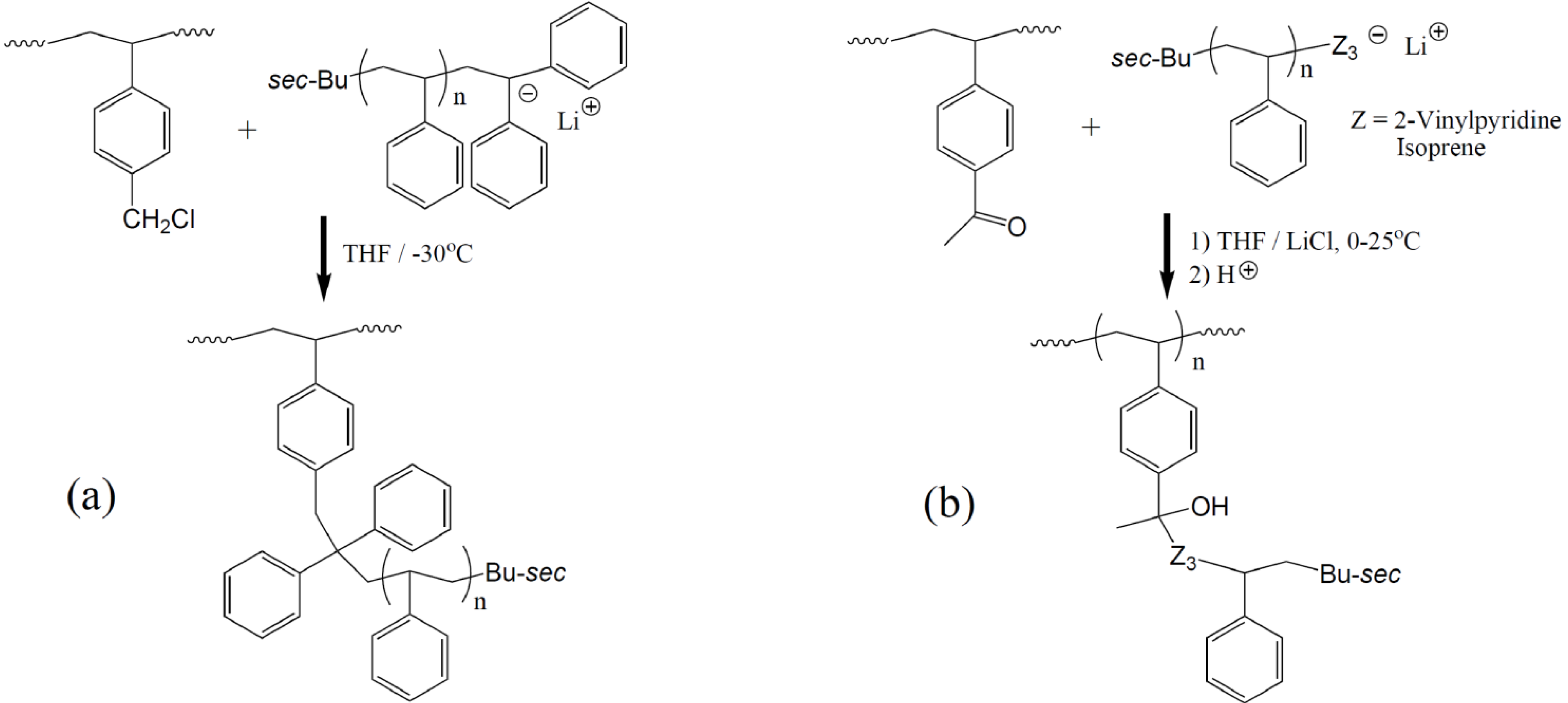
Arborescent polystyrene is of particular importance, because it served as grafting substrate in the synthesis of many phase-separated copolymers. Consequently the synthesis of the homopolymers will be discussed first, but it will be shown that even these macromolecules display characteristics typical of heterogeneous copolymer systems due to differences in segmental density within the core and the outer shell (corona) of the molecules. The synthesis of arborescent polystyrene starts with the nucleophilic attack of “living” polystyryl anions onto a linear polystyrene substrate, randomly functionalized with suitable electrophilic substituents on a fraction (typically 25–30%) of the structural units, to generate the G0 polymer. Both chloromethyl [
8] and acetyl [
17] functionalities have been successfully applied as electrophilic coupling sites in the synthesis of arborescent structures (
Scheme 2). In each case it was important to match the reactivity of the macroanions and the electrophile, in order to minimize side reactions and maximize the yield of the grafting reaction. It was thus necessary to “cap” the polystyryl anions with a single 1,1-diphenylethylene unit to suppress metal-halogen exchange reactions competing with coupling for the chloromethyl sites. The grafting yield (defined as the fraction of side-chains generated in the polymerization reaction becoming attached to the substrate) was maximized when grafting was carried out in tetrahydrofuran (THF) at −30 °C [
8]. Acetyl groups, on the other hand, are susceptible to proton abstraction by the macroanion acting as a strong base. In this case the yield was maximized in THF in the 0–25 °C range, after capping the chains with a few units of relatively unhindered or more reactive monomers such as isoprene or 2-vinylpyridine, and the addition of LiCl to the reaction to suppress chain end ionization [
17]. Irrespective of the grafting path selected, the functionalization and grafting reaction cycle can be repeated to obtain arborescent polystyrene structures of generations G1 and above.
Scheme 2.
Synthesis of arborescent polystyrene by grafting polystyryl anions onto (a) chloromethylated and (b) acetylated polystyrene substrates.
Scheme 2.
Synthesis of arborescent polystyrene by grafting polystyryl anions onto (a) chloromethylated and (b) acetylated polystyrene substrates.
An important characteristic of arborescent polymers is that their apparent molecular weight, determined by size exclusion chromatography (SEC) analysis using a linear polystyrene standards calibration curve, is strongly underestimated (
Table 1) [
17]. Comparison of these values with the absolute weight-average molecular weights (
Mw) determined from static light scattering measurements reveals discrepancies between both sets of results, the magnitude of the discrepancies increasing for the higher generations. This is a direct consequence of the very compact structure of arborescent polymers, which display an increase in average segmental density over successive generations, in contrast to randomly coiled linear polymers for which the hydrodynamic density decreases exponentially as the molecular weight increases. The absolute molecular weight and branching functionality of the molecules (determined from light scattering measurements) otherwise follow the expected trend, with roughly geometric increases over successive generations, while a low polydispersity index is maintained.
Table 1.
Characterization data for a series of arborescent styrene homopolymers synthesized from acetylated polystyrene substrates.a
Table 1.
Characterization data for a series of arborescent styrene homopolymers synthesized from acetylated polystyrene substrates.a
| Sample | Generation | Mwb | Mw /Mnb | fwc | Mwd |
|---|
| PS-PS5 | 0 | 5.3 × 104 | 1.08 | 11 | 3.3 × 104 |
| G0PS-PS5 | 1 | 4.3 × 105 | 1.08 | 84 | 1.3 × 105 |
| G1PS-PS5 | 2 | 3.9 × 106 | 1.09 | 690 | 4.5 × 105 |
| G2PS-PS5 | 3 | 2.5 × 107 | | 3800 | |
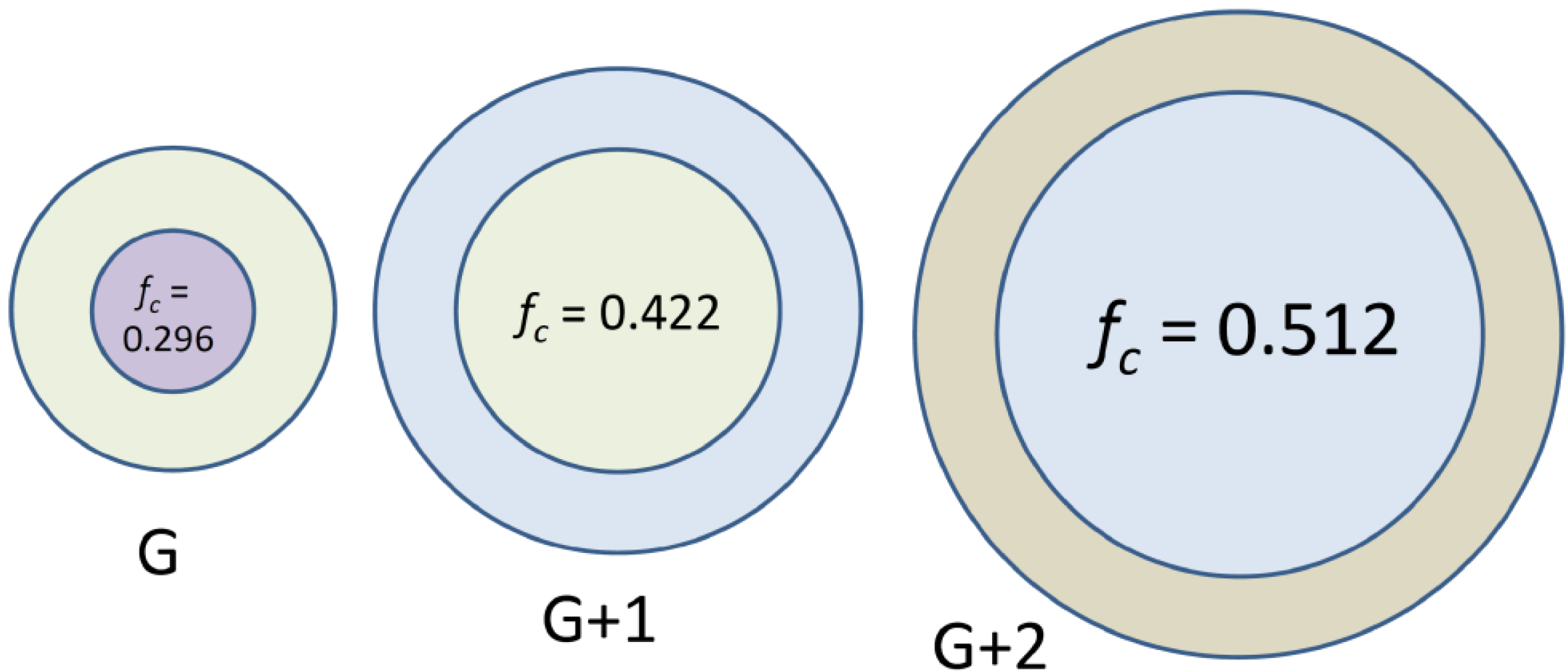
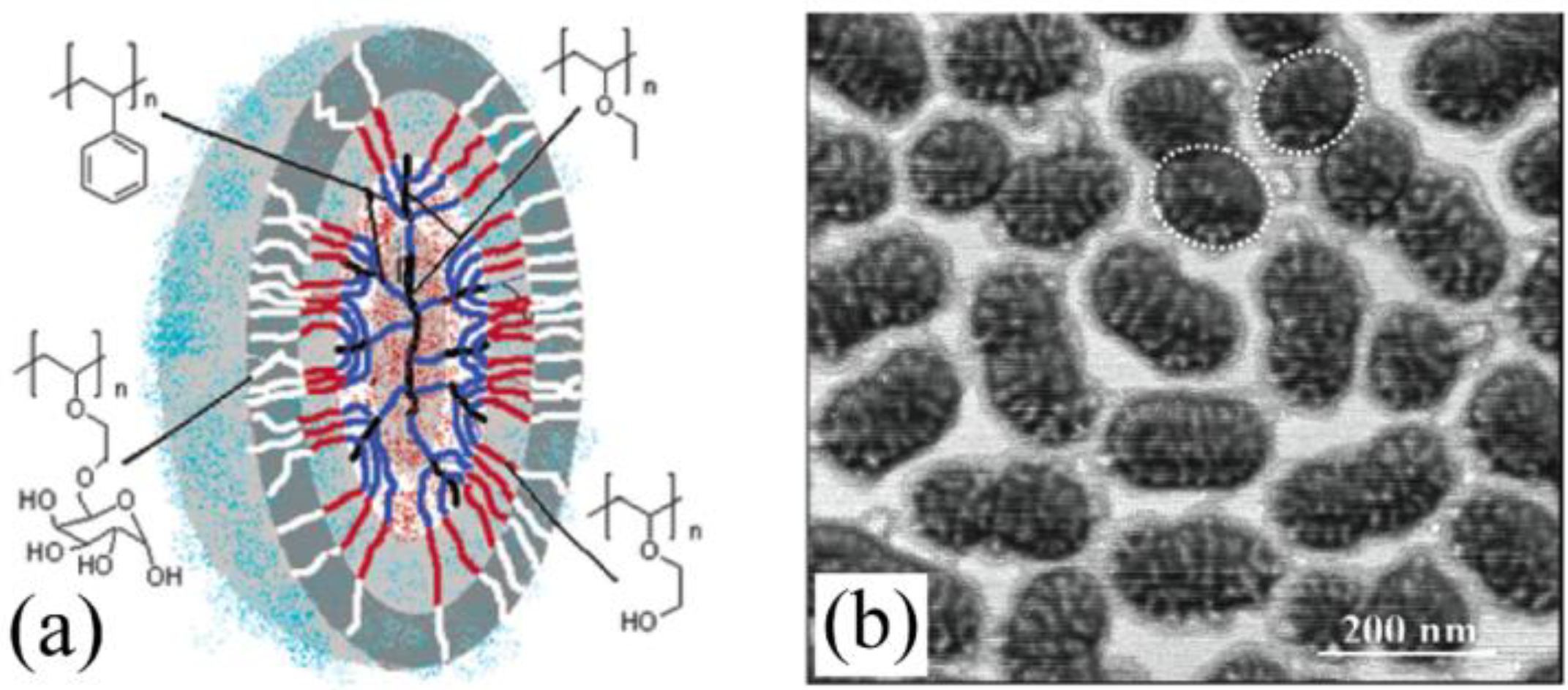
Arborescent macromolecules, including even the homopolymers, may be regarded as having a dual phase morphology. This is because of the “diffuse layer” growth mechanism of the molecules over successive generations, represented in a cartoon fashion in
Scheme 1(a): A high segmental density (low chain mobility) region is expected in the core portion of the molecules, due to the coupling reactions leading to the formation of branching points, while the chains added in the last grafting cycle are only tethered to the substrate at one end. These remain relatively flexible and form a mobile shell (corona) of linear chains on the outside of the molecule. The corona should be rather diffuse not only because of the higher mobility of the chains, but also due to fluctuations in the position of the coupling sites along the side-chains of the substrate as shown in
Scheme 1(a). To a first approximation, the growth of arborescent molecules over successive generations may thus be represented by the addition of successive layers of chains onto a rigid core substrate (
Figure 1).
The heterogeneous character of arborescent polystyrene molecules was first quantified by labeling a series of polymers with short branches (
Mw ≈ 5,000) with pyrene and observing their fluorescence quenching behavior when exposed to small molecule (nitrobenzene) and macromolecular (nitrated polystyrene) quenchers in solution [
18]. The diffusion coefficients measured for the small molecule quencher within the arborescent polystyrene molecules decreased as a function of increasing generation number. Quenching experiments with linear nitrated polystyrene led to downward curvature in Stern-Volmer plots characteristic for protective quenching,
i.e., reduced accessibility of a portion of the chromophores to the quencher groups. The application of a fractional quenching model allowed the determination of the quenching rate constant and the fraction of accessible chromophores for each sample. These parameters decreased for the upper generation polymers, in agreement with the diffuse layer growth model of
Figure 1: When adding successive layers of constant thickness (corresponding to the chains added in the last grafting reaction) on the surface of a sphere, the volume fraction of the core (fraction of inaccessible material,
fc) should increase over successive generations.
Figure 1.
Schematic representation of the diffuse layer growth model for arborescent polymers. The volume fraction of inaccessible (core) material in the molecules, for a core of initial radius R = 2 onto which layers of thickness d = 1 are added, is represented by fc.
Figure 1.
Schematic representation of the diffuse layer growth model for arborescent polymers. The volume fraction of inaccessible (core) material in the molecules, for a core of initial radius R = 2 onto which layers of thickness d = 1 are added, is represented by fc.
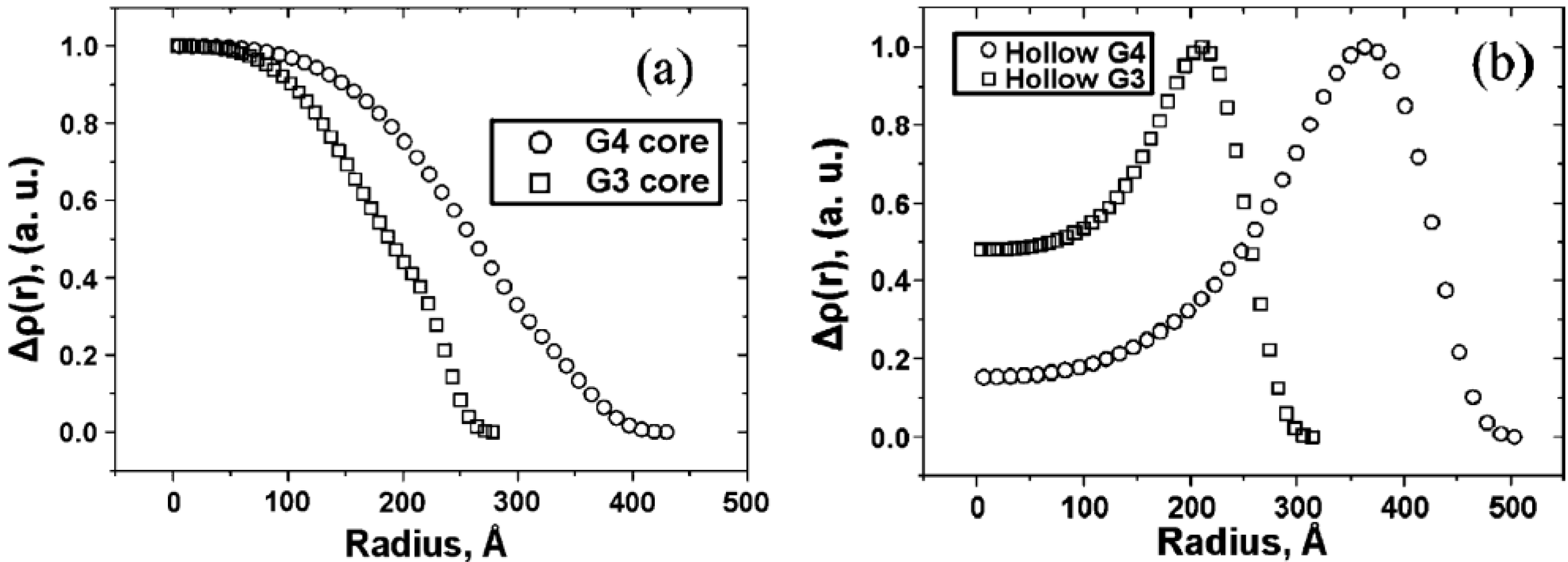
The morphology of arborescent polystyrene molecules was also the topic of a detailed investigation using small-angle neutron scattering (SANS) experiments with contrast matching for polymers derived from arborescent polystyrene substrates grafted with deuterated polystyrene chains forming the shell [
19]. Most interestingly, radial SANS contrast density profiles (proportional to the local concentration of scattering segments) could be generated for the core and the shell portions of the molecule under matched contrast conditions, and for polymers of two different generations (G3
vs. G4). It is clear from the profiles obtained (
Figure 2) that the core and the shell components are segregated, even for systems expected to display minimal phase separation such as copolymers with a hydrio-polystyrene core and a deutero-polystyrene shell. The core-shell interface is nonetheless diffuse due to significant mixing between the core and the shell components, in agreement with the diffuse layer growth model proposed above.
Figure 2.
Radial SANS contrast profiles generated from measurements on arborescent polystyrenes with a deuterated polystyrene shell under (a) shell and (b) core contrast matching conditions. Adapted from Reference 19 by permission from the American Chemical Society.
Figure 2.
Radial SANS contrast profiles generated from measurements on arborescent polystyrenes with a deuterated polystyrene shell under (a) shell and (b) core contrast matching conditions. Adapted from Reference 19 by permission from the American Chemical Society.
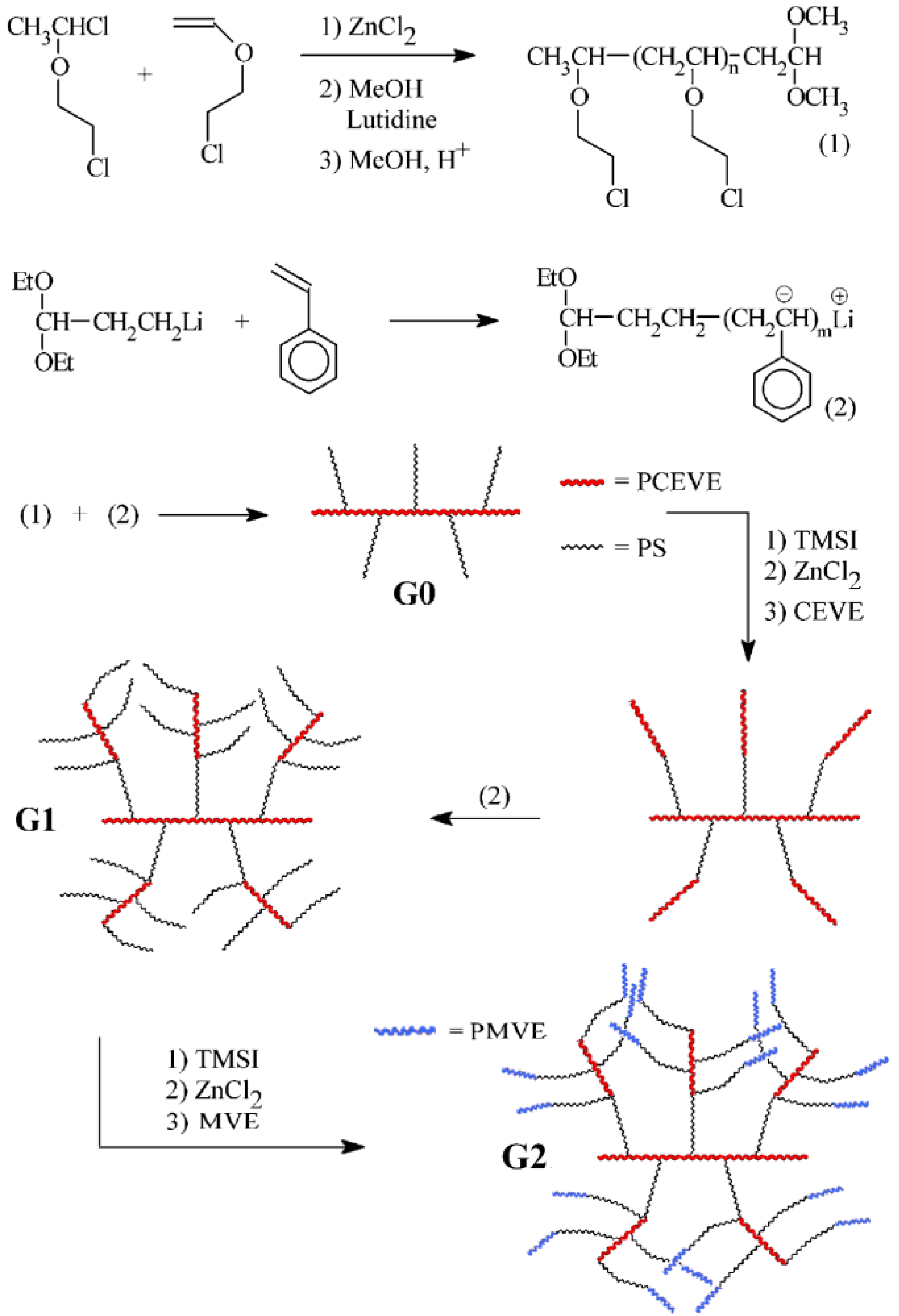
2.2. Poly(ethylene oxide) Copolymers
The first example of an arborescent copolymer structure with a clearly heterogeneous morphology, published in 1996, was for polymers incorporating an arborescent polystyrene core grafted with poly(ethylene oxide) (PEO) segments at the chain termini [
20]. The synthesis of micelle-like core-shell macromolecules with different core sizes (generations) and varying PEO contents or shell thicknesses was demonstrated.
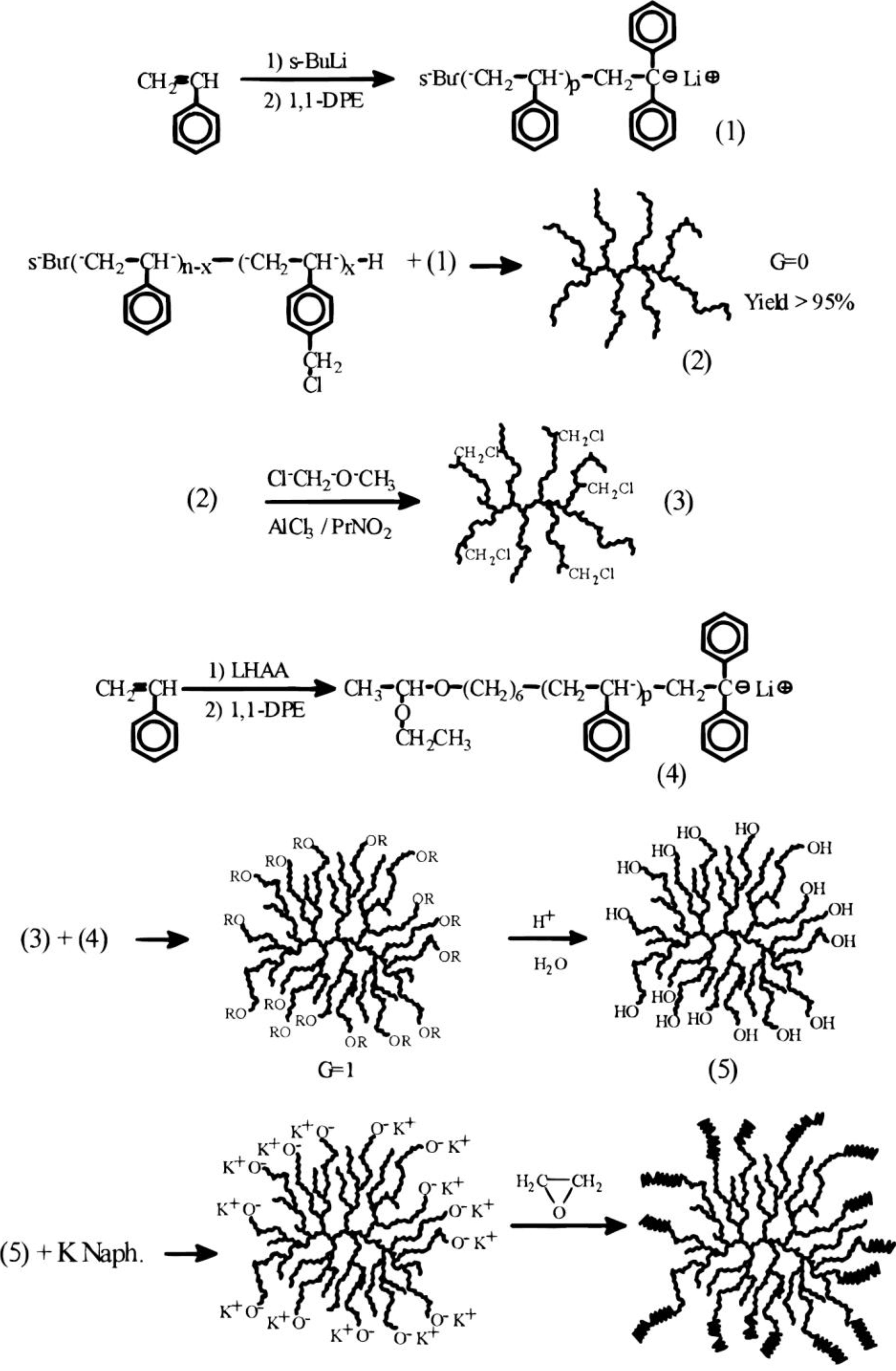
The preparation of the arborescent polystyrene core relied on the grafting technique with chloromethyl coupling sites [
8], but the addition of a PEO shell required a modification of the procedure in the last step. The synthesis of a G1 copolymer is illustrated in
Scheme 3 as an example. A comb-branched (G0) polystyrene substrate was prepared in the usual fashion, by initiating the polymerization of styrene with
sec-butyllithium and capping with 1,1-diphenylethylene, followed by titration of the “living” anions with a solution of chloromethylated linear polystyrene. A twice-grafted (G1) substrate with protected hydroxyl end-groups was obtained by using (6-lithioexyl)acetaldehyde acetal (LHAA in
Scheme 3) to initiate the polymerization of styrene, followed by capping and coupling with the chloromethylated G0 polymer. The acetal functionalities were then hydrolyzed and the hydroxyl end-groups were deprotonated with potassium naphthalide before adding ethylene oxide to the substrate. To maintain a narrow molecular weight distribution in the reaction, it was necessary to eliminate residual chloromethyl sites on the substrate via metal-halogen exchange prior to shell growth. The core-shell copolymers derived from a G1 core with
Mw = 7 × 10
5 had PEO contents of 19% and 66% by weight, and apparent polydispersities
Mw/
Mn = 1.07–1.21. Another sample containing 36% PEO by weight was obtained from a G4 core having
Mw ~ 10
8.
Scheme 3.
Synthesis of arborescent polystyrene-graft-poly(ethylene oxide) amphiphilic copolymers. Reproduced from Reference 20 by permission from the American Chemical Society.
Scheme 3.
Synthesis of arborescent polystyrene-graft-poly(ethylene oxide) amphiphilic copolymers. Reproduced from Reference 20 by permission from the American Chemical Society.
Comparison of the hydrodynamic radii determined from dynamic light scattering measurements for the core homopolymers and the core-shell copolymers demonstrated that the hydrophilic poly(ethylene oxide) chains on the surface of the molecules essentially adopted a randomly coiled conformation, with minimal stretching of the chains. The solubility characteristics of the macromolecules were consistent with a core-shell morphology: The copolymers were freely soluble in methanol, in contrast to the bare polystyrene cores.
The self-assembly of the amphiphilic arborescent polystyrene-
graft-poly(ethylene oxide) copolymers at the air-water interface was investigated as a function of the composition and the structure of the molecules [
21,
22]. The types of superstructures formed by the amphiphiles strongly depended on characteristics including their composition, branching functionality, and structural rigidity. Thus copolymers with low PEO contents simply dewetted from the water surface and formed large island-like clusters, while molecules with a high PEO content had little tendency to self-assemble in the absence of compression [
Figures 3(a,c)] [
21]. However copolymers of intermediate compositions (
ca. 20–35% PEO content by weight) self-assembled into ribbon-like superstructures [
Figure 3(b)] mainly via end-to-end aggregation, the extent of aggregation (ribbon length) being determined not only by the composition but also by the flexibility of the molecules (branching functionality and molecular weight of the polystyrene side-chains within the core). This end-to-end aggregation phenomenon was rationalized through sideways rearrangement of the linear PEO chains in the shell leading to an increase in the magnitude of van der Waals interactions between the hydrophobic cores along the long axis of the ribbons, while thickening of the stabilizing PEO layer on the sides of the ribbons hindered further aggregation of the molecules [
Figure 3(d)]. It was also shown that the length of the ribbon-like superstructures increased under the influence of compression in Langmuir force balance experiments, or through temperature variations influencing the degree of hydration of the stabilizing PEO chains [
22].
Figure 3.
Self-assembly of arborescent polystyrene-graft-poly(ethylene oxide) copolymer molecules at the air-water interface without compression: (a) low branching functionality, low PEO content G1-30PS-LB-15, (b) low branching functionality, intermediate PEO content G1-30PS-LB-31, and (c) high branching functionality, high PEO content G1-30PS-HB-43. Each picture has a width of 1.5 μm. (d) End-to-end aggregation mechanism proposed to explain the formation of ribbons in (b). Adapted from Reference 21 by permission from Wiley-VCH.
Figure 3.
Self-assembly of arborescent polystyrene-graft-poly(ethylene oxide) copolymer molecules at the air-water interface without compression: (a) low branching functionality, low PEO content G1-30PS-LB-15, (b) low branching functionality, intermediate PEO content G1-30PS-LB-31, and (c) high branching functionality, high PEO content G1-30PS-HB-43. Each picture has a width of 1.5 μm. (d) End-to-end aggregation mechanism proposed to explain the formation of ribbons in (b). Adapted from Reference 21 by permission from Wiley-VCH.
2.3. Poly(2-vinylpyridine) Copolymers
The synthesis of arborescent polystyrene-
graft-poly(2-vinylpyridine) copolymers was initially achieved by coupling poly(2-vinylpyridinyl)lithium with chloromethylated polystyrene substrates of generations up to G2 in tetrahydrofuran with N,N,N′,N′-tetramethylethylenediamine (TMEDA) [
23]. This reaction is analogous to
Scheme 2(a), where the 1,1-diphenylethylene-capped polystyryllithium species are replaced with the “living” poly(2-vinylpyridinyl)lithium chains without capping. Copolymers with branching functionalities ranging from 14–3,880 and molecular weights of 8.2 × 10
4 to 6.7 × 10
7 were thus obtained while maintaining low apparent polydispersity indices (
Mw/
Mn ≈ 1.06–1.15). The polystyrene content of these copolymers was rather low and varied from
ca. 3 to 19% by weight, depending on the arborescent polystyrene substrate generation and the molecular weight of the side-chains used in the reaction. Comparable results were subsequently obtained when coupling poly(2-vinylpyridinyl)lithium with acetylated polystyrene substrates in the presence of LiCl, similarly to
Scheme 2(b) (in this case the side-chains correspond to a long segment of “Z” units without styrene) [
24].
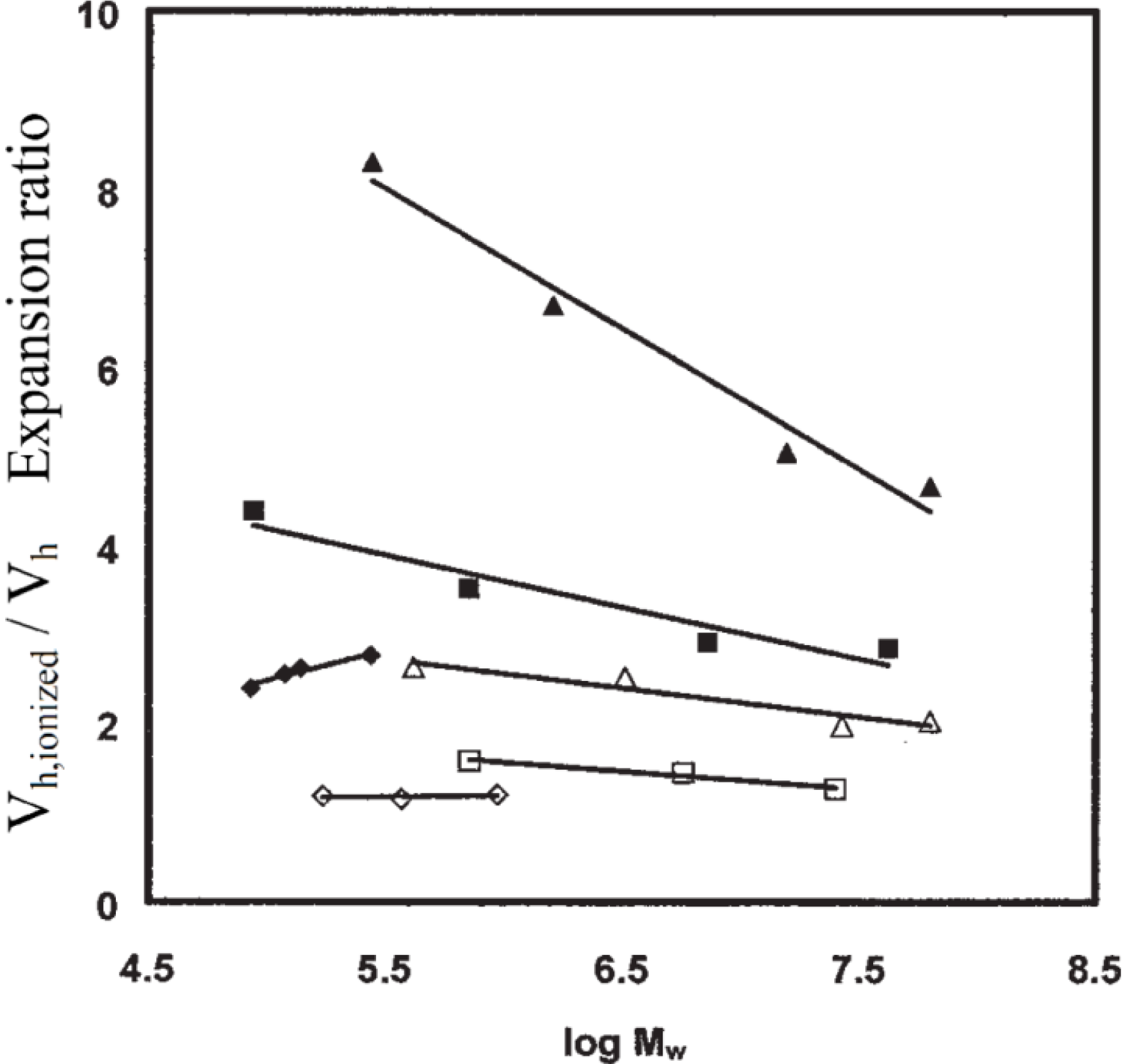
Dynamic light scattering measurements demonstrated that arborescent poly(2-vinylpyridine) (P2VP) copolymers, when protonated with excess HCl (at high ionic strengths), expand much more in solution than their linear homologues (
Figure 4). This effect was attributed to the higher charge density (charge/volume) attained in these branched polyelectrolytes [
25], which can be viewed as P2VP homopolymers to a first approximation due to their low polystyrene contents. Nevertheless the expansion ratio of the copolymers, expressed as the ratio of hydrodynamic volumes in the ionized and the neutral states, decreased as the generation number (structural rigidity) of the copolymer increased. It was also larger for copolymers containing longer, more flexible side-chains.
Figure 4.
Hydrodynamic volume expansion for arborescent polystyrene-graft-poly(2-vinylpyridine), polystyrene-graft-poly(methacrylic acid), and their linear homologues upon ionization. Linear P2VP (◊), arborescent P2VP copolymers with short (Mw ~ 5,000; □) and long (Mw ~ 30,000; ∆) side-chains dissolved in MeOH/H2O 95/5 with 0.10 N HCl. Linear PMAA (♦), arborescent PMAA copolymers with short (Mw ~ 5,000; ■) and long (Mw ~ 30,000; ▲) side-chains dissolved in MeOH/H2O 95/5 with 0.05 N NaCl and neutralized with NaOH. Adapted from Reference 25 by permission from Wiley.
Figure 4.
Hydrodynamic volume expansion for arborescent polystyrene-graft-poly(2-vinylpyridine), polystyrene-graft-poly(methacrylic acid), and their linear homologues upon ionization. Linear P2VP (◊), arborescent P2VP copolymers with short (Mw ~ 5,000; □) and long (Mw ~ 30,000; ∆) side-chains dissolved in MeOH/H2O 95/5 with 0.10 N HCl. Linear PMAA (♦), arborescent PMAA copolymers with short (Mw ~ 5,000; ■) and long (Mw ~ 30,000; ▲) side-chains dissolved in MeOH/H2O 95/5 with 0.05 N NaCl and neutralized with NaOH. Adapted from Reference 25 by permission from Wiley.
An investigation on the dilute-solution structure of aggregated, partially ionized arborescent P2VP copolymer molecules at low ionic strengths was completed by Yun
et al. using SANS measurements [
26]. Solutions of charged G1 arborescent polystyrene-
graft-poly(2-vinylpyridine) copolymers in methanol-d4 and in D
2O were characterized in the dilute concentration regime (mass fraction f = 0.005–0.05). Upon addition of less than one equivalent of HCl per 2-vinylpyridine unit (degree of ionization 0 < α < 0.4), a peak appeared in plots of the scattering intensity
I(
q)
vs. q (where
q = sin(θ)4π/λ is the scattering vector, θ is the observation angle, and λ is the wavelength) due to aggregation of the molecules. The intermolecular distance within the aggregates, calculated from the peak position, corresponded to the expected value for the formation of superstructures having a uniform liquid-like distribution of molecules. The lower dielectric constant of methanol-d4 resulted in long-range Coulombic interactions persisting to lower polymer concentrations than in D
2O. Dynamic light scattering measurements displayed two diffusive relaxation processes under these conditions, the slow diffusion mode reflecting aggregate formation within the solutions. The SANS scattering peak and the slow diffusion mode both disappeared upon addition of NaCl or excess HCl to the solutions, due to screening of the electrostatic interactions promoting aggregate dissociation. Furthermore, the G1 copolymer grafted with long P2VP side-chains (
Mw = 30,000) formed a gel upon addition of HCl for volume fractions
ϕ > 0.01. This result is consistent with enhanced molecular expansion promoting gelation for these branched polyelectrolytes, the aggregated network breaking up upon addition of NaCl or excess HCl due to screening of the electrostatic interactions [
26].
The ability to control the solution properties of arborescent polyelectrolytes by varying parameters such as the length and the number of P2VP segments in the molecules is interesting for the design of pH-sensitive reversible gels with tunable properties such as the sol-gel transition point and the gel modulus [
23].
The arborescent polystyrene-
graft-poly(2-vinylpyridine) copolymers were also investigated for their potential application as unimolecular micelles, to take advantage of their heterogeneous morphology combining a hydrophobic core and a corona of polyelectrolyte chains. The copolymers derived from a linear polystyrene substrate aggregated when dissolved in aqueous HCl solutions, but those incorporating G0–G2 substrates yielded unimolecular (non-aggregated) micellar solutions with interesting solubilization properties for various polycyclic aromatic hydrophobes [
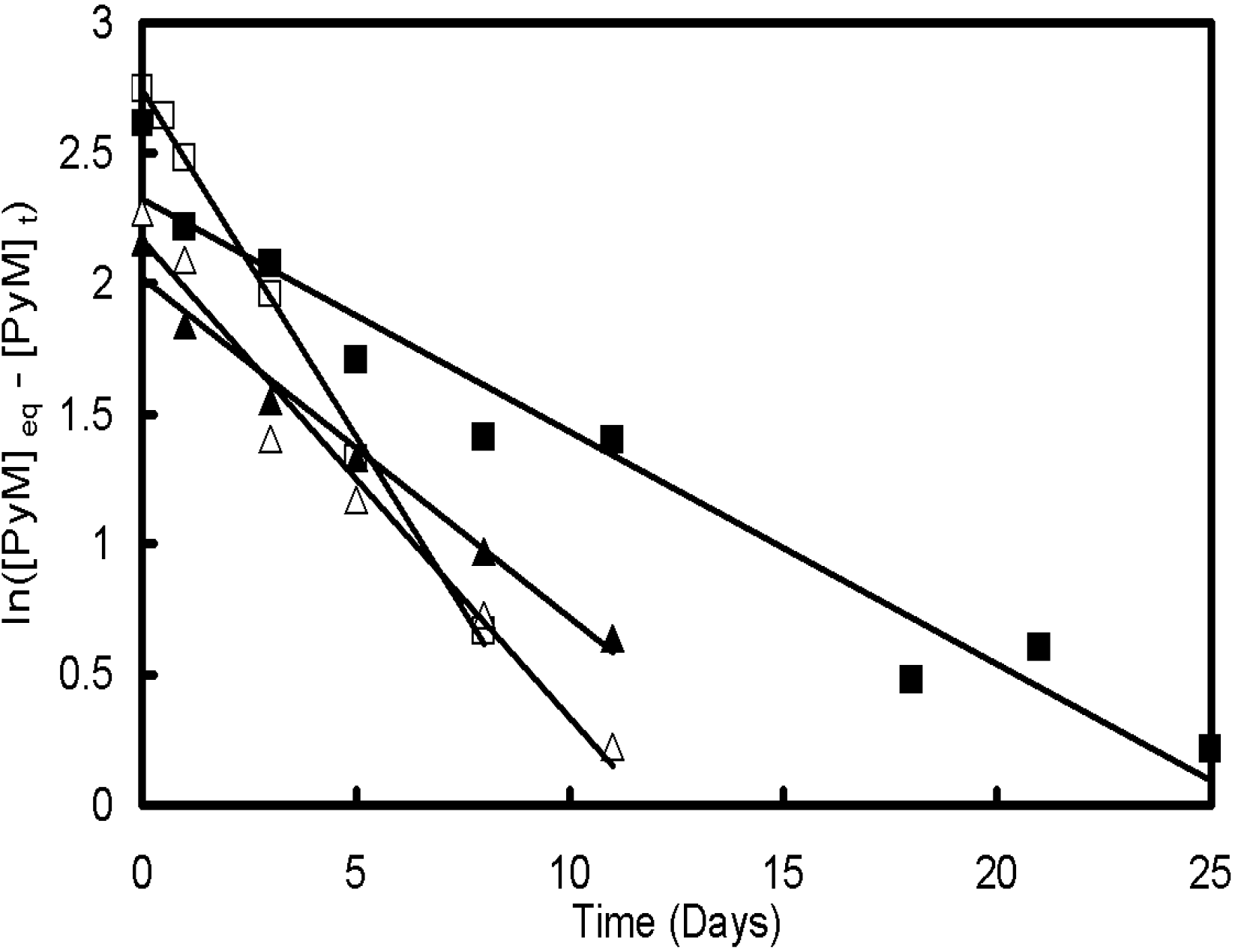
27]. It was shown that the solubilization kinetics and capacity of the micelles varied with their composition (polystyrene content), but also with their structure (generation number, molecular weight of the P2VP side-chains). The kinetics for pyrene solubilization were found to obey a first order kinetic model where the time-dependent concentration of pyrene within the micelles, [
PyM]
t, could be expressed in terms of the equilibrium concentration [
PyM]
eq as [
PyM]
t = [
PyM]
eq(1 −
e−kt) (
Figure 5), where the solubilization rate constant k varied
ca. 3-fold among the different samples investigated. While the copolymers used in the investigation were not specifically designed to serve as drug delivery vehicles (being non-biocompatible and having low polystyrene weight fractions), the study still demonstrates that the solubilization or release characteristics of arborescent unimolecular micelles can be tailored through variations in structure and composition. The solubilization capacity of the micelles also varied with the type of hydrophobic probe solubilized, significantly higher capacities being observed for probes with non-specific solubilization within the polystyrene core and the P2VP shell of the molecules rather than exclusively within the core [
27]. Similar observations were reported in a release study using lidocaine and indomethacin as model drugs, a higher loading capacity being observed for indomethacin due to strong interactions between its carboxylic acid group and the P2VP component of the copolymers in the side-chains [
28].
Figure 5.
Analysis of the micellar solubilization kinetics of pyrene by arborescent polystyrene-graft-poly(2-vinylpyridine) copolymers. The following rate constants were obtained: Δ G0PS-P2VP5-12, k = (0.18 ± 0.01) d−1; ▲ G1PS-P2VP5-13, k = (0.13 ± 0.01) d−1; □ G1PS-P2VP5-20, k = (0.266 ± 0.006) d−1; ■ G2PS-P2VP5-18, k = (0.089 ± 0.007) d−1. Reprinted from Reference 27 by permission from Elsevier.
Figure 5.
Analysis of the micellar solubilization kinetics of pyrene by arborescent polystyrene-graft-poly(2-vinylpyridine) copolymers. The following rate constants were obtained: Δ G0PS-P2VP5-12, k = (0.18 ± 0.01) d−1; ▲ G1PS-P2VP5-13, k = (0.13 ± 0.01) d−1; □ G1PS-P2VP5-20, k = (0.266 ± 0.006) d−1; ■ G2PS-P2VP5-18, k = (0.089 ± 0.007) d−1. Reprinted from Reference 27 by permission from Elsevier.
2.5. Polyisoprene Copolymers
Arborescent copolymers containing polyisoprene (PIP) segments were obtained by coupling polyisoprenyllithium “living” chains with either chloromethylated [
29] or acetylated [
30] polystyrene substrates according to the procedures outlined in
Scheme 2. The synthesis from chloromethylated substrates proceeded most efficiently after capping of the chains with 1,1-diphenylethylene and coupling at −30 °C. For grafting on acetylated polystyrene substrates the yield was maximized at 25 °C, in the presence of 5 equiv of LiCl per “living” end to attenuate the reactivity of polyisoprenyllithium. Isoprene was polymerized with
sec-butyllithium either in cyclohexane or in tetrahydrofuran, to yield predominantly
cis-1,4 or mixed microstructures, respectively. However when the polymerization was carried out in cyclohexane, the addition of tetrahydrofuran before the coupling step was still preferable to increase the grafting yield. Arborescent copolymers were synthesized by grafting PIP side-chains having a
Mw of either 5,000 (PIP5) or 30,000 (PIP30) onto linear, G0, G1, and G2 polystyrene. The copolymers with short (PIP5) side-chains had a polyisoprene content of 80–90% by weight, increasing to 94–98% for long (PIP30) side-chains. The expected geometric increases in branching functionality and molecular weight were observed, while a narrow molecular weight distribution was maintained over successive generations.
Film formation by the isoprene copolymers on mica surfaces was investigated using tapping mode atomic force microscopy (AFM) after spin-casting from different solvents [
30]. Measurements in the phase contrast mode are ideal to probe phase separation in these systems, because the glassy polystyrene core and the rubbery polyisoprene shell should lead to significant phase changes in the signal. A heterogeneous morphology was indeed detected for these copolymer systems (
Figure 6) and, surprisingly, even for the copolymers with very low polystyrene contents (down to
ca. 3% by weight). It was interesting that when heptane, a solvent selective for the polyisoprene segments, was used to prepare the films, phase separation between the polystyrene core and the polyisoprene shell was significantly enhanced in phase contrast imaging, particularly for copolymers with longer PIP side-chains (low polystyrene contents). Conversely, in nonselective solvents (toluene and chloroform) the phase contrast was reduced, presumably due to enhanced mixing of the polystyrene and polyisoprene components. This clearly confirms the occurrence of phase separation between the polystyrene core and the polyisoprene shell of the molecules on the nanometric scale, but also that the extent of phase separation achieved strongly depends on the selectivity of the solvent used in film preparation [
30].
Figure 6.
AFM phase contrast images for films of arborescent polystyrene-graft-polyisoprene obtained by spin-casting (concentration ~1 mg/mL): G0PS-PIP30 in heptane (A) and chloroform (C); G1PS-PIP30 in heptane (B) and chloroform (D). The insets of (A) and (B) are the height images, showing the topology of the monolayers, and the 100-nm scale bar shown in (D) is the same for all the images. Reprinted from Reference 30 by permission from the American Chemical Society.
Figure 6.
AFM phase contrast images for films of arborescent polystyrene-graft-polyisoprene obtained by spin-casting (concentration ~1 mg/mL): G0PS-PIP30 in heptane (A) and chloroform (C); G1PS-PIP30 in heptane (B) and chloroform (D). The insets of (A) and (B) are the height images, showing the topology of the monolayers, and the 100-nm scale bar shown in (D) is the same for all the images. Reprinted from Reference 30 by permission from the American Chemical Society.
While AFM imaging provides direct evidence for phase segregation within the individual styrene-isoprene graft copolymer molecules, characterization results obtained by other methods were also consistent with a heterogeneous morphology. Thus breakdown of the time-temperature superposition principle was observed in an investigation of the rheological properties of arborescent polystyrene-
graft-polyisoprene copolymers, when constructing modulus-frequency master curves for copolymers with sufficiently large (G1 and G2) polystyrene cores [
31]. This type of behavior is typical for heterogeneous polymer systems.
The arborescent polystyrene-
graft-polyisoprene copolymers were very recently investigated for their potential use as polymer processing additives in linear low density polyethylene (LLDPE) resins [
32]. The copolymers, after modification with fluorinated substituents on a fraction of the isoprene units, were shown to suppress sharkskin formation and delay the onset of cyclic melt fracture when used at a concentration of 0.5% by weight in the extrusion of LLDPE monofilaments. Pressure reductions reaching up to 29% as compared with the unmodified LLDPE resin were also observed in the extrusion experiments.
2.6. Layered Copolymer Architectures
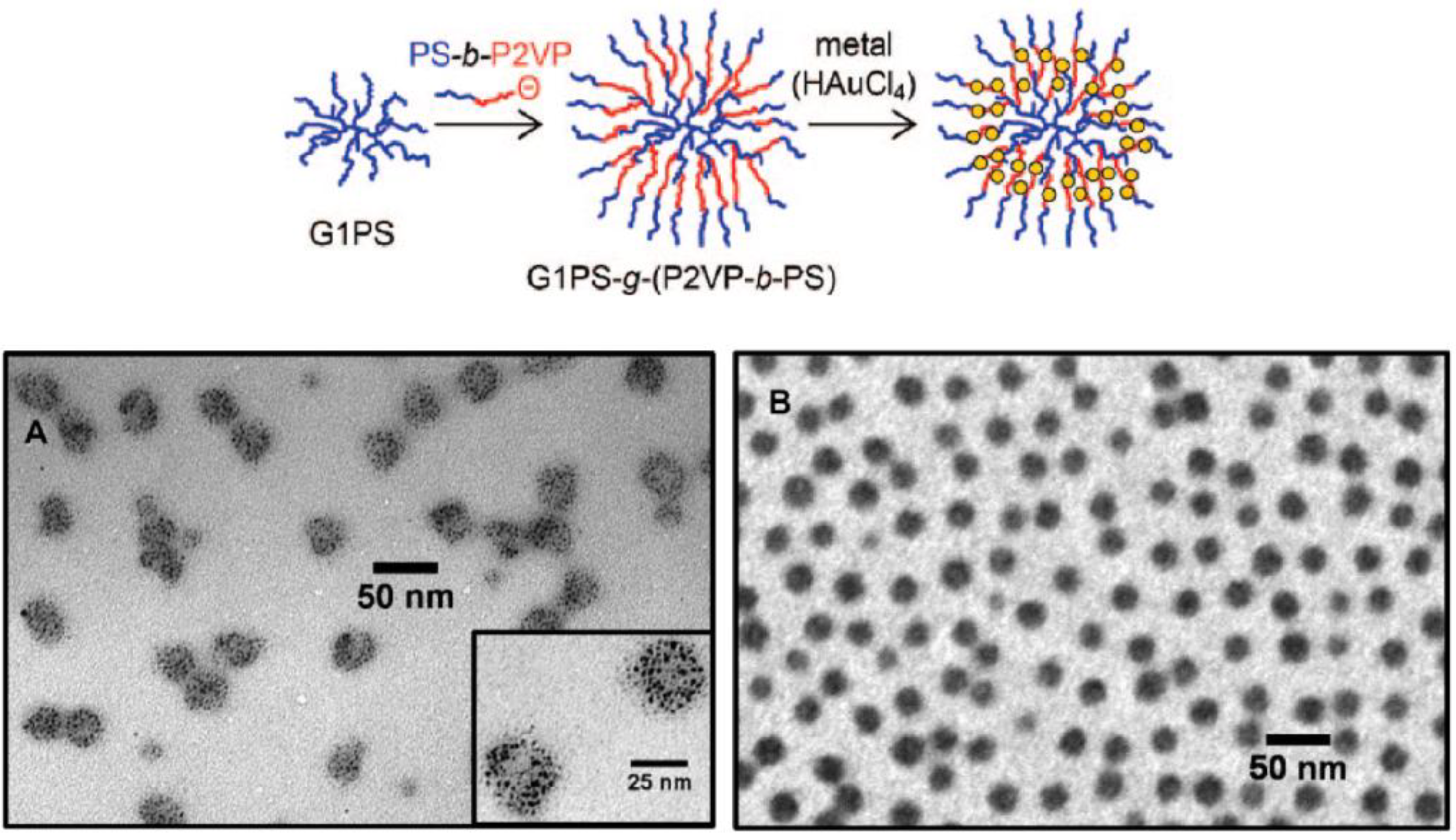
All the examples discussed so far are for copolymers with core-shell morphologies, derived from arborescent polystyrene substrates grafted with side-chains having a different chemical composition and forming the shell. Multilayered core-shell-corona systems have also been obtained recently by a similar approach. Thus the coupling reaction of “living” poly(2-vinylpyridine)-
block-polystyrene carrying the macroanion at the P2VP chain end yielded a layered morphology with a polystyrene core, an inner shell of P2VP, and a corona of polystyrene chains on the outside (
Figure 7) [
33]. The usefulness of these complex macromolecular architectures lays in their ability to complex various transition metal salts within the P2VP shell, while the polystyrene corona ensures good solubility of the metal-loaded micelles with minimal aggregation in non-polar solvents such as toluene. These copolymers may thus serve as templates for the formation of metallic nanoparticles, in analogy to block copolymer micelles [
34], but are potentially much more powerful: Spherical micelles are only formed by block copolymers under specific conditions (solvent selective for the shell component, appropriate volume fraction of each block), but these restrictions are relaxed for arborescent core-shell-corona copolymers. The covalently bonded architecture of these molecules also precludes extensive rearrangements of the side-chains, including their dynamic exchange (dissociation) from the micellar structure. Consequently, the micellar morphology of the arborescent copolymer molecules is preserved even under non-selective solvency conditions (e.g., tetrahydrofuran, chloroform) that hinder the self-assembly of poly(2-vinylpyridine)-
block-polystyrene copolymers into micelles. A preliminary investigation on the core-shell-corona copolymer systems demonstrated that intricate metallic nanoparticle morphologies could be obtained upon loading metallic compounds such as tetrachloroauric acid (HAuCl
4) in these template molecules (
Figure 7).
Figure 7.
Top: Schematic representation of the synthesis of a core-shell-corona arborescent copolymer structure by grafting “living” block copolymer chains onto an arborescent polystyrene substrate. Bottom: Examples of TEM micrographs obtained for G2 (A) and G0 (B) templates loaded with HAuCl4 at 0.25 and 0.50 equiv relative to 2VP residues, respectively. Adapted from Reference 33 by permission from the American Chemical Society.
Figure 7.
Top: Schematic representation of the synthesis of a core-shell-corona arborescent copolymer structure by grafting “living” block copolymer chains onto an arborescent polystyrene substrate. Bottom: Examples of TEM micrographs obtained for G2 (A) and G0 (B) templates loaded with HAuCl4 at 0.25 and 0.50 equiv relative to 2VP residues, respectively. Adapted from Reference 33 by permission from the American Chemical Society.
2.7. One-pot Synthetic Procedures
The stepwise or generation-based synthesis is clearly advantageous in terms of the extent of control achieved over the reaction and the characterization of the products obtained: The molecular weight of the side-chains and the substitution level of the coupling substrates can be easily controlled. A sample of side-chains can be removed from the reactor prior to the grafting reaction and analyzed by SEC to determine their exact molecular weight and size uniformity (polydispersity index). Finally, since the absolute molecular weight of the substrate and the graft polymer can be measured, the exact number of side-chains grafted onto the substrate can be determined for each generation. On the down side, product isolation after functionalization of the substrate, and the generation of “living” chains for each grafting cycle are time-consuming and relatively expensive procedures. To circumvent these problems, a one-pot process has been developed by Yuan and Gauthier for the synthesis of arborescent polymers which does not require the isolation and purification of reaction intermediates [
35,
36]. The procedure is applicable to the preparation of G0 and G1 homo- and copolymer architectures from successive functionalization and grafting reactions carried out sequentially in the same reactor, and yields products with fairly low polydispersity indices (
Mw/
Mn ≈ 1.2–1.3 typically).
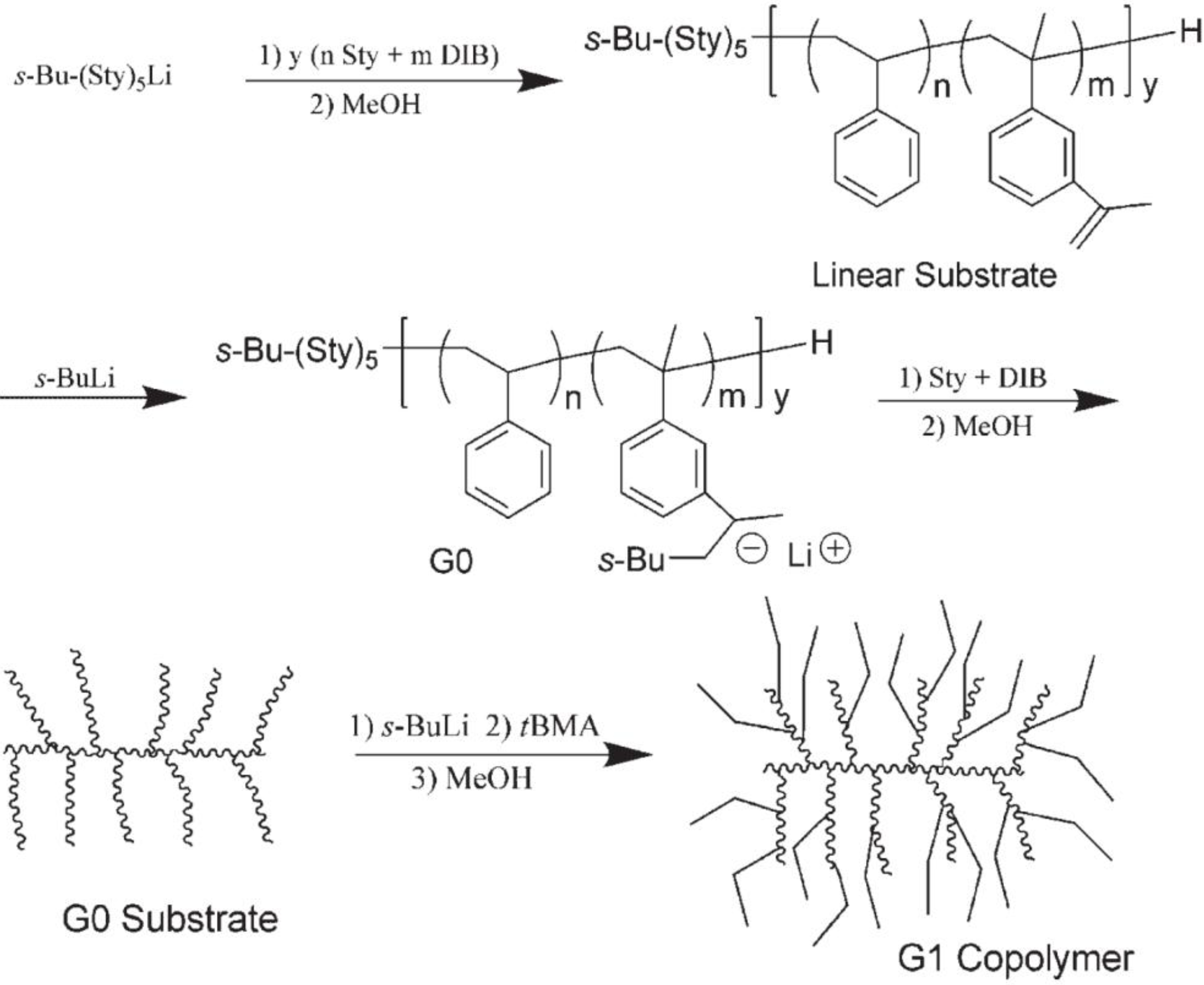
The one-pot method starts with the anionic copolymerization of styrene and
m-diisopropenylbenzene as shown in
Scheme 4. To compensate for the unfavorable copolymerization behavior of the two monomers, a semi-batch monomer addition protocol was developed to minimize segregation of the
m-diisopropenylbenzene units along the chains. After the preparation of the linear copolymer, the “living” chains are terminated by titration of the solution with methanol. The pendent isopropenyl moieties of the copolymer are then activated with
sec-butyllithium to generate initiating sites along the linear substrate, and a G0 styrene-
m-diisopropenylbenzene copolymer is obtained by successive additions of a styrene-
m-diisopropenylbenzene monomer mixture. Upon termination of the “living” G0 copolymer, the pendent isopropenyl groups on the side-chains are again activated and used to initiate the polymerization of styrene to obtain a G1 styrene homopolymer, or other monomers such as 2-vinylpyridine or
tert-butyl methacrylate (as shown in
Scheme 4) to give the corresponding copolymer. This approach, while not yielding products as well defined as the stepwise method, is much more convenient when large samples are required for less demanding applications. The formation of linear homopolymer in the procedure outlined in
Scheme 4 (due to incomplete reaction of the
sec-butyllithium used to activate the initiating sites) varied from 6–34% of the total sample weight, depending on the monomer and the substrate used in the reaction. Unfortunately, the exact characterization of the branched polymer structure obtained (branching functionality and molecular weight) is difficult in this case, as for all anionic
grafting from procedures in general. It was indeed pointed out that in these anionic
grafting from procedures, the branching functionality is usually approximated from the molecular weight of the graft polymer and the linear contaminant formed. Unfortunately this calculation method can lead to an underestimated branching functionality, because chain growth is faster for the linear polymer contaminant than for the graft polymer [
35,
36].
Scheme 4.
Synthesis of arborescent polystyrene-graft-poly(tert-butyl methacrylate) copolymers by a one-pot process. Reprinted from Reference 36 by permission from Wiley-VCH.
Scheme 4.
Synthesis of arborescent polystyrene-graft-poly(tert-butyl methacrylate) copolymers by a one-pot process. Reprinted from Reference 36 by permission from Wiley-VCH.
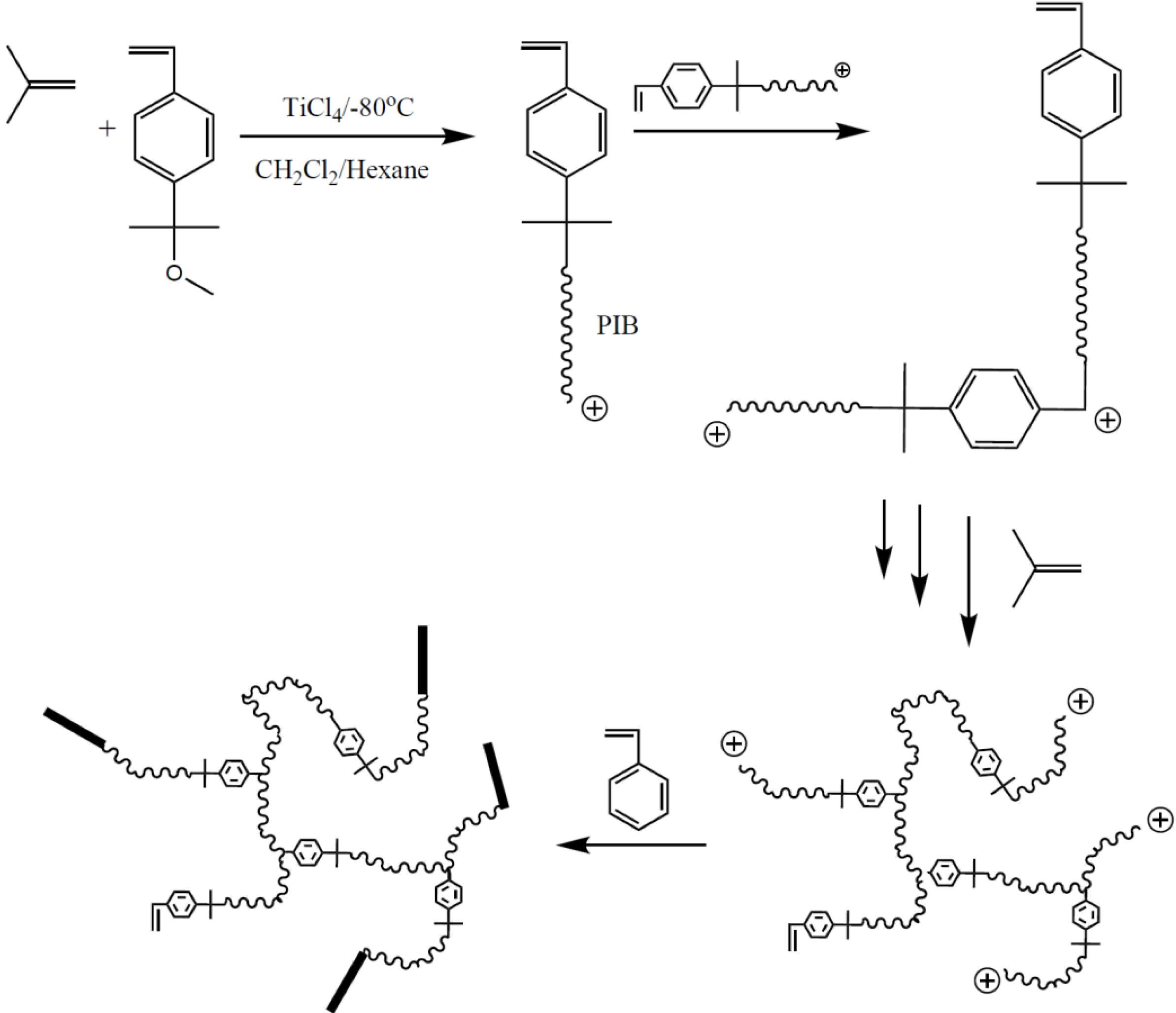
The one-pot synthesis of arborescent polyisobutylene structures by cationic polymerization in the presence of inimers (monomers containing a latent initiating site within their structure) has received much attention by the group of Puskas [
37]. This technique, initially developed for the preparation of arborescent isobutylene homopolymers, was subsequently extended to the preparation of copolymers with polystyrene or poly(
p-methylstyrene) segments as shown in
Scheme 5. For example, the cationic copolymerization of isobutylene with 4-(2-methoxyisopropyl)styrene serving as inimer produces “living” macromonomers which can self-condense
in situ and copolymerize with isobutylene in a
grafting through scheme to give dendritic branched structures carrying multiple cationic active sites at the chain ends. The addition of styrene or
p-methylstyrene to the “living” branched substrate leads to the growth of glassy styrenic segments (
grafting from process). The main interest in these copolymers lays in their elastomeric properties, as the glassy segments aggregate into phase-separated domains, giving rise to thermoplastic elastomers (TPE) with excellent tensile properties [
38,
39,
40]. The biomedical application of these systems as coatings for the sustained release of drugs in stents has also received attention [
41], considering that the analogous linear block copolymer TPE are already FDA-approved.
Scheme 5.
One-pot synthesis of arborescent polyisobutylene with polystyrene end-blocks by the cationic inimer method according to Puskas.
Scheme 5.
One-pot synthesis of arborescent polyisobutylene with polystyrene end-blocks by the cationic inimer method according to Puskas.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}