Abstract
Hybrid block copolymers based on peptides and synthetic polymers, displaying different types of topologies, offer new possibilities to integrate the properties and functions of biomacromolecules and synthetic polymers in a single hybrid material. This review provides a current status report of the field concerning peptide-synthetic polymer hybrids. The first section is focused on the different synthetic approaches that have been used within the last three years for the preparation of peptide-polymer hybrids having different topologies. In the last two sections, the attractive properties, displayed in solution or in the solid state, together with the potential applications of this type of macromolecules or supramolecular systems are highlighted.
1. Introduction
Peptide-synthetic polymer hybrid architectures have received great interest in materials science within the last years. The incorporation of peptide blocks into a synthetic polymer has opened up new challenges in areas as diverse as nanotechnology (e.g., biosensors or medical diagnostics) and biotechnology (e.g., drug delivery systems, tissue engineering or implants) [1,2,3,4].
The most important aspect of peptides is their inherent capacity to adopt stable conformations and self-assemble into highly organized nanoscale structures due to their non-covalent interactions. Specifically, depending on the amino acid lateral groups, peptides are able to adopt several secondary structures. The corresponding backbone conformation is stabilized according to the specific intra- or intermolecular interactions (i.e., hydrogen bonds, hydrophobic (van der Waals), coulombic and dipolar) that can establish the involved amino acid sequence. This kind of secondary structures are mainly constituted by helices, sheets or turns which can self-assemble into tertiary structures (e.g., the β-strand-helix-β-strand unit found in β-barrels), and quaternary assemblies (e.g., collagen microfibrils) [5,6].
Hybrids or molecular chimeras based on amino acid sequences and synthetic segments might have the advantageous properties characteristic of each of the building blocks. Namely, the economy, solubility, melt processability, versatility, or rubber elasticity of the synthetic block and the defined structure, functionality, mutual recognition, high mechanical performance or biodegradability of the peptide block. Moreover, these hybrids can be useful to stabilize the interface between polymer systems and biosystems. An additional point of interest derives from the stable amide backbone of peptides which make them rather inert against hydrolysis and more suitable for materials science applications compared to other highly interesting bioorganic macromolecules such as oligonucleotides or oligosaccharides.
The main individual characteristics of peptides and synthetic polymers are summarized in Scheme I. It is clear that the combination of these block elements is an interesting approach to synergistically join the properties of both macromolecular families.
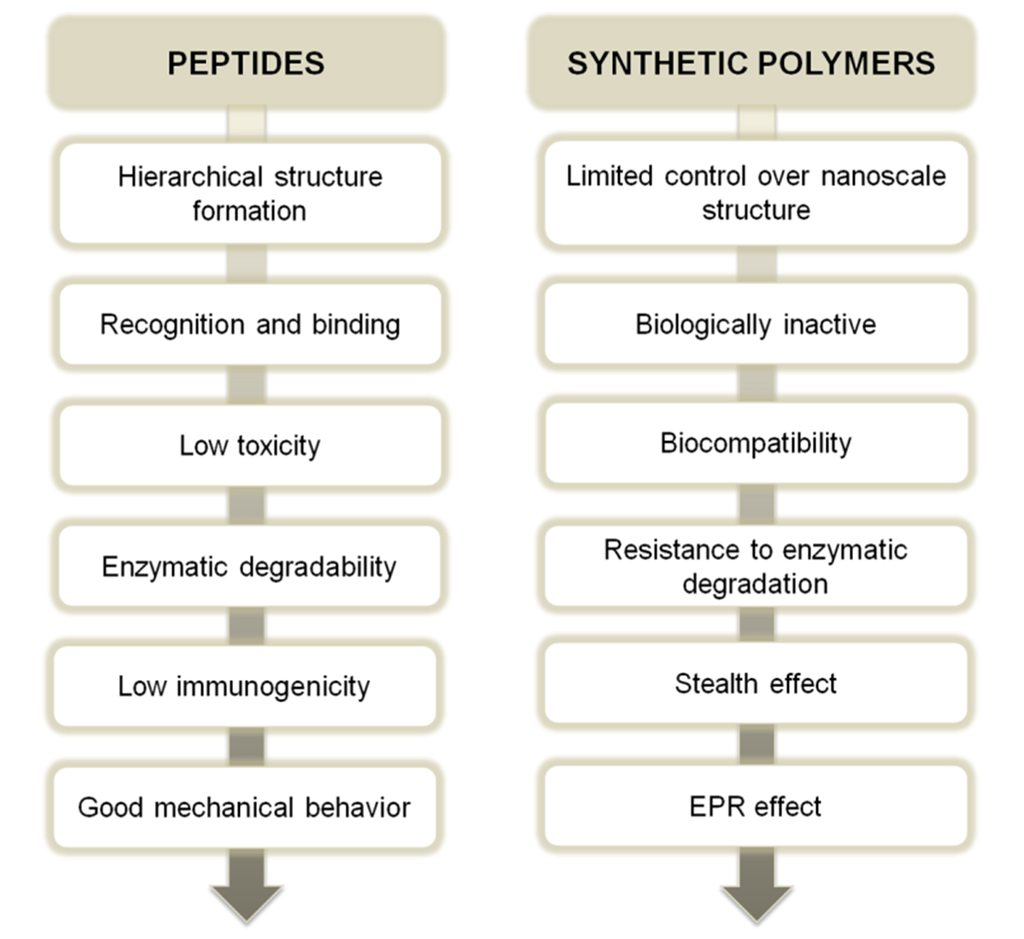
Scheme I.
Characteristic properties of peptides and synthetic polymers as potential benefits of these different macromolecular families. Stealth effect is related to the increased blood circulation time of a substance; EPR (enhanced permeability and retention) effect is related to capability of certain substances to accumulate in the inflamed tissue.
In summary, the possibility to combine the structural and functional control of peptides with the versatility of synthetic polymers has given access to a novel class of macromolecules that can take advantage of their complex solution behavior and their self-organization in the bulk. These materials can find as above indicated highly specific applications, including also for example their use as smart materials.
The increased activities in bio-hybrid materials science can largely be explained by the development of new synthetic methodologies that allow for the build-up of well-defined peptide/protein-polymer hybrid materials. Techniques from organic and peptide chemistry, polymer chemistry and molecular biology have enriched the materials scientist’s platform. The combination of solid phase peptide synthesis with polymer chemistry has proven to be a versatile method for the preparation of peptide-polymer hybrids. Introduction of native ligation methods even allows the synthesis of polymer modified peptides and proteins via an entire organic chemistry approach.
In the field of polymer chemistry besides the advances in α-NCA (α-amino acid-N-carboxy-anhydrides) polymerization, controlled radical polymerization (CRP) has been shown to be a robust technique, capable of creating well-defined biofunctional polymer architectures. Through protein engineering, several methods have been established that enable the construction of tailor-made proteins, which can be functionalized with synthetic polymer chains in a highly defined manner. In this way, there is a wide array of approaches which draws the world of biomacromolecules and the world of synthetic polymers together. Strategies for the preparation of peptide/protein–polymer conjugates can for example be split into those formulated for producing peptides/proteins bearing chemoselective groups and those formulated to introduce chemoselective handles into synthetic polymers. For more detailed information on the synthesis of polymer bioconjugates via the ligation of peptides, proteins, oligonucleotides/nucleic acids or carbohydrates on synthetic polymers the reader is directed to reviews of Gauthier and Klok [7], Lutz and Börner [4], and Le Drournaguet and Velonia [8]. For the different synthetic approaches towards peptide-polymer and protein-polymer block copolymers, reviews of Klok [9], and Marsden and Kros [10], or the book chapter of Deming [11] are specially recommended.
Because of their tunable conformational structure and hierarchical self-assembly, peptide-polymer hybrid block copolymers have been reviewed, both in solution and in the solid state, with an emphasis on the supramolecular structure formation [3,12,13]. The current research trends are involved in the combination of their special organization and behavior with other kind of properties. Concretely, peptide-polymer hybrids are nowadays investigated as stimuli-responsive biomaterials (e.g., pH, temperature, or target species) which can make more attractive the applicability of these substances in the field of nanotechnology.
This review highlights current activities towards the generation of functional peptide-polymer hybrids and is constituted by three sections. The first one is focused on different synthetic approaches that have been used within the last three years for the preparation of peptide-polymer block copolymers with linear or different types of topologies. Subsequently, the second part is focused on the attractive properties which this type of supramolecular systems displays in solution or in the solid state due to their particular organization. Finally, the last part explains the most recent applications of this new class of materials which are currently under investigation. The applications selected for the last section represent a general assortment of the most original and interesting advances in this field.
2. Synthetic Approaches for Peptide-Synthetic Polymer Hybrids
A number of techniques are presently available for the synthetic design of such peptide-polymer conjugates, including coupling a peptide to a premade polymer using one or multiple reactive sites (the “grafting to” approach), polymerizing a peptide-functionalized (macro)monomers (the “grafting through” approach), or performing a polymerization using a peptide macroinitiator (the “grafting from” approach), which are summarized in Scheme II. However in some cases, the “grafting through” and “grafting from” approaches can be used equally but using the synthetic polymer as a synthetic-functionalized monomer or macroinitiator, respectively. In addition, the three different strategies can be performed using either solution-phase chemistry or in a solid-phase supported manner. When compared to grafting to, grafting from and grafting through have the potential to facilitate an efficient synthesis of bioconjugates, because at first only a small component needs to be coupled to the protein or peptide after which the polymer component is grown in a stepwise fashion.
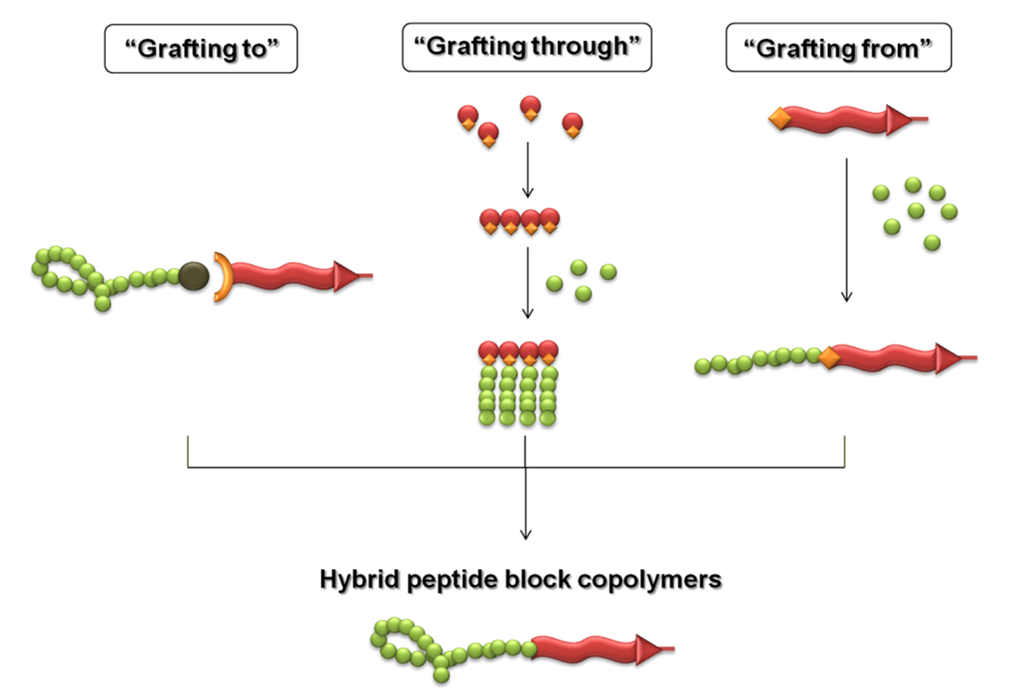
Scheme II.
Schematic illustration of the principal synthetic approaches to get peptide-synthetic polymer hybrids.
2.1. Liquid-Phase Peptide Synthesis. Preparation of Peptide-Synthetic Polymer Hybrids in Liquid-Phase
α-N-carboxyanhydride ring-opening polymerization (α-NCA ROP) enables the efficient generation of homopolypeptides or block copolypeptides possessing desired chemical functionality to a targeted molecular weight in a controlled manner. This technique allows multi-gram scale synthesis, but has the disadvantage that NCA’s are very sensitive monomers. Thus, polymerizations proceed via multiple competitive chain growth pathways (different initiating sites and termination reactions), which restrict control over polymer architecture as well as molecular weight and molecular weight distribution. Over the past decade new strategies were developed so as to enable the “living” behavior of NCA’s polymerization. The control over the growing polymer chains was achieved using primary amines or ammonium salts, thiol groups and silazanes, by means of the use of transition metal complexes based principally on nickel, cobalt and platinum or using high vacuum techniques [14,15].
In spite of the fact that the research on the synthesis of peptide hybrid block copolymers began in the mild 1970s by Ito et al. [16], this area is constantly under evolution due to its potential to obtain different hybrid peptide-polymer architectures. Recently, Zhang et al. [17], proposed the synthesis of well-defined core-shell molecular bottlebrushes (polydispersity index, PDI, between 1.02 and 1.03) with a helical poly(L-glutamate) backbone and polylactide-b-poly(ethylene glycol) block copolymer as side chains through the “grafting to” strategy and using the azide-alkyne Huisgen 1,3-dipolar cycloaddition. These core-shell brushes exhibited a wormlike conformation related to the retention of the α-helical conformation in both solution and solid state.
A related work focused on hybrid peptide-polymer brushes, prepared by means of the “grafting through” approach (Scheme III) [18], was based on polynorbornene (PN) as backbone with grafted poly(γ-benzyl-L-glutamate) (PBLG) or poly(ε-benzyloxycarbonyl-L-lysine) (PLL) as side chains. The selected synthetic methodology led to a small library of polymer brushes with variable PN and peptide lengths and grafting densities. Relevant information related to the helical structure of these complex structures characterized by a high “local” concentration of extensive inter-chain interactions was consequently provided.
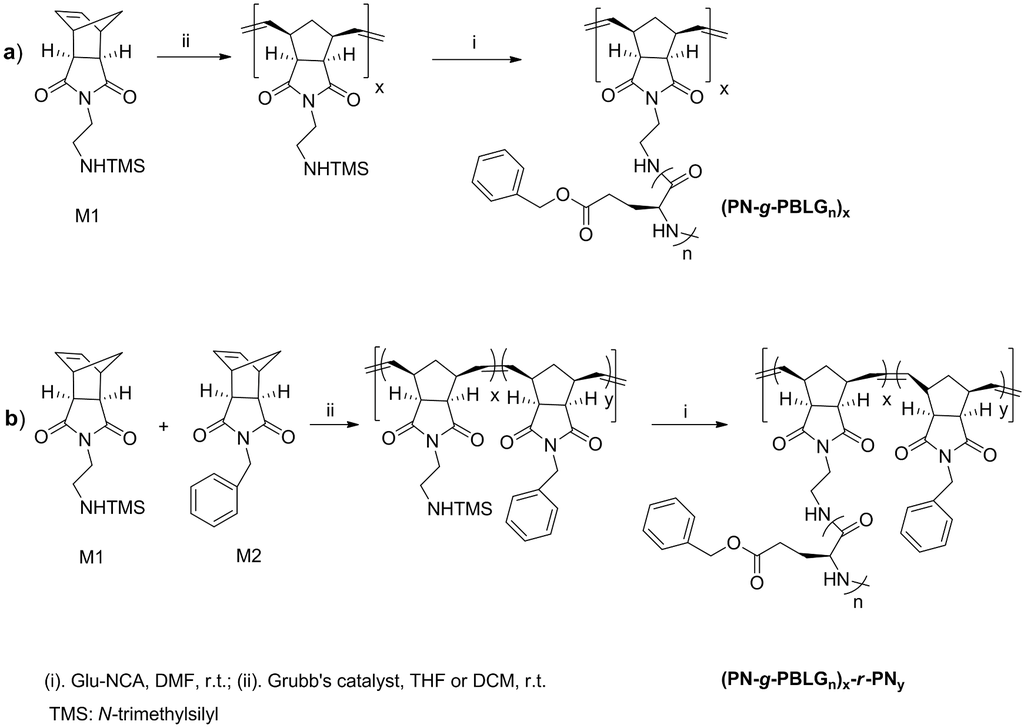
Scheme III.
Synthetic approaches based on [18] towards PBLG grafted on a homopolymer backbone of polynorbonene (PN-g-PBLGn)x (a), and PBLG grafted on random copolymer backbone of polynorbonene (PN-g-PBLGn)x-r-PNy (b).
Also addressing control over the polymer architecture, Zhang et al. [19], proposed the “grafting from” route to obtain a well-defined polymer brush with a narrow molar mass dispersity (≈1.2). In this study an oligolysine (n = 8) possessing a terminal double bond was employed as macroinitiator to grow methyl methacrylate (MMA) by a conventional radical polymerization that resulted in a PMMA backbone having oligolysine side chains. This molecular brush architecture afforded the short peptide side chains a constrained environment to adopt an enhanced chirality. In this way, the ordered secondary structures of oligopeptides were favored and stabilized.
An elegant work based also on polymer brushes was carried out by Jeong et al. [20] by combining the “grafting from” and the “grafting to” approaches. This study proposed the synthesis of a pH/temperature sensitive poly(ethylene glycol)-poly(L-alanine) grafted chitosan (CS-g-(PA-PEG)) in two steps. Firstly, an amine-terminated PEG was used as macroinitiator to polymerize alanine NCA and afterwards, reacted with succinic anhydride (SA) to obtain carboxylic acid-terminated PA-PEG chains. Secondly, the grafting of PA-PEG chains onto chitosan (7500 g/mol) was performed using EDC/NHS as catalysts in the coupling reaction. A grafting ratio of nearly 50% was achieved and the copolymer became sensitive to pH and temperature through conformational changes of their constitutive chitosan, polyalanine and PEG blocks.
Parallel to efforts to research into polymer brushes, multiarm star polymers are one of the most investigated topologies due to the possibility to possess a great number of peptides per molecule by using hyperbranched polymers or dendrimers as macroinitiators. The “grafting from” is normally the most common synthetic methodology employed to obtain this architecture. For example, well-defined star polypeptides were successfully synthesized by initiation of γ-benzyl-L-glutamate N-carboxyanhydride from polypropylene imine (PPI) dendrimers. The dendrimer generation and the dendrimer to NCA ratio were systematically varied to afford a range of star shaped architectures with a maximum of 8 to 64 poly(γ-benzyl-L-glutamate) (PBLG) arms. High molecular weights up to 500,000 g/mol were obtained, which could not otherwise be achieved from the analogous linear polypeptides in the absence of the dendrimer core. The resulting hybrid materials were employed as a drug delivery system (DDS) providing highly specific temporal and spatial controlled release [21]. A related work consisted in the synthesis of amphiphilic multiarm star polymers based on a polyethylene imine (PEI) as a core molecule and polyphenylalanine (PPhe) arms of different degree of polymerization (i.e., 40–120), which were obtained by means of the corresponding NCA polymerization. The multiarm stars were subsequently used as gene carriers on studying the transfection efficiency with different cell lines [22].
Despite of the expanding research on complex polymer architectures, the synthetic strategies on linear hybrid copolymers are being day by day updated so as to get smart structures based on different synthetic blocks with a different role inside the macromolecule. Dong et al. [23], have recently designed different photoresponsive poly(S-(o-nitrobenzyl)-L-cysteine)-b-poly(ethylene glycol) block copolymers taking advantage of photocleavable behavior of o-nitrobenzyl groups by means of UV irradiation (Scheme IV).
The obtained block copolymers showed self-assembling into spherical nanoparticles in aqueous solution and had a photoresponsive self-assembling behavior together with a size reduction of nanoparticles after irradiation. Anticancer drugs (e.g., doxorubicin) could be loaded in the polymer matrix and released in a controlled manner by changing the light irradiation time. Thus, the proposed strategy appears suitable for nanomedicine applications such as anticancer therapy.
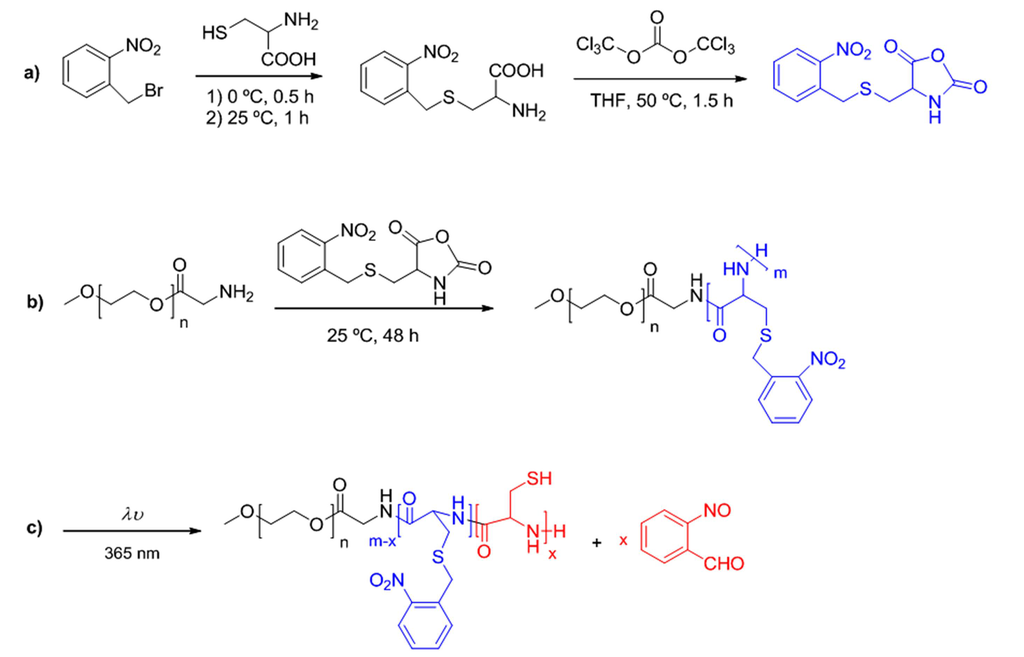
Scheme IV.
Syntheses of photoresponsive NCA monomer derived from cysteine (a), the related block copolymer based on poly(ethylene glycol) (b) and characteristic photocleavage reaction (c).
Another recent study involved the synthesis of an amphiphilic poly(ethylene glycol)-block-poly(γ-cholesterol-L-glutamate) diblock copolymer by means of the “grafting from” approach. Specifically, an amino-terminated poly(ethylene glycol) (Mn 2200 g/mol) was used as macroinitiator for the ROP polymerization of γ-cholesterol-L-glutamate-N-carboxyanhydride. In this way, a well-defined structure with a molecular weight of 8600 g/mol and a narrow molar-mass dispersity (≈1.1) was obtained. The copolymer was used to prepare nanoparticles with low cellular toxicity and potential applicability as a drug delivery system (DDS) [24]. A related work, based on the same synthetic methodology, was carried out to get biodegradable poly(L-lactide)-block-(Nξ-carbobenzyloxy-L-lysine) copolymers [25]. Interestingly, the synthesis of poly(L-lactide) macroinitiators was performed using novel metal complexes of Li, Na or K, which led to controlled polymerizations and products with narrow molar-mass dispersity indices.
The “grafting to” methodology results also efficient towards the synthesis of hybrid amphiphilic block copolymers. For example, Klumperman et al. [26] have recently documented a system based on poly(N-vinylpyrrolidone) (PVP) as a hydrophilic component and poly(γ-benzyl-L-glutamate) (PBLG) as a hydrophobic peptide, which led to a pH-sensitive bioconjugate. A modular approach was used for the synthesis of these conjugates. Optimized procedures were developed to obtain ω-aldehyde end-functional PVP and to synthesize cysteine end-functional PBLG. The latter makes use of a modified deprotection step for the thiol-protecting acetamidomethyl group. Finally, the conjugates were obtained by means of a thiazolidine linkage between both parts. The self-assembly behavior and pH dependence of the PVP-PBLG bioconjugate was demonstrated and opened the potential applications of the system as a smart drug delivery vehicle.
Generally the accessible synthetic approaches can be combined so as to obtain more complex block copolymers constituted by different peptide or synthetic polymer segments in the same structure. Recently, amphiphilic triblock copolymers based on poly(L-lactic acid)-block-poly(L-lysine)-block-poly(ethylene glycol) (PLA-b-PLL-b-MPEG) were synthesized in three steps. Firstly, the diblock copolymer PLA-b-PZLL was synthesized by ROP of Lys(Z)-NCA using α-amino-functionalized PLA (PLA-NH2) as macroinitiator. The triblock copolymer was synthesized by amidation of PLA-b-PZLL using MPEG-COOH and then by removing the side chain protected groups with HBr. The obtained copolymers were able to form spherical polymeric micelles in water exhibiting a low cytotoxicity [27].
Another recent study combined the “grafting to” and the “grafting from” approaches in order to obtain dendron-like triblock copolymers (Scheme V) [28]. Concretely; an azide-terminated polycaprolactone was obtained and used to couple; by click chemistry; an alkyne-terminated poly(amidoamide) (PAMAM) dendron (Dm) containing from 1 to 8 primary amine groups. Subsequently; by means of NCA polymerization; poly(γ-benzyl-L-glutamate) (PBLG) chains were grown. Molecular weights of the final structures ranged between 7800 and 45,700 g/mol as estimated by NMR analyses.
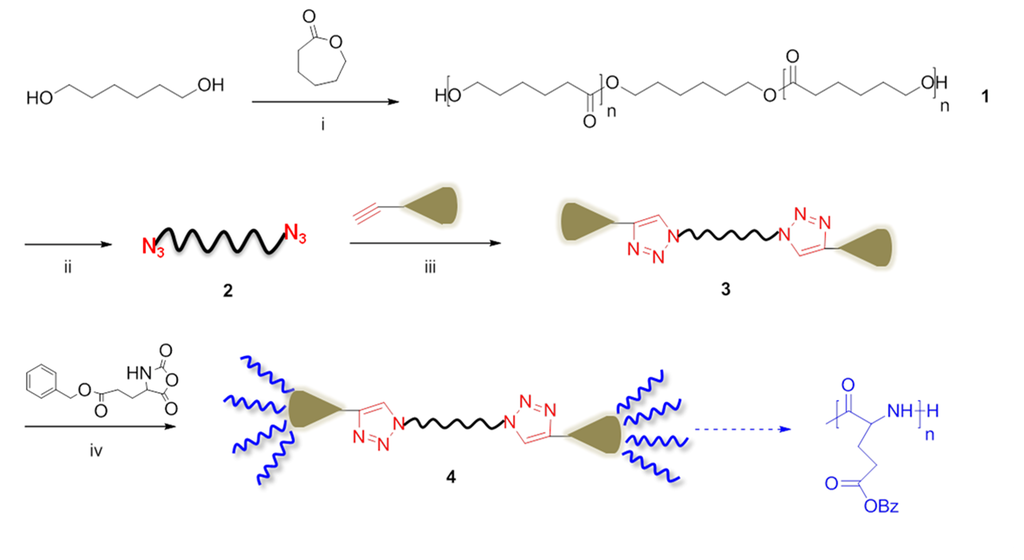
Scheme V.
Synthesis of PBLG-Dm-PCL-Dm-PBLG triblock copolymers by combining ROP and click chemistry based on [28]. Reagents and conditions: i) Sn(Oct)2, 115 °C; ii) TsCl, 24 h, r.t., NaN3, 24 h, r.t.; iii) [2]/[Dm]/[CuBr]/[PMDETA] = 2.4/0.4/0.4, 24 h, 35 °C; iv) 24 h, r.t.
The crystallinity of the central PCL segment within these copolymers was increasingly suppressed by the flanking of PBLG chains, whereas the PBLG segments gradually changed from a β-sheet to α-helix conformation on increasing the degree of polymerization of PBLG wedges. In addition, these triblock copolymers formed thermoreversible organogels in toluene with critical gelation concentrations controlled by the dendritic topology of the PBLG wedges.
Easier synthetic methodologies can also be used to obtain stimuli-responsive peptide-based triblock copolymers. Recently, ABA triblock copolymers were obtained by employing diamine-functionalized polypropylene oxides (average degree of polymerization of 33 or 68) as macroinitiators to polymerize Z-lysine NCA. In this way, triblock copolymers with a degree of polymerization of the polylysine segments ranging from 27 to 46 were obtained and used to study the micelle-to-vesicle or micelle-to-disk transitions as function of pH. The unique responsive behavior of such system could be exploited in areas such as drug delivery, whereby a delivered drug could be spontaneously released upon pH-driven morphology transitions [29].
The “grafting from” approach can also be exploited in order to obtain ABC triblock copolymers as Zhang et al. have recently documented [30]. In this work, PEG–peptide hybrid drug carriers, specifically poly(ethylene glycol)-b-poly(L-cysteine)-b-poly(L-phenylalanine) (PEG-PCys-PPhe) triblock copolymers, were prepared via the ring-opening polymerization of amino acid N-carboxyanhydrides. The copolymers were able to self-assemble to form core–shell–corona micelles in aqueous solutions. The shell of these micelles had the ability to self-cross-link by the oxidation of thiol groups in the PCys segments. The in vitro drug release was also studied, which demonstrated that the cross-linked shell could be helpful to reduce the drug loss in the extracellular environment.
The “grafting from” is one of the most employed routes to the synthesis of linear peptide-polymer hybrids. Moreover nowadays this frequently pathway is being exploited by combining it with new controlled, “living” radical polymerization techniques, which are expanding the scope of these “grafting” methodologies [10]. Techniques such as atom transfer radical polymerization (ATRP) [31], reversible addition-fragmentation chain transfer (RAFT) polymerization [32,33], and nitroxide mediated polymerization (NMP) [34], allow for the synthesis of low-dispersity polymers.
The combination of controlled radical polymerization and N-carboxyanhydride (NCA) ring-opening polymerization enables the generation of well-defined block copolymers. A clear example is the synthesis of amphiphilic hybrid block copolymers based on block or random γ-benzyl-L-glutamate/L-alanine peptide sequences and polystyrene or poly(n-butyl acrylate) as synthetic segments [35]. A bifunctional initiator was employed so as to conduct NMP and NCA ROP polymerizations. This strategy led to block copolymers with low molar mass dispersities (≈1.1) and moderate molecular weights (7500–25,200 g/mol). It was also shown that the elastase-induced degradation was related to the composition of the hydrophilic peptide block (i.e., the higher the L-alanine content the faster degradation of the micelle) and the composition of the hydrophobic non-degradable block.
This strategy was also employed to prepare cross-linked star polymers based on amphiphilic diblock copolymers of poly(γ-benzyl-L-glutamate-b-styrene) (PBLG-b-PS) [36]. Firstly, a PBLG macroinitiator was synthesized by means of NCA polymerization with nitroxide terminal-groups. In a second step, styrene was polymerized by NMP resulting in block copolymers. The cross-linking of the terminal groups of the block copolymer chains was conducted in a last step by using divinylbenzene (DVB) as cross-linker. Using this strategy, high molecular weights (i.e., Mn ranged from 47,000 to 548,000 g/mol) and relatively narrow dispersities (1.3–1.9) were attained. After cleavage of the PBLG chains, pH responsive particles with a poly(L-glutamic acid) shell and polystyrene core were obtained (Scheme VI).
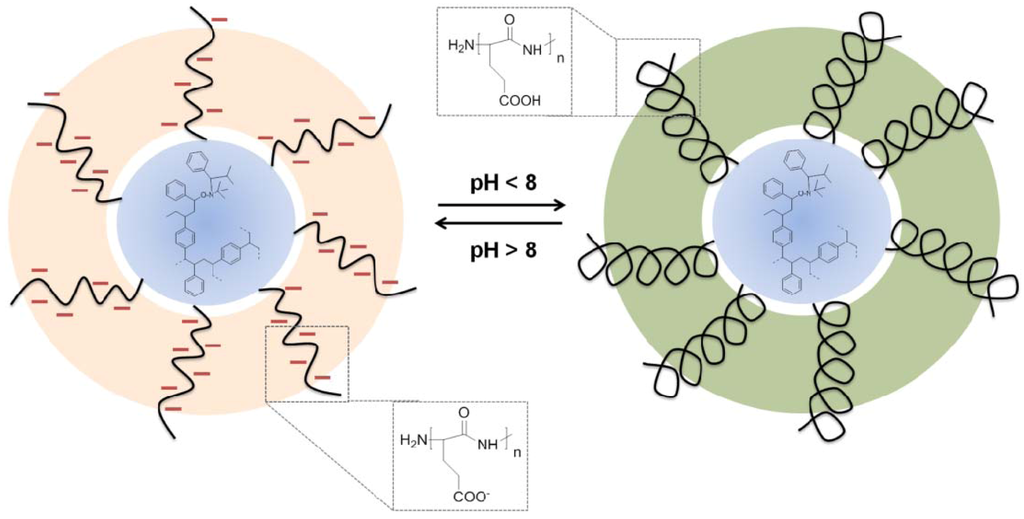
Scheme VI.
Depiction of pH responsive peptide-based core-shell nanoparticles assuming a helix-coil transition induced by a pH change [36].
2.2. Solid-Phase Peptide Synthesis. Preparation of Peptide-Synthetic Polymer Hybrids in Liquid- or Solid-Phase
Solid phase peptide synthesis (SPPS) is nowadays a routine technique that allows access to peptides containing up to 50 amino acid residues with perfect control over the chain length and monomer sequence [37,38,39]. This is a special feature in comparison to NCAs polymerization because the latter remains limited to the preparation of homopolypeptides thus lacking the ability to play with biomolecular sequence. SPPS allows versatile modifications of the peptides, ranging from single position mutants, where one amino acid is substituted, to the synthesis of pseudo-peptides comprising of non-peptidic backbones.
In the early of 1980s, a simple method of using SPPS to access peptide-polymer block copolymers was developed. Concretely, the technique consisted in using polystyrene (PS) beads loaded with amino-terminated poly(ethylene glycol) (PEG) through an acid labile benzyl-ether linker [40,41,42,43,44]. Subsequently, SPPS was successfully conducted with high efficiency from PEG according to the so-called “divergent approach” or alternatively a selected pre-formed peptide was chemically linked to the synthetic polymer chain giving rise to the “convergent approach”. Both methodologies were able to liberate the peptide-PEG block copolymer by a subsequent cleavage of the ether linker.
A recent example of the use of these pre-loaded resins is the preparation of a peptide-polymer block copolymer in which a β-sheet forming peptide (βAβAKLVFF) was attached to a hydrophilic PEG chain (3000 g/mol) supported on a resin. Specifically, a PEGylated TentaGel PAP resin was used and the peptide was assembled from the C-terminus towards the N-terminus. Attachment of the peptide to the solid support at the C-terminal was achieved through the α-carboxyl group of the amino acid. The obtained hybrid molecules were able to self-assemble into micelles comprising a modified amyloid peptide core surrounded by a PEG corona. The modified amyloid peptide was able to form helical ribbons based on a β-sheet motif and contained β-amino acids that were excluded from the β-sheet structure, thus being potentially useful as fibrillization inhibitors. In the model peptide-PEG hybrid system, the enzymatic degradation using R-chymotrypsin led to selective cleavage close to the PEG-peptide linkage, breaking up the micelles, and releasing peptides in unassociated form. This concept has considerable potential in the targeted delivery of peptides for therapeutic applications [45].
Unfortunately, only resins pre-loaded with PEG are commercially available, and the range of molecular weights is limited. For this reason, other strategies are being nowadays more exploited so as to achieve higher structural versatility. A recent work carried out by Gigmes et al. [46], reported a straightforward route to a peptide-synthetic polymer hybrid by means of the synthesis of a protected peptide by automatic SPPS containing a terminal initiating for nitroxide-mediated polymerization (NMP) of styrene (Scheme VII). After trifluoroacetic acid cleavage the peptide-polystyrene copolymer was obtained. The NMP polymerization of styrene was controlled on changing the time. In this way, the molar mass could be tuned resulting in macromolecules with a molecular weight ranging from 20,500 to 25,000 g/mol and with a polydispersity index between 1.22 and 1.32.
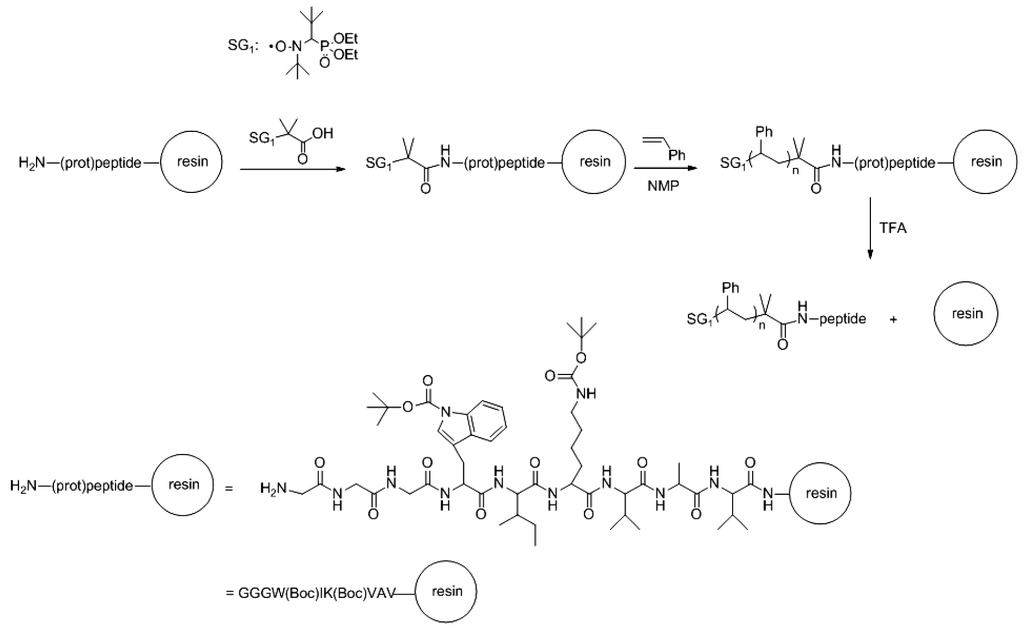
Scheme VII.
Strategy of preparation of SG1-functionalized resin peptide for the initiation of nitroxide-mediated polymerization (NMP); (Prot)peptide: GGGW(Boc)IK(Boc)VAV, with the N-Boc-protected Lysine and Tryptophan (M = 1099 g/mol); Peptide: GGGWIKVAV (M = 885 g/mol) based on [46].
One of the mostly employed strategies is based on the synthesis of peptide-polymer hybrids by using a functionalized peptide, synthesized by SPPS, and performing a “grafting from” by employing the peptide as a macroinitiator in bulk or solution. For example, Hennink et al. [47], have recently proposed the synthesis of ABC block copolymers by growing two different polymer chains, poly(oligo(ethylene glycol) methyl ether methacrylate) (pOEGMA) and the thermosensitive block of poly(N-isopropylacrylamide) (pNIPAm), from a native peptide using atom transfer radical polymerization (ATRP). Specifically, these two different ATRP initiators were coupled via orthogonal methods to the N- and C-terminus of the peptide Ser-Gly-Pro-G ln-Gly-Ile-Phe-Gly-Gln-Met-Gly, a substrate for matrix metalloproteases. Above the cloud point of one of the blocks, the polymers self-assembled into micelles, which were organized with the hydrophilic block on their outer side. The peptide linkage between the polymer blocks could be cut by a metalloprotease (collagenase), leading to “shedding” of the corona of the micelles. This technology offers new possibilities for enzyme-triggered drug delivery.
Other related works, in terms of the selected synthetic methodology, were conducted by Paira et al. [48,49]. Oligopeptide initiators were designed and used to polymerize ε-caprolactone or methyl methacrylate by means of ring-opening polymerization or ATRP, respectively. The obtained hybrid polymers presented narrow molar mass dispersities (ranging from 1.20 to 1.50 in the case of PMMA blocks and from 1.3 to 2.2 for PCL blocks) and relatively high molecular weights, especially those containing PMMA blocks. Both block copolymer structures self-assembled into micelles depending on the solvent. These micelles could undergo a secondary aggregation as function of time resulting in micro or nanospheres. The obtained hybrid spheres are interesting due to their potential to be used as micro- or nanocarriers for drug delivery.
Finally, another common synthetic strategy, in which SPPS is also employed, is based on the conjugation of a designed peptide to the synthetic polymer chain by means of bioorthogonal coupling reactions such as, amide formation, thioether formation, Pd°-catalyzed coupling reactions, Staudinger ligation, cycloaddition reactions (Diels-Alder, 1,3-dipolar cycloaddition), reductive alkylation or amination, oxime and hydrazone formation, thiol addition reactions, or oxidative coupling [7,50]. Looking to the wide variety of coupling reactions it is obvious that they provide ample opportunities for the synthesis of complex peptide-synthetic polymer conjugates without the need for possible complex protective group strategies.
For example, a well-defined protected glycopolymer (i.e., poly(2-(2,3,4,6-tetra-O-acetyl-β-D-glucosyloxy)ethyl methacrylate) (PAcGlcEMA)) was recently synthesized by RAFT polymerization. In the presence of ethanolamine and 2,2'-dithiodipyridine, PAcGlcEMA-PDS with a terminal thiol-reactive pyridyldisulfide (PDS) group was generated by one-pot approach. After deprotection of PAcGlcEMA, a tripeptide reduced glutathione (GSH) was conjugated to PGlcEMA-PDS by a thiol-disulfide exchange reaction under mild conditions. The specific recognition with protein Concanavalin A and its antioxidant activity justify that the proposed peptide-glycopolymer bioconjugate has a great interest for biomedical applications such as biodetection, biomimetics and targeted antioxidant delivery [51].
Related to the amide bond formation an original work appeared in which this coupling reaction was used to obtain complex polymer architectures (i.e., star polymer). Concretely, a thermo-sensitive elastin-like oligopeptide was successfully assembled onto a poly(amidoamine) dendrimer surface [52]. The obtained hybrid peptide/dendrimer was found to exhibit low critical superior temperature (LCST) behavior at physiological temperature range under neutral pH variation. This dual-responsive character can play an important role in biological applications based on the ability to storage and render a controlled-release of small molecules as a response to specific environmental conditions.
A related work, focused on the use of a elastin-like peptide to obtain hybrid brush copolymers was the one proposed by Klok et al. [53]. In this study, poly(pentafluorophenyl methacrylate), obtained by ATRP, was first modified with allylamine to give polymers with pendant alkene groups. Subsequently, the modified polymer was conjugated with a thiol-terminated elastin-derived peptide sequence by means of a thiol-ene addition (Scheme VIII). The achieved grafting density was about 50%. The use of this tandem postpolymerization modification can present the opportunity to synthesize complex copolymers as smart/responsive materials, peptidic drug carriers, or as precursors for nanocarriers. Furthermore, this versatile strategy should extend the scope of controlled radical polymerization and “click”-type reactions.
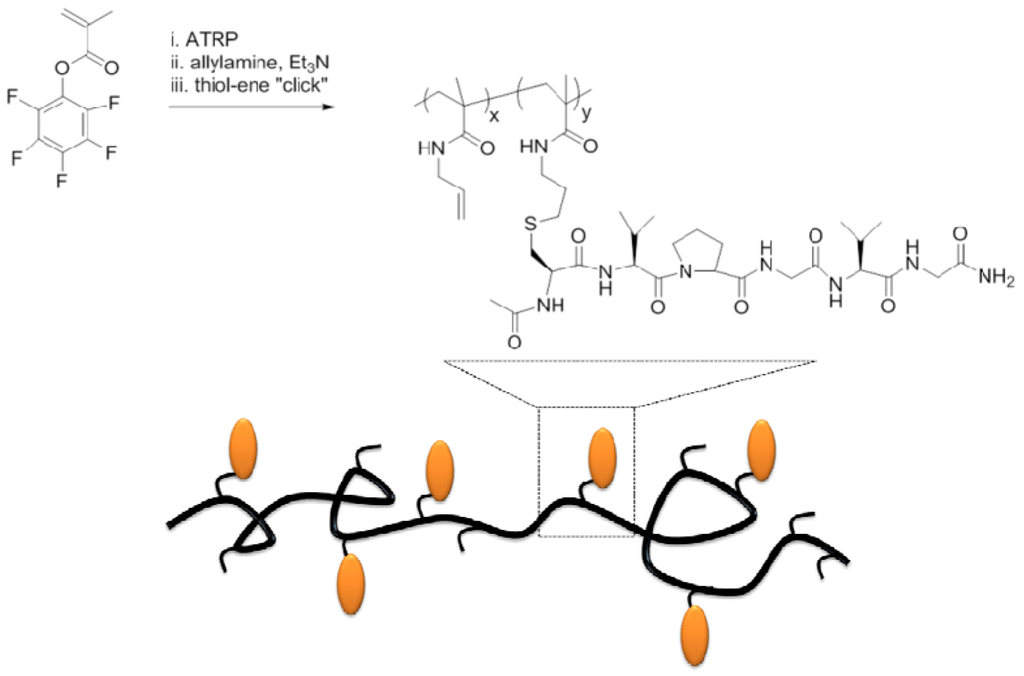
Scheme VIII.
Biofunctionalized polymethacrylate via post-polymerization modification, based on [53].
As it has been observed, the concrete design of the peptide sequence is an important feature because it is the main responsible of the special characteristics of the final material. Introducing specific successions can provide the peptide structure with different behaviors at the same time. One recent example of this case is the work presented by Werner et al. [54], which proposed the synthesis of a peptide linker sensitive to the presence of matrix metalloproteinases (MMP), thus permitting the degradation and remodeling of the material by cells secreting this enzyme. Moreover, this strategical synthetic methodology involved a short amino acid sequence encoded as RGD, one of the most important ligands of cellular adhesion receptors which are found in several ECM proteins. Afterwards, this peptide was conjugated to a acrylated-modified star poly(ethylene glycol) (starPEG) through Michael-type addition between an acrylate groups and a cysteine residue of the custom-designed peptide. A key of the proposed work was the use of heparin, a naturally occurring highly anionic polysaccharide which has a high affinity to many important signalling molecules (e.g., growth factors, cytokines, ECM molecules). In this way, carboxylic groups of heparin were activated with carbodiimide/sulfo-N-hydroxysuccimide to create a hydrogel network by the formation of amide bonds with N-terminal amino groups of the starPEG-peptide conjugate. The obtained gelled materials were shown to stimulate three-dimensional growth and matrix remodeling by human endothelial cells, which are important requirements for pro-angiogenic processes. Thus, the gels can be expected to support the vascularization of engineered tissues.
3. Supramolecular Organization of Peptide-Synthetic Polymer Hybrids
As long as structure formation of block copolymers is solely driven by the counterbalance of thermodynamic de-mixing and chain elasticity, the size and shape of superstructures can be controlled by the relative composition of polymer chains and the number of repeating units [55]. The most commonly morphologies of block copolymers observed in solution are spherical and cylindrical micelles and vesicles, which can be originated from the non-equilibrium or the equilibrium states. Different theories have been developed in order to describe the equilibrium states of block copolymers in solution [56]. However, in case of peptide-polymer hybrids theoretical models based on rod-coil systems are the most used to explain their organization behavior. Polymer chains may exhibit a stiff conformation, “rod-like” conformation, due to extended π-conjugation [57,58], aromatic groups along the polymer backbone or presence of secondary structures as in the case of peptides or other kind of biomolecules [59,60]. Rods possess low conformational entropy which is directly related to their predisposition to form liquid crystalline alignments. The structural packing in the rod domain is the consequence of the competition between the deformation energy of coil stretching and the rod/coil interfacial energy. The prevalence of one of these parameters is based on the relation of the lengths of both domains (Scheme IX). Normally, for rod-coil block copolymers (RC-type) the interfacial energy is dominant and it results in smectic A-aggregates (the arrangement is characterized by a slippage angle greater than 67.5°). In contrast, for coil-rod-coil copolymers (CRC-type) the deformation energy dominates in smectic C-aggregates (denoted as smectic CI when the slippage angle varies between 67.5° and 60°, and smectic CII when the angle is lower than 60°) [61].
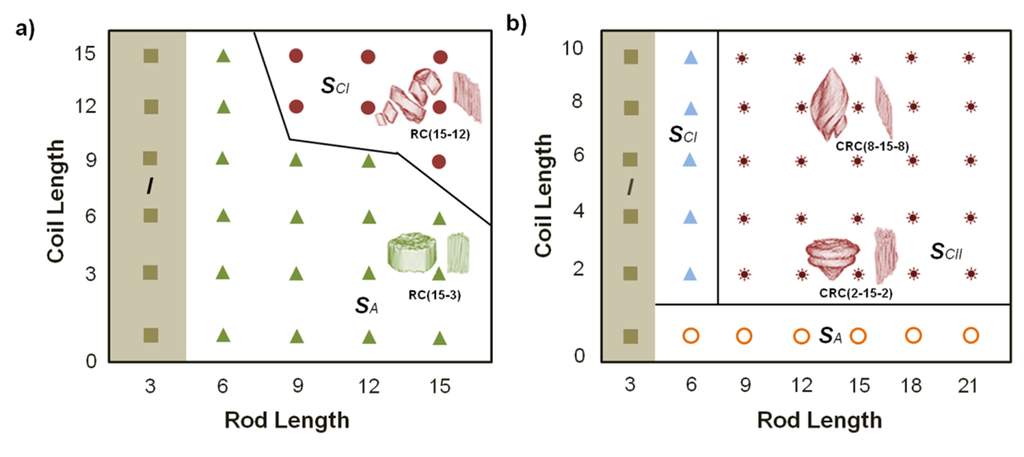
Scheme IX.
Phase diagram, based on [61], of the structure of the rod domain for rod-coil (RC) (a) and coil-rod-coil (CRC) (b). I, SA, and SC represent the isotropic, smectic A, and smectic C arrangements of the rod domains. The corresponding morphologies are also shown.
The elastic contribution from stretching of the rods is usually negligible compared to the rod/coil interfacial free energy. As a consequence, bilayers are often preferred over micelles in rod-coil systems [62]. The smectic or nematic order leads to high bilayer stiffness and high surface tensions, and makes these equilibrium vesicles very large and stable. Alternatively, “disk-shaped” micelles or “crew-cut” micelles with a planar rod-coil interface were also observed when the rod is the soluble segment. Similarly to other rod-coil block copolymers, the spontaneous formation of vesicles has been often observed in hybrids containing helical peptide segments [63,64,65]. Since the secondary structures of the peptide segment strongly influence the selection of the shape of the aggregate, helix to coil transitions can be used to induce dramatic shape changes such as micelle-vesicle transitions or vesicle inversions [66].
The stiffness asymmetry in rod-coil diblock copolymers results in an increase in the Flory-Huggins parameter and consequently these copolymers might self-assemble into phase-separated structures at relatively low degrees of polymerization. Thus, whereas phase separation of coil-coil diblock copolymers typically gives rise to 5–100 nm domain sizes, rod-coil diblock copolymers have the potential to render phase-separated structures of only several nanometers.
Recently, a series of novel rod-rod diblock copolymers containing poly{2,5-bis[(4-methoxyphenyl)-oxycarbonyl]styrene} (PMPCS) and poly(γ-benzyl-L-glutamate) (PBLG) have extensively been studied. It was shown that for an fPBLG close to 0.5 the in bulk structure was lamellar. Specifically, PMPCS was arranged in a columnar nematic phase whereas PBLG gave rise to a hexagonally packed-cylinder structure. In this system, a stacked bilayer structure in a hexagon in lamella morphology was proposed (Scheme Xb,c). On the contrary, on increasing fPBLG to nearly 0.7 a micro-separated hexagon in cylinder morphology was found.
Basically, PMPCS domains constituted the core of the cylinders which were surrounded by the PBLG matrix where helices were arranged in a hexagonal structure. Both PMPCS and PBLG rods were packed in an interdigitated way in their own domains (Scheme X, d and e). These results obtained from TEM observation and 2D WAXD patterns [67] are significant since indicate that the development of the liquid crystal phases did not destroy the self-assembling structures from microphase separation. Thus, both mesogen-jacketed liquid crystalline polymers and polypeptides obtained from controlled polymerization methods are excellent rod-like building blocks to form rod-rod block copolymers with nanoscale hierarchical structures.
The most part of the works focused on the supramolecular organization of peptide-synthetic polymer hybrids have selected poly(γ-benzyl-L-glutamate) (PBLG) or poly(Z-lysine) (PZlys) as the peptide block. These blocks have a capability to easily adopt α-helices or β-sheets by just adjusting the distribution of D/L units in their sequences. Moreover, both peptides have the possibility to be hydrolyzed resulting in poly(L-glutamic acid) (PLGA) and poly(L-lysine) (PLL), which show pH-responsive behavior displaying coil to helix transitions depending on the pH of the medium. Specifically, PLGA adopts α-helical conformations at low pHs (i.e., pH ≤ 6.3), while PLL becomes α-helical at high pHs (i.e., pH ≥ 8.4) [17].
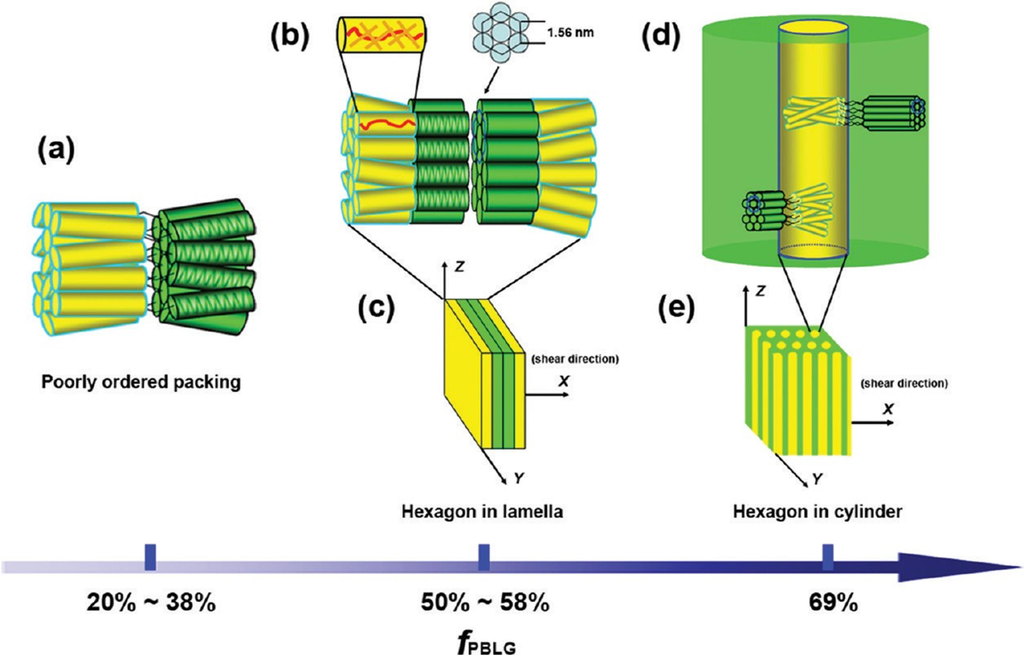
Scheme X.
Schematic representation of the proposed model for the self-assembly of the rod-rod diblock copolymer in poorly ordered packing (a), hexagonal in lamella (b), and cylinder (d) structures, respectively. Schematic drawings of the shearing geometry with the X-axis being the shear direction are shown in (c) and (e) for lamellar and cylinder structures, respectively. Reprinted with permission from [67]. Copyright (2010) American Chemical Society.
Schlaad et al. [68], have recently studied the influence on the secondary structure of different predefined D/L stereosequences by considering three block copolymers with the same composition and constituted by poly(ethylene oxide) and monodisperse peptide segments of 18 (Nε-benzyloxycarbonyl)lysine (ZLys) units. Secondary structures (i.e., random coil, β-sheet or α-helix) of these bioconjugate samples were determined by 1H NMR, FTIR and CD spectroscopies and evaluated according to their ability to gelate tetrahydrofuran at room temperature (using viscosimetry and scanning force microscopy). The tendency for organogelation was gradually increased when the secondary structure of the peptide block changed from random coil (peptide with a close to random distribution of 8 and 10 D and L units, respectively), α-helix (peptide sequence constituted by only L units) to β-sheet (peptide sequence constituted by two blocks having 7 L units each one and a middle block with 4 D units), which was the one exhibiting the highest tendency to form a gel. Microscopy images showed the formation of fibrils (1 nm in height and 10–14 nm in width) constituted by β-sheet assemblies of congener peptide-PEO conjugates. This fibrillation phenomenon seemed to occur via antiparallel β-sheet formation since the width of the fibrils obtained were in agreement with the estimated value found in the literature (i.e., 11.1 nm). On the other hand, the hybrid conjugate possessing a peptide block with α-helix secondary structure presented fibrils with a lower width (i.e., close to 6–10 nm), which was explained by a structure of interdigitated α-helices. PEO-peptide molecules were postulated to be disposed orthogonally to the fibril axis, which is the contrary to the usual packing of α-helix homopolypeptides. The observed behavior was rationalized on the basis of the steric repulsion of the second block segment. However, the direct assembly into fibrils could not be fully understood.
A related current work based on the control of fibrillation of as β-sheet forming peptide was conducted by Perrieret al. [69]. In this study, a water soluble poly(hydroxyethyl acrylate) [P(HEA)] was conjugated to an azide functionalized β-sheet forming peptide. In terms of supramolecular behavior it was observed that the fibrillation (i.e., the β-sheet formation) diminished severely by P(HEA) conjugation. In this case, the polarity and hydrogen bonding of the conjugated polymer appeared to have a major effect on the self-assembly of the hybrid copolymers. Highly polar polymers tended to adopt a random coil conformation as observed by circular dichroism (CD) spectroscopy. The conjugation of P(HEA) to peptides that have a tendency to form aggregates is therefore an interesting potential route towards the control of peptide fibrillation.
As it has been previously commented, new packing frustration is introduced by the fact that one or more copolymer blocks are rod-like. In particular, the α-helical conformation of peptides leads to mesogenic rods which possess dipole moments, potentially include kinks, and have extensively been employed in peptide/polymer hybrid systems [70,71,72,73]. The polymer topology and conformational asymmetry can also influence the final structure. The most recent works exploring both parameters have been conducted by Hadjichristidis et al. [74]. Concretely, they focused on the study of ABC miktoarm star terpolymers based on an α-helical mesogenic polypeptide, poly(ε-tert-butyloxycarbonyl-L-lysine) (PBLL), and polyisoprene (PI)/polystyrene (PS) as coil-like arms with a wt% composition of the PS, PI, PBLL arms of 30.1, 38.6 and 31.3, respectively. They found that the (PS)(PI)(PBLL) miktoarm formed a hierarchical smectic self-assembly where the α-helical rod-like PBLL blocks stacked to form independent peptide lamellae while PS and PI blocks constituted lamellae of rectangular cylinders. SAXS measurements led to determine the lamellar periodicity between alternating PBLL and PS/PI lamellae which was of 26 nm. According to the value obtained, their hypothesis was the interdigitation of PBLL α-helices in an antiparallel manner. This fact was the responsible to control the self-pattern observed by transmission electron and atomic force microscopies.
A more recent work, conducted by the same research group, was based on the study of the influence of the side chains of peptide miktoarm star polymers on the solubility, conformation and, therefore, self-assembly in the solid state [75]. These chimeras were constituted by poly(ε-tert-butyloxycarbonyl-L-lysine) (PBLL), as the peptide block (A), and polystyrene (PS), as the synthetic segment (B), using A2B and A2B2 topologies. It was found that PS2PBLL and PS2PBLL2, with protected L-lysine arms, adopted a lamellar block self-assembly showing a better organization (i.e., lower steric frustration) for the compound containing four arms (Scheme XI).
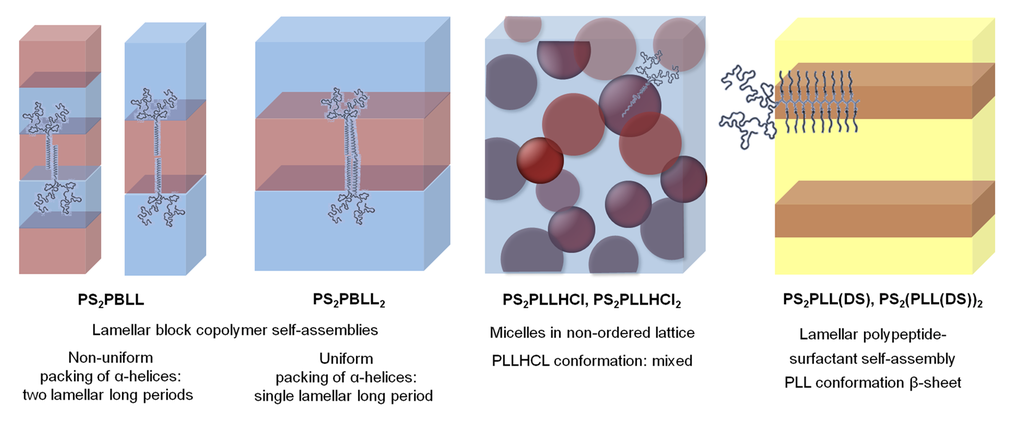
Scheme XI.
Schematic illustration of the packing of the miktoarm molecules in bulk (pink, PS; yellow, polypeptide; blue, surfactant) based on [75].
When the amine groups of poly(L-lysine) were transformed to ammonium acid salts, the supramolecular organization changed from a lamellar self-assembly to micelles in a non-ordered lattice, which was observed by transmission electron microscopy (TEM). By infrared spectroscopy, a number of different amide I bands suggested a mixture of conformations involving α-helices and β-sheets. In case of modifying the amine groups with DS surfactants (e.g., sodium 1-dodecanesulfonate, SDS) a pronounced small-scale ordering of the polypeptide-surfactant complex arms, which dominated over the formation of block copolymer scale structures, was obtained. The supramolecular organization was extracted from TEM micrographs and wide angle X-ray scattering (WAXS) measurements since the lamellar periodicity fitted well with the thickness of a single PLL-DS β-sheet surrounded on each side by a long alkyl chain of the surfactant. Results suggest that control of the extent of frustration will be possible by tunable self-assemblies caused by the steric packing constraints, imposed by the miktoarm architecture, together with the polypeptide conformation and possible intermolecular interactions.
As it has been previously described, poly(γ-benzyl-L-glutamate) (PBLG) or poly(Z-lysine) (PZlys) have the possibility to be hydrolyzed resulting in poly(L-glutamic acid) (PLGA) and poly(L-lysine) (PLL) displaying coil to helix transitions depending on the pH of the medium. In aqueous solution, asymmetrical block copolymers constituted by poly(L-lysine) and polystyrene displayed rod-like micelles in an energetically trapped state. However, in a recent study, changing the solvent and temperature, micelles were reorganized towards their intrinsically favored morphology as a consequence of the effects of the secondary structure of the peptide segment and/or the thermal properties of the synthetic block [76]. The results of this study underlined the complexity of the self-assembly behavior of peptide–synthetic hybrid block copolymers, which is determined not only by the tendency of the constituent blocks to phase separate, but also by effects of the secondary structure of the peptide segment and/or the thermal properties of the synthetic block.
An additional work towards the effect of organic acid concentration (i.e., trifluoroacetic acid (TFA) in chloroform) on the conformational stability of hybrid block copolymers based on poly(γ-benzyl-L-glutamate) (PBLG) with polystyrene (PS) attached to the N or C terminus was recently performed [77]. Although the helix-coil transformation of PBLG is a well-investigated subject, it was found that deformation of the helical structure at the C-terminal side was predominantly caused by PS attachment instead of the TFA concentration, whereas the effect of TFA addition on the helical structure was greater on the N-terminal side. Results were experimentally determined from the ratio of the helix C(α)H proton peaks to integral of all C(α)H proton peaks by 1H nuclear magnetic resonance (NMR). The work allows us to assume that the interaction of free NH groups with the side-chain carbonyl groups at the N terminus of the helix was the responsible for the different sensitivities of the PBLG chain to PS attachment and TFA addition.
In spite of the fact that the most part of the studies concerning supramolecular organization involved poly(γ-benzyl-L-glutamate) (PBLG) or poly(Z-lysine) (PZlys) as the peptidic block, other peptides with special features, derived also from natural amino acids, are currently investigated as new rod-type segments. One of these studies is related to synthesis of water soluble poly(L-proline) (PLP) to be employed to get AB or ABA block copolymers [78]. From circular dichroism (CD), infrared spectroscopy (IR) and X-ray diffraction (XRD) measurements, the two characteristic forms of poly(L-proline) were detected (i.e., form I with a low crystallinity and form II with a well-organized macromolecular structure). The transition between both forms was found to be cooperative and reversible on changing the solvent characteristics. From XRD powder data, in case of poly(ethylene oxide) diblock copolymers (i.e., PLP-PEO), the PLP peptide fully adopted the form I whereas PEO could not crystallize, implying an interfacial mixing. On the other hand the ABA triblock copolymer (i.e., A, PLP and B, PEO) mainly showed the form II of PLP and a significant crystallinity of PEO. In any case, PLP displayed the form II indicative of a left-handed helix when both architectures were dissolved in water. This form was also the most favorable for PLP hybrids with poly(L-lysine) or poly(L-glutamic acid) as blocks when they were dissolved in water or trifluoroacetic acid (TFA). The synthesis of amphiphiles containing PLP II, a hydrophilic helix independent of pH and temperature within the physiological range, was postulated to have significant applications on the formation of nanoconstructs for drug and gene delivery.
Stimuli-responsive polypeptide block copolymers, as a type of block copolymers, are very appealing systems due to their polymorphism of morphologies in selective solvents (e.g., micelles, star micelles, vesicles, tubules, fibrils or other kind of supramolecular aggregates). The most frequently used stimuli in these kinds of systems are pH, light, temperature, redox potential, magnetic field or ultrasounds [79]. The last efforts in this area have been made so as to develop new photo- or thermoresponsive systems which will be discussed below.
Mezzenga et al. [80], have recently presented a photosensitive diblock copolymer constituted by poly(glutamic acid) (PLGA) and poly(ethylene oxide) (PEO) containing spiropyran (SP) as side-chain motifs. These side-chains could undergo conformational changes (from α-helix to random coil and vice versa) with UV and visible light, respectively (Scheme XII).
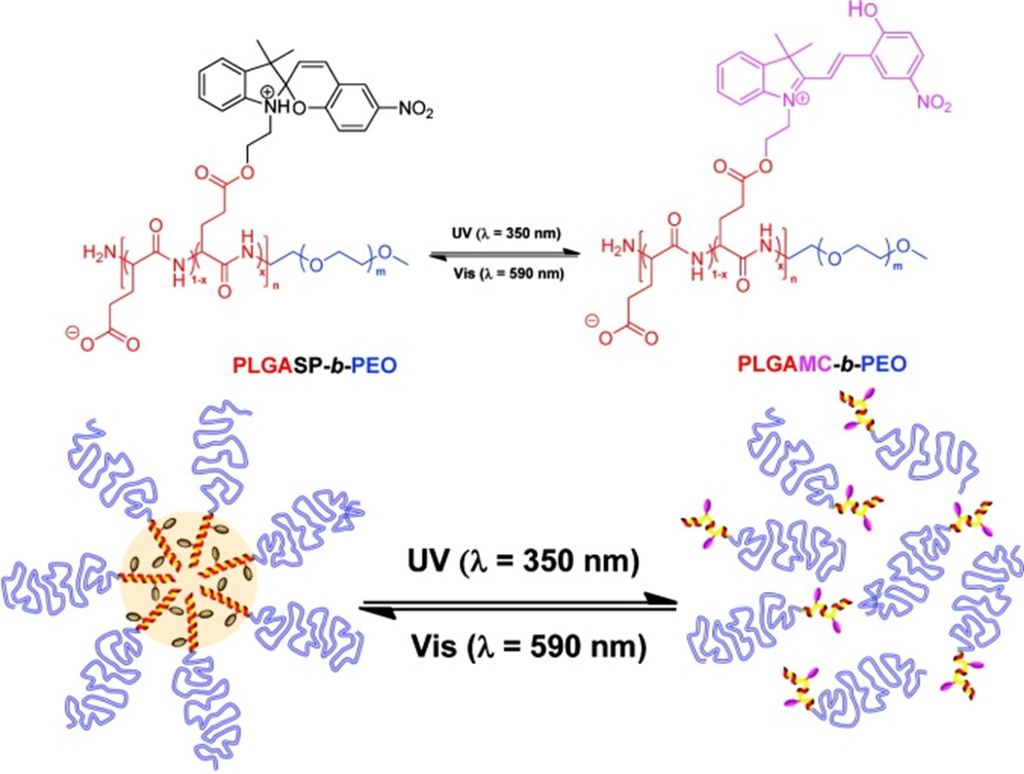
Scheme XII.
Schematic illustration of photoresponsive micellization/dissolution process for the PLGASP-b-PEO block copolymer. Reprinted with permission from [80]. Copyright (2011) American Chemical Society.

Figure 1.
First raw: cryo-TEM images of the PLGASP20-b-PEO460 sample before UV irradiation (a), after after 5 min of UV irradiation (b) and after 180 min of visible irradiation (c). Second raw: cryo-TEM images of the sample PLGASP10-b-PEO235 sample before UV irradiation (d), after 5 min of UV irradiation (e) and after 180 min of visible irradiation (f). Reprinted with permission from [80]. Copyright (2011) American Chemical Society.
The reversible aggregation-dissolution-aggregation process in water solutions in response to a suitable light exposure history can be visualized in Figure 1. Before UV irradiation, when the peptide block was mostly hydrophobic due to the close form of the spiropyran (SP), the sample showed flowerlike micellar structures composed of dark regions (i.e., PLGASP domains) and bright domains corresponding to PEO. After UV exposure (λ = 350 nm), the aggregates disrupted entirely resulting in single soluble copolymer molecules due to the switch associated with the SP form to MC form (merocyanine open form). After a period in visible light irradiation, flower-like micellar aggregates were recovered again in sizes comparable as UV prior exposure. Because the stimulus used was noninvasive UV source and the block copolymer had a bioinspired architecture, this photoresponsive rod-coil block copolymer can be used as model system to study photoinduced drug release processes or light-controlled biomedical applications.
Other recent works involved temperature as stimuli to achieve responsive systems basically based on gel-sol transformations. One of these examples was the study conducted on reverse thermal gelation of poly(alanine) end-capped poly(propylene glycol)-poly(ethylene glycol)-poly(propylene glycol) copolymers (PA-PLX-PA) [81]. The structure-property relationship of the sol-gel transition was investigated by varying PA and PLX lengths and L-Ala/DL-Ala ratio. It was found that changes in the secondary structure of PA (i.e., from random coil to β-sheet) and a decrease in molecular motion of PLX were observed by increasing the temperature. Moreover, the molecular weight of each block and the composition of PA played a critical role in determining the sol-gel transition temperature. Therefore, it was observed from the phase diagrams that when the PA block length increased, at a fix PLX length, the sol to gel transition temperature decreased. On the other side, as the hydrophilic PLX length increased at a similar PA length, the sol to gel transition temperature increased due to the increase in hydrophilicity. Moreover, when the proportion of L-Ala was higher the sol to gel transition temperature decreased, this suggested that L-Ala, constituting the hydrophobic block, was particularly effective for the gelation and the materials containing a high L-Ala/DL-Ala ratio formed easily preassembled β-sheet structures that facilitated the gel formation.
An analogous work was based on rod-coil-rod peptide-based triblock copolymers consisting of poly(γ-benzyl-L-glutamate)-b-poly(dimethylsiloxane)-b-poly(γ-benzyl-L-glutamate) (PBLG-PDMS-PBLG) [82]. In terms of sol to gel process, it was found that the critical gel concentration was 1.5 wt% in toluene at room temperature (25 °C) but around 50 °C the organogels underwent gel-solution transition. Infrared spectroscopy measurements were used to understand the influence of these compounds on the gel properties. The initial spectrum of all triblock copolymers in the solid state clearly showed absorption peaks characteristic of α-helix and β-sheet secondary structures and also to random coil or turn population. The α-helix content was stabilized on increasing the volume fraction of PBLG blocks, while the β-sheet and the random coil or turn population decreased. This behavior was also observed for the gelled samples. Interestingly, the presence of gel-like and sol-like structures was studied by diffusion experiments, where a reduced mobility of an azobenzene dye molecule and silica tracer nanoparticles was observed in the gel state compared to the solution state. This mobility was increased with the volume fraction of PBLG in the compound. Moreover the organogels had a nanofibril-like morphology with an average thickness in the range of 6–12 nm as observed by transmission electron microscopy (TEM). Taking together the results, it was postulated a supramolecular organization where PBLG rods were confined within the core of the nanofibrils, whereas PDMS coils remained exposed to and swollen by the toluene solvent molecules. The proposed packing mechanism regardless of the volume fraction of PBLG was the head-to-head morphology.
A related work towards the study of the sol to gel process in peptide hybrid block copolymers was conducted by Li et al. [83]. Specificaly, they worked with pegylated (PEG) poly-L-glutamates and studied the effect of the length of the PEG side chain on the secondary structure. The main relevant result was that the α-helix secondary structure was empowered on increasing the length of the PEG chain. Thus, the incorporation of such side chains had a significant stabilizing effect on the helical structure
Kiick et al. [84] have recently proposed the study of the morphological transformation in a dually thermoresponsive coil-rod-coil bioconjugated based on poly(diethylene glycol methyl ether methacrylate) (PDEGMEMA) linked to both ends of a triple-helix forming collagen-like peptide. The ability of the middle block to assemble as a triple helix in the hybrid conjugate was confirmed by circular dichroism since a local positive maximum around 227 nm was clearly detected. However, this triple helix conformation was gradually lost on increasing the temperature, especially over 35 °C. Above the low critical solution temperature (~35 °C), the collapse of the thermoresponsive PDEGMEMA at the ends of the peptide domain resulted in a concomitant increase in the conformational stability of the peptide domain towards thermal denaturation. Cryogenic scanning and regular transmission electron microscopies revealed the formation of spherical aggregates that increased on size at temperatures up to 65 °C and a morphological transformation into fibrils at 75 °C. It was also suggested that the transition to a fibrillar morphology occurred as a result of simple desolvation of the collagen domains, coupled with their increased conformational freedom upon unfolding, which would respectively drive and permit such molecular rearrangement. In fact, the synergistic effect of dual thermoresponsive behavior from peptide and synthetic polymer block in the hybrid appears crucial to explain the observed conformational and assembly changes.
Another recent smart strategy was based on the use of calcium ions as bioinspired trigger to reversibly control the coil to helix transition in peptide-poly(ethylene oxide) conjugates having different block lengths [85].To realize a peptide segment capable to perform a reversible coil-to-helix transition, established rules for the design of amphipathic α-helices were applied. A specific pattern of polar (i.e., glutamic acid, p) and hydrophobic (i.e., leucine, h) amino acids in a concrete sequence (i.e., [hpphppp]x) was in this case selected. Consistent with the design concept, the peptide, as well as, PEO conjugates, adopted a statistical chain segment conformation (i.e., negative Cotton effect at 198 nm) prior to the addition of calcium ions as observed by circular dichroism. However, the coil to α-helix transition (i.e., positive at 192–195, negative at 208 and 222 nm) was stimulated by the addition of different concentrations of calcium chloride. Moreover, strong competitive Ca2+ cations were added to prove the reversibility of the coil-to-helix transition. An immediate decrease in the molar ellipticities at 222 nm and 208 nm was observed which proved the rapid conformational change from α-helix to random coil (Scheme XIII). In terms of the effect of the synthetic polymer length, it was observed that short PEO chains disturbed the formation of the ordered α-helix via molecular stress effects and that on increasing the molecular weight of PEO block the random coil structure was more favored.
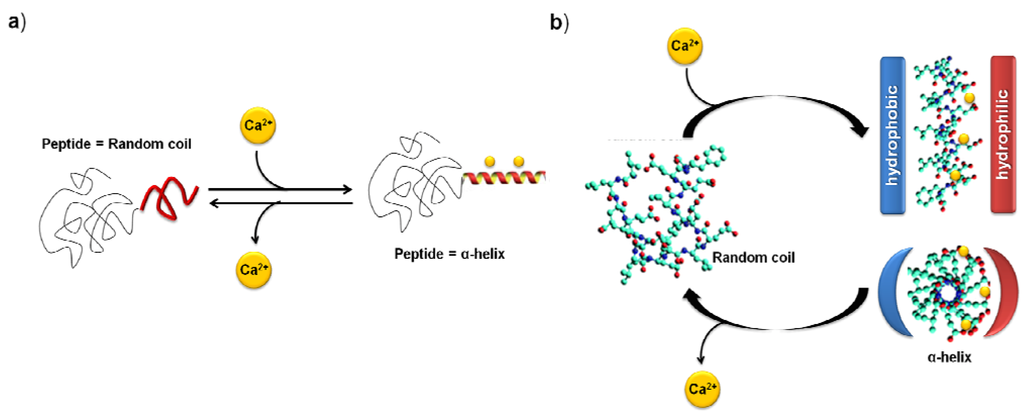
Scheme XIII.
(a) Schematic representation of the calcium ion induced coil-to-helix transition in peptide-polymer conjugates. (b) Idealized mechanism for the calcium-ion regulated coil-to-helix transition of the peptide segment conformation, based on [85].
Finally it should be indicated that the control over the secondary structure can also be addressed taking advantage of other kind of special stimuli or chemical interactions creating smart unique hybrid systems. One of these examples is the work presented by Manners et al. [86] who proposed the study of the self-assembly in organic media of tetrapeptide conjugates with polyferrocenylsilanes (PFSs) of different length. With the exception of the conjugates with longer PFS chains, all the materials exhibited only anti-parallel β-sheet structures in the solid state. Moreover, upon dissolution in toluene, all the materials with shorter metallopolymer segments, self-assembled to afford fibrous networks with peptidic cores and PFS coronas as observed from transmission electron microscopy and energy dispersive X-ray spectra. After an oxidizing treatment of the PFS with Magic Blue (i.e., [p-BrC6H4)3N][SbCl6]) or I2 and subsequent addition of Ag[PF6], the patterning of silver nanoparticles on the top of the fibrous networks was achieved.
4. Potential Applications of Peptide-Synthetic Polymer Hybrids
Mimicking the functions that biology is capable to realize such as, (i) transport, (ii) activity regulation, (iii) defined packing and unpacking of cargo, (iv) control of crystal morphogenesis, or (v) communication in complex systems, would be attractive for synthetic polymer science and would allow progress in diverse fields. So far the number of applications of biohybrid copolymers has remained to date rather limited, most of the work being focused on drug delivery applications. However the potential of such materials has been only weakly explored and important developments are emerging in other biomedical fields like tissue engineering and adaptive materials design and even in other areas like chemistry (e.g., chirality and programmable reactions) and material science (e.g., biomineralization) [50,87,88,89].
4.1. Biological Evaluations (Cytotoxicity)
The final hybrid conjugates can be classified as non-toxic and biocompatible depending on the nature of the synthetic polymer. Thus, multiple works involved methodologies so as to evaluate the cytotoxicity of the obtained synthetic copolymers in order to be sure that they can be used for biomedical applications. A recent review reported by Oh [90], was precisely focused in the recent advances concerning the synthesis and self-assembly of PLA-containing amphiphilic block copolymers (e.g., including poly(meth)acrylates, poly(ethylene glycol), polypeptides, polysaccharides, and polyurethanes) and their bio-related applications (i.e., drug delivery and imaging platforms of self-assembled nanoparticles, and tissue engineering of crosslinked hydrogels). These kind of hybrid materials appeared as a promising approach to solve the problems associated to the inherent hydrophobicity of polylactide.
Other recent studies, described in Section 2, performed also cytotoxicity evaluations of synthetic peptide-polymer hybrids. This is the case of amphiphilic poly(ethylene glycol)-block-poly(γ-cholesterol-L-glutamate) diblock nanoparticles which were tested using a MTT assay (a standard colorimetric test for the evaluation of cell viability) [24]. The test showed that for two different cell lines (i.e., human embryonic kidney cell line HEK-293 and adenocarcinomic human alveolar basal epithelial cell line A549) the viability was practically the same and close to 100%, although a slight decrease (i.e., 80%–90%) was detected when the nanoparticles concentration was high (i.e., 43.75 mg/mL). This effect was related to the fact that positive charges of conjugates may induce cytotoxicity by disrupting and solubilizing cell membranes, and even that cationic polymers may interfere with critical intracellular processes of cells.
Cytotoxicity studies using Bel7402 human hepatoma cells were also conducted on amphiphilic nanoparticles based on triblock copolymers constituted by hydrophobic polylactide and hydrophilic polylysine-poly(ethylene glycol) segments (i.e., poly(L-lactic acid)-block-poly(Z-lysine)-block-monomethoxypoly(ethylene glycol) and poly(L-lactic acid)-block-poly(L-lysine)-block-mono-methoxypoly(ethylene glycol) abbreviated as AZLE and ALE, respectively) [27]. It was found that cytotoxicity was dose- and time-dependent for both compounds. Although the cell activity decreased when the concentration of the sample increased, the degree of apoptosis caused by the deprotected ALE copolymer was low, and even at the high concentration of 100 mg/mL the cell death rate was only about 20%. The cell death rate of AZLE was relatively higher, suggesting that the use of cleavable polylysine has more advantage in biocompatibility than the protected one and that it should be useful for biological research. In fact, polylysine was positive charged in aqueous solution when deprotected and had a slight influence on the cell metabolism.
4.2. Self-Assembled Nanoparticles/Micelles for Drug/Enzyme/Gene Delivery
Polymeric micelles as drug delivery systems (DDS) offer many advantages [91,92], the most important being: i) colloidal stability with low critical micellar concentration (CMC), ii) tunable sizes with narrow size distribution, iii) ability to preserve the activities of drugs during circulation in blood, iv) high physical loading efficiency of drugs, v) bioconjugation for active targeting to specific diseased cells, and vi) controllable release of drugs upon external stimuli and degradation. Due to their size range (i.e., 10–100 nm) they are sufficiently large to avoid renal excretion, yet small enough to bypass filtration by the spleen. However, the applicability of peptide/polymer hybrids in this field has been only weakly explored since conventional block polymers where used instead of them. However as it has been commented along this review, peptides possess numerous advantages for creating new and advanced systems. For example, it is possible to have an accurate triggering and control over the different type of substance delivery (e.g., drugs, enzymes, genes) by taking into account the stimuli responsive behavior that some peptide blocks display in solution. Some of the recent advances in this field have also been commented in Section 2 [21,22,23,26,29,30,47,49,52,53,80].
Landfester et al. [93] have proposed an interesting procedure to monitor in real time the cargo release from hybrid nanocapsules induced by enzyme cleavage. Thus, polyurea/peptide hybrid nanocapsules containing a water-soluble dye (i.e., sulforhodamine 101) efficiently encapsulated were studied. The peptide sequences were specially prepared in such a way that they contained several features: the recognition sequence (Gly-Phe-Phe, GFF) for a peptidase and a surrounding FRET (Förster resonance energy transfer) system, which was attached to ε-amino groups of lysine residues. Specifically, 7-methoxycoumarin-4-yl-acetic acid (Mca) was employed as the fluorophore and 2,4-dinitrophenyl (Dnp) as a suitable quencher. In this system, emission upon excitation of the donor (fluorophore) remains unimpaired as long as the distance to the acceptor (quencher) is significantly greater than the Förster radius (1–8 nm). Decrease of the distance between donor and acceptor will result in nonradiative energy transfer from the donor to the acceptor (quenching). In the intact peptide sequence, the mutual distance is below the Förster radius, and thus FRET, which is a common tool in the detection of enzymatic activity, is highly effective. Cleavage of the peptide sequence will lead to a fluorescence signal due to the increasing distance between donor and acceptor that can be detected in situ.
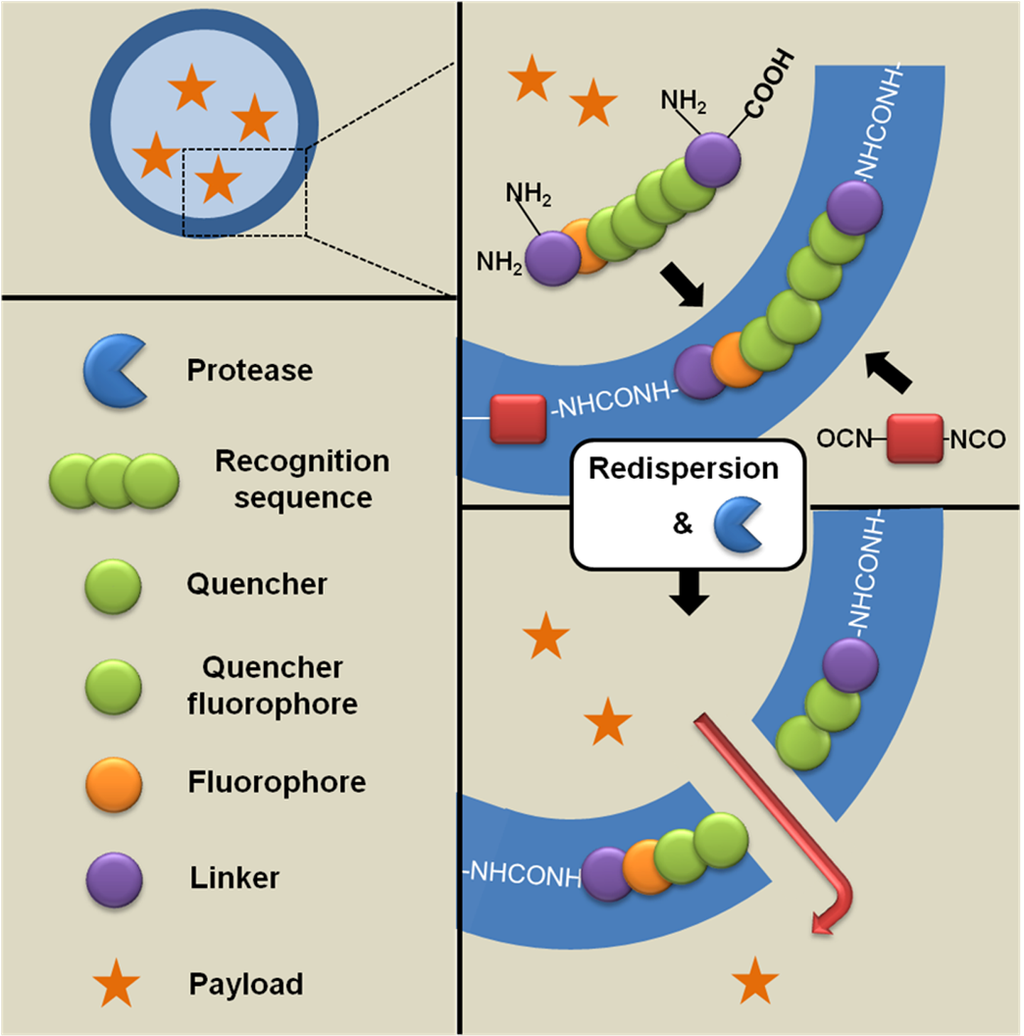
Scheme XIV.
Cargo release from peptide-based hybrid nanocapsules induced by enzyme cleavage based on [93].
The cleavage of the peptide sequence by using trypsin as an enzymatic triggering was effectively monitored (by means of the FRET system), while the liberation of encapsulated dye was simultaneously detected by fluorescence recovery upon release to aqueous environment (Scheme XIV).
The influence of the synthetic parameters on the size of the polymer chains, the morphology of the capsules and the encapsulation efficiency could be effectively studied by the given approach. Moreover, results clearly indicated that the fluorescence of the dye increased simultaneously to the Mca fluorescence only when the enzyme was added. Therefore, it was possible to infer that the dye was released from the capsules, and that its fluorescence was no longer quenched because of encapsulation. The proposed approach opens interesting perspectives for specific enzyme-triggered drug delivery using nanostructured materials.
The same methodology was also used for studying hydrophobic crosslinked nanoparticles (i.e., 230 nm of diameter), based on peptide-functionalized polystyrene [94]. Again the peptide sequences contained the recognition sequence (Gly-Phe-Phe, GFF) for the peptidase and a FRET (Förster resonance energy transfer) system in order to detect the enzymatic degradation.
Klok et al. [95] provided another interesting contribution towards the design of polymer therapeutics. In this case, the cargo (i.e., dye) was attached to a polymer backbone, based on poly(N-(2-hydroxypropyl)methacrylamide (PHPMA), via a noncovalent, biologically inspired, coiled coil linker. Specifically, two complementary peptide sequences were linked to the polymer carrier and the dye, respectively. This approximation differs from those based on polymer-drug conjugates, in which the drug was typically attached via an enzymatically or hydrolytically cleavable linker. It was demonstrated that coiled coil based peptide linkers may not only be useful to bind and release guests but may also play an active role in enhancing and directing intracellular transport and trafficking. These constructs may have particular interest for the cytosolic delivery of biomolecular therapeutics (i.e., the delivery of peptide, protein, and oligonucleotide drugs that act in the cytosol) because coiled coil peptide sequences are known to be involved in various intracellular vesicle and viral membrane fusion processes, which may facilitate cytosolic escape. The proposed new noncovalent polymer therapeutics may offer several potential advantages, including facile access to combination therapeutics and rapid production of compound libraries to screen for structure-activity relationships.
Interestingly, the work was performed using peptide sequences (instead of real drug molecules) that were functionalized with various dye labels to allow FRET experiments. Thus, binding and release in model experiments and in cells could be monitored and the cell uptake visualized and studied. Concretely, 5-((2-aminoethyl)amino)naphthalene-1-sulfonic acid and 4-((4-(dimethylamino)phe-nyl)azo)benzoic acid functionalized peptides were prepared. FRET experiments pointed out that guest molecules were effectively bounded to the PHPMA backbone during cell uptake, and that guest release started at ~60 min incubation time. Because enzymatic degradation was not found to be significant on the time scale of experiments, release could only be attributed to intracellular unfolding of the linker.
4.3. Crosslinked Hydrogels and Scaffolds for Tissue Engineering
Hidrogels are three-dimensional networks crosslinked with hydrophilic polymers. They possess biocompatibility, the possibility to store high water content and can be tuned chemically and physically exhibiting good mechanical properties. Due to these unique properties, hydrogels have great potential in the development of useful biomaterials, principally for biological and biomedical applications [96]. In addition, peptides, proteins, poly(amino acid)s, and hybrid macromolecules spontaneously undergo biorecognition-driven self assembly into hydrogel structures. Developments that may be expected in the near future include: design of hydrogels with multiple functions, hydrogels sensitive to stimuli, hydrogels with programmable responses, and hydrogels translating substrate recognition into mechanical action [97].
Peptide-synthetic polymer hybrids are also gaining attention in three dimensional cell culture applications taking into account, for example, that reverse thermal gelling polymer aqueous solutions undergoes sol-to-gel transition as the temperature increases. Therefore, these systems can be used for the formation of drug or cell encapsulating 3D hydrogel matrix by mixing drugs or cells in a low viscous sol state, followed by increasing temperature. Growth factors can be also simply incorporated, and by injecting the polymer aqueous solution, it can easily take the shape of the cavity or the disease site, avoiding the use of surgical procedures.
Related to the last studies performed on peptide-synthetic polymer hybrid hydrogels usable as injectable scaffolds, two recent works developed by Jeong et al. [98,99] merit attention. In this way, a thermogelling system, in which the solution to gel transformation occurred from 10 °C to 37 °C, was used as an injectable in situ gelling system into the subcutaneous layer of rats. The system consisted on poly(ethylene glycol)-poly(L-alanine-co-L-phenylalanine) grafted chitosan (CS-g-(PAF-PEG)) where specifically phenylalanine units were incorporated in the peptide segment to strengthen its hydrophobicity. This polymer was able to form micelles with diameters ranging from 10 to 50 nm at low temperatures and large aggregates ranging from hundreds to thousands of nanometers in size as the temperature increased. In vivo assays demonstrated that the polymer formed a gel depot in situ in the subcutaneous layer. The apparent size (by volume) of the gel decreased to 30%–40% of the first day to seventh day, and the gel was completely cleared in 14 days from the implanted site. From H&E (hematoxylin and eosin) stained images, it was possible to observe blood capillaries inside the gel as well as collagen capsules around the implant, indicating marginal responses to the gel and good tissue compatibility of the gel.
A similar thermogelling system (PAF-PEG) [99] was used to evaluate the effect of the stereochemistry (i.e., D-alanine/D-phenylalanine (PAF-D-PEG), non-natural amino acids or L-alanine/L-phenylalanine, (PAF-L-PEG) naturally occurring amino acids) on the in vitro/in vivo degradation profiles, and tissue compatibility. Stereochemistry was found to play an important role in the polymer degradation from both in vitro assays using enzyme solutions and in vivo assays considering the duration of the gel in the subcutaneous layer of rats. Thus, PAF-L-PEG was degraded in vitro by cathepsin B, and elastase, whereas PAF-D-PEG was quite stable against all the studied proteases (including collagenase). For both compounds, a transparent hydrogel was again formed upon the subcutaneous injection of the PAF-PEG aqueous polymer solution due to the temperature-sensitive sol-to-gel transition. The PAF-L-PEG gel was decreased in size after 15 days of implantation whereas a tight gel of PAF-D-PEG remained after this period of time suggesting the preferential biodegradation of PAF-L-PEG. Moreover, the histocompatibility around the in situ formed gels of PAF-L-PEG and PAF-D-PEG was also compared by the H&E staining method. Collagen capsules were observed around the gel for both systems. However, the collagen capsule was thicker for PAF-D-PEG than PAF-L-PEG, indicating milder tissue irritation or acute inflammation for material constituted by natural L-amino acids.
Polyalanine-poloxamer-polyalanine (PA-PLX-PA) block copolymers have been developed for this kind of application [100]. Specifically hybrids were constituted by a segment where a central hydrophilic moiety of poly(ethylene oxide) was flanked by two hydrophobic moieties of poly(propylene oxide). Peptides derived from L-alanine, DL-alanine, and their mixture (L/DL-alanine) were used in order to to control the secondary structure as well as their resulting fibrous nanostructure. The resulting hydrogel was proved to be an excellent 3D system for the proliferation and differentiation of chondrocytes. With a reverse thermal gelling property, current polypeptide aqueous solution appears a very promising minimally-invasive injectable tissue engineering scaffold. Jeong and coworkers [101] observed that long range nanofibrous orientation was attained by a self-assembling ionic complex between the above explained (+)-charged amphiphilic peptide block copolymer (PA-PLX-PA) and a (-)-charged hyaluronic acid (HA). Cell proliferation and biomarker expression for articular cartilage were significantly improved when the PA-PLX-PA/HA complex system was employed. In particular, noticeable cell clustering was observed in the PA-PLX-PA/HA complex system with the long range nanofibrous structure. This point is highly interesting since suggests a new method for developing a nanofibrous structure using an amphiphilic peptide block copolymer and demonstrates its potential uses as a unique biomimetic cell-culture matrix.
A poly(ethylene glycol)-b-poly(L-alanine) (PEG-L-PA) gel encapsulating fibroblasts was investigated for wound healing as a new thermogel application [102]. The fibroblasts were encapsulated by the temperature sensitive sol-to-gel transition of the polymer aqueous solution. Under the in vitro three-dimensional (3D) cell culture condition, the new thermogel was comparable with a commercially available control (i.e., a soluble extract of basement membrane proteins derived from the rat tumor tissue named Matrigel) for cell proliferation and was significantly better than this control for collagen types I and III formation. Furthermore, wound healing experiments demonstrated that PEG-L-PA not only accelerated the wound closure but also improved epithelialization and the formation of skin appendages such as keratinocytelayer (epidermis), hair follicles, and sebaceous glands.
Other related works based on peptide/poly(ethylene glycol) (PEG) or other copolymer hydrogels have been carried out lately. For example, thermoresponsive hydrogels constituted of a telechelic PEG end-capped with hydrophobic dipeptides (i.e., phenylalanine-phenylalanine (Phe-Phe) or tyrosine-tyrosine (Tyr-Tyr) were proposed to be used in responsive delivery or encapsulation for biomedical applications [103]. In another study, PEG was replaced with an amphiphilic ABA-block copolymer (Pluronic F127) consisting in PEG end blocks and a poly(propylene oxide) (PPO) center block, which was able to self-assemble into micelles with a water-incompatible compartment that was segregated from the aqueous exterior by the PEG shell. An alanine (A)-based peptide (AK2) doped with charged lysines (K) with a sequence of (AKA3KA)2 was selected from the crosslinking regions of the natural elastin as a starting point for the hydrogel formation. It was prepared via covalent linkages between the terminal vinyl sulfone groups of the synthetic block copolymer and the lysine amines of the peptide [104]. By heating up to 60 °C the Michael-type addition reaction was accelerated and led to the formation of stable viscoelastic hydrogels. Furthermore, the indicated temperature enhanced the fibril assembly of peptide via an antiparallel arrangement of the β-sheets through hydrophobic interactions. These interactions were established between the alanine residues laterally stabilized by hydrogen bonding between lysine and peptide carbonyls of adjacent polypeptide chains. To assess the applicability of the hybrid hydrogels for vocal fold tissue engineering, the stiffness (i.e., elastic modulus) and the degree of elasticity (i.e., loss tangent) of the gels were tested by torsional wave analysis showing good mechanical resistance and stability. However the major drawback was the incompatibility of the crosslinking process with mammalian cells.
The indicated limitation was subsequently overcome by just adjusting the peptide sequence [105] and specifically by adding arginylglycylaspartic acid groups (RGD) that enhanced cell adhesion. Thus, elastin-mimetic hybrid polymers (EMHPs) with an alternating multiblock architecture were again obtained by a step-growth polymerization method. In this case, PEG was employed as a synthetic polymer that was covalently linked to different AK2 peptides that incorporated the indicated RGD sequences. Crosslinking between lysine amine groups was performed by using hexamethylene diisocyanate and hydrogels with a compressive modulus of 1.06 ± 0.1 MPa were subsequently derived. Interestingly, EMHPs having RGD groups exhibited increased helical structure compared with the peptides alone, likely owing to H-bonding between the arginine residues with the oxygen atoms in PEG. It was also observed that neonatal human dermal fibroblasts (NHDFs) were able to adhere to the hydrogels within 1 h of culture and to spread and develop F-actin filaments after 24 h. Furthermore, NHDF proliferation was only observed on hydrogels containing RGD domains, demonstrating the importance of integrin engagement for cell growth. In this way, the synthesized cell-instructive hybrid copolymers are promising candidates as elastomeric scaffolds for tissue engineering since not only were strong and elastic but also could support cell adhesion and growth.
Finally, another interesting and recent work related to smart biohydrogels was conducted by Guan et al. [106], who reported the preparation of peptide-saccharide hydrogels through a vinyl sulfone-thiol conjugated addition under mild conditions. In order to evaluate the viability of mesenchymal stem cells (MSCs) in the hydrogels a qualitative Live/Dead staining analysis (Invitrogen) was performed. After 1, 7 and 14 days in vitro at pH 7, the viability of MSCs encapsulated in the hydrogels was found to be greater than 90%, which was directly related to differentiation potential and metabolic activity. Thus, due to the non-cytotoxicity (i.e., good cell viability) of the 3D samples and biodegradability (i.e., they were composed of natural building blocks), the peptide-saccharide hydrogels can be used as a platform for culturing MSCs. Furthermore, the new hydrogels have the potential to be further functionalized to provide biophysical and biochemical cues to encapsulated cells and precisely control cell function and fates.
5. Conclusions
Design and synthesis of peptide-synthetic polymer block copolymers constitute nowadays a highly dynamic field since new applications and properties are sought out for such hybrid materials. Recent developments in peptide-polymer block copolymer synthesis are mainly focused to cover two main goals: i) to increase the molecular level control of the hybrids (i.e., control over the block length and molar mass dispersity of both synthetic polymer and peptide segments); and ii) to broaden the available architectures of the hybrid copolymers. The control over intra- and intermolecular interactions of the designed peptide blocks is also crucial to get conjugates able to interact with other (bio)molecules in a well defined manner.
Just as there are many polymers and peptides with widely differing properties available to make up these chimera molecules, there are likewise many different synthetic approaches to create the hybrid conjugates. Over the past decade, impressive advances have been made in the synthesis of such chimeras. Specifically, new strategies for the ring-opening polymerization of amino acid N-carboxy-anhydrides now provide an easy access to peptide-synthetic polymer hybrids and other more complex architectures with a precise control over both polymer molecular weight and chain architecture. The examples given in this paper only supply a flavor of the possibilities offered by these synthetic advances, being obvious that these new developments have not yet been fully explored.
The importance of producing site-specific modifications on both peptides and synthetic polymers, as well as, controlling the supramolecular organization and the drastic changes or enhancements should be clear when applications related to stimuli-responsiveness, self-assembly and bioactivity are considered. In particular, new peptide-polymer hybrids are crucial for the development of novel biomolecular drugs, new biosensor materials and bioactive hybrid hydrogels that can aid in wound healing and tissue regeneration. The enhanced possibilities given for the highly precise synthesis of peptide-synthetic polymer hybrids provide new and unique opportunities to control the structure and final properties of these materials, which can result in new characteristics with contemporary and unforeseen relevance.
The capability of peptide-polymer hybrids to spontaneously undergo biorecognition-driven self assembly into hydrogel structures is probably the basis of the most promising biomedical applications. Next developments that may be expected concern to the design of hydrogels with multiple functions, hydrogels sensitive to stimuli, hydrogels with programmable responses, and hydrogels translating substrate recognition into mechanical action. Furthermore, peptide-synthetic polymer hybrid hydrogels appear an ideal system to get minimally-invasive injectable tissue engineering scaffolds.
Acknowledgments
This work has been supported by grants from MINECO/FEDER and AGAUR (MAT2009-11503, MAT2012-36205, 2009SGR-1208).
References
- Schlaad, H.; Antonietti, M. Block copolymers with amino acid sequences: molecular chimeras of polypeptides and synthetic polymers. Eur. Phys. J. E 2003, 10, 17–23. [Google Scholar] [CrossRef]
- Klok, H.A.; Ayres, L. Peptide Hybrid Polymers; Klok, H.A., Schlaad, H., Eds.; Springer: Berlin, Germany, 2006. [Google Scholar]
- Börner, H.G. Strategies exploiting functions and self-assembly properties of bioconjugates for polymer and materials sciences. Prog. Polym. Sci. 2009, 34, 811–851. [Google Scholar] [CrossRef]
- Lutz, J.F.; Börner, H.G. Modern trends in polymer bioconjugates design. Prog. Polym. Sci. 2008, 33, 1–39. [Google Scholar] [CrossRef]
- Gallot, B. Comb-like and block liquid crystalline polymers for biological applications. Prog. Polym. Sci. 1996, 21, 1035–1088. [Google Scholar] [CrossRef]
- Langer, R.; Tirrell, D.A. Designing materials for biology and medicine. Nature 2004, 428, 487–491. [Google Scholar]
- Gauthier, M.A.; Klok, H.A. Peptide/protein-polymer conjugates: Synthetic strategies and design concepts. Chem. Commun. 2008, 23, 2591–2611. [Google Scholar] [CrossRef]
- Le Droumaguet, B.; Velonia, K. Click Chemistry: A powerful tool to create polymer-based macromolecular chimeras. Macromol. Rapid. Commun. 2008, 29, 1073–1089. [Google Scholar] [CrossRef]
- Klok, H.A. Peptide/Protein-synthetic polymer conjugates: Quo Vadis. Macromolecules 2009, 42, 7990–8000. [Google Scholar] [CrossRef]
- Marsden, H.R.; Kros, A. Polymer-peptide block copolymers—An overview and assessment of synthesis methods. Macromol. Biosci. 2009, 9, 939–951. [Google Scholar] [CrossRef]
- Deming, T.J. Polypeptide and polypeptide hybrid copolymer synthesis via NCA polymerization. In Peptide Hybrid Polymers; Klok, H.A., Schlaad, H., Eds.; Springer: Berlin, Germany, 2006. [Google Scholar]
- König, H.M.; Kilbinger, A.F.M. Learning from nature: β-Sheet-mimicking copolymers get organized. Angew. Chem. Int. Ed. 2007, 46, 8334–8340. [Google Scholar] [CrossRef]
- van Hest, J.C.M. Biosynthetic-synthetic polymer conjugates. Polym. Rev. 2007, 47, 63–92. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Iatrou, H.; Pitsikalis, M.; Sakellariou, G. Synthesis of well-defined polypeptide-based materials via the ring-opening polymerization of α-amino acid N-carboxyanhydrides. Chem. Rev. 2009, 109, 5528–5578. [Google Scholar] [CrossRef]
- Habraken, G.J.M.; Heise, A.; Thornton, P.D. Block copolypeptides prepared by N-carboxyanhydride ring-opening polymerization. Macromol. Rapid Commun. 2011, 33, 272–286. [Google Scholar] [CrossRef]
- Yamashita, Y.; Iwaya, Y.; Ito, K. Block copolymerization, 9. Polymerization of the NCA of methyl D-glutamate by telechelic polystyrene having glycyl groups as active chain ends. Makromol. Chem. 1975, 176, 1207–1216. [Google Scholar] [CrossRef]
- Tang, H.; Li, Y.; Lahasky, S.H.; Sheiko, S.S.; Zhang, D. Core-shell molecular bottlebrushes with helical polypeptide backbone: Synthesis, characterization, and solution conformations. Macromolecules 2011, 44, 1491–1499. [Google Scholar] [CrossRef]
- Wang, J.; Lu, H.; Ren, Y.; Zhang, Y.; Morton, M.; Chen, J.; Lin, Y. Interrupted helical structure of grafted polypeptides in brush-like macromolecules. Macromolecules 2011, 44, 8699–8708. [Google Scholar] [CrossRef]
- Li, W.; Zhang, X.; Wang, J.; Qiao, X.; Liu, K.; Zhang, A. Peptidic molecular brushes with enhanced chirality. J. Polym. Sci. A Polym. Chem. 2012, 50, 4063–4072. [Google Scholar] [CrossRef]
- Jang, J.H.; Choi, Y.M.; Choi, Y.Y.; Joo, M.K.; Park, M.H.; Choi, B.G.; Kang, E.Y.; Jeong, B. pH/temperature sensitive chitosan-g-(PA-PEG) aqueous solutions as new thermogelling systems. J. Mater. Chem. 2011, 21, 5484–5491. [Google Scholar]
- Byrne, M.; Thornton, P.D.; Cryan, S.A.; Heise, A. Star polypeptides by NCA polymerisation from dendritic initiators: synthesis and enzyme controlled payload release. Polym. Chem. 2012, 3, 2825–2831. [Google Scholar] [CrossRef]
- Xia, J.; Chen, L.; Chen, J.; Tian, H.; Li, F.; Zhu, X.; Li, G.; Chen, X. Hydrophobic polyphenylalanine-grafted hyperbranched polyethyleneimine and its in vitro gene transfection. Macromol. Biosci. 2011, 11, 211–218. [Google Scholar] [CrossRef]
- Liu, G.; Dong, C.M. Photoresponsive Poly(S-(o-nitrobenzyl)-L-cysteine)-b-PEO from a L-cysteine N-carboxyanhydride monomer: Synthesis, self-assembly, and phototriggered drug release. Biomacromolecules 2012, 13, 1573–1583. [Google Scholar] [CrossRef]
- Wu, Y.; Ma, Q.; Song, X.R.; Zheng, Y.; Ren, W.; Zhang, J.K.; Ouyang, L.; Wu, F.B.; He, G. Biocompatible poly(ethylene glycol)-poly(γ-cholesterol-L-glutamate) copolymers: Synthesis, characterization, and in vitro studies. J. Polym. Sci. A Polym. Chem. 2012, 50, 4532–4537. [Google Scholar] [CrossRef]
- Peng, Y.L.; Huang, Y.; Chuang, H.J.; Kuo, C.Y.; Lin, C.C. Synthesis and characterization of biodegradable polylactides and polylactide-block-poly(Z-lysine) copolymers. Polymer 2010, 51, 4329–4335. [Google Scholar] [CrossRef]
- Jacobs, J.; Pound-Lana, G.; Klumperman, B. Poly(N-vinylpyrrolidone-b-(gamma-benzyl-L-glutamate))—synthesis and self-assembly into pH-sensitive micelles. Polym. Chem. 2012, 3, 2551–2560. [Google Scholar] [CrossRef]
- Xiang, L.; Shen, L.J.; Long, F.; Yang, K.; Fan, J.B.; Li, Y.J.; Xiang, J.N.; Zhu, M.Q. A convenient method for the synthesis of the amphiphilic triblock copolymer poly(L-lactic acid)-block-poly(L-lysine)-block-poly(ethylene glycol) monomethyl ether. Macromol. Chem. Phys. 2011, 212, 563–573. [Google Scholar] [CrossRef]
- You, Y.; Chen, Y.; Hua, C.; Dong, C.M. Synthesis and thermoreversible gelation of dendron-like polypeptide/linear poly(ε-caprolactone)/dendron-like polypeptide triblock copolymers. J. Polym. Sci. A Polym. Chem. 2010, 48, 709–718. [Google Scholar] [CrossRef]
- Ray, J.G.; Naik, S.S.; Hoff, E.A.; Johnson, A.J.; Ly, J.T.; Easterling, C.P.; Patton, D.L.; Savin, D.A. Stimuli-responsive peptide-based ABA-triblock copolymers: Unique morphology transitions with pH. Macromol. Rapid Commun. 2012, 33, 819–826. [Google Scholar] [CrossRef]
- Wang, K.; Luo, G.F.; Liu, Y.; Li, C.; Cheng, S.X.; Zhuo, R.X.; Zhang, X.Z. Redox-sensitive shell cross-linked PEG-polypeptide hybrid micelles for controlled drug release. Polym. Chem. 2012, 3, 1084–1090. [Google Scholar] [CrossRef]
- Pintauer, T.; Matysjaszewski, K. Atom transfer radical addition and polymerization reactions catalyzed by ppm amounts of copper complexes. Chem. Soc. Rev. 2008, 6, 1087–1097. [Google Scholar] [CrossRef]
- Boyer, C.; Bulmus, V.; Davis, T.P.; Ladmiral, V.; Liu, J.; Perrier, S. Bioapplications of RAFT polymerization. Chem. Rev. 2009, 11, 5402–5436. [Google Scholar]
- Gregory, A.; Stenzel, M.H. Complex polymer architectures via RAFT polymerization: From fundamental process to extending the scope using click chemistry and nature’s building blocks. Prog. Polym. Sci. 2012, 37, 38–105. [Google Scholar] [CrossRef]
- Grubbs, R.B. Nitroxide-mediated radical polymerization: Limitations and versatility. Polym. Rev. 2011, 51, 104–137. [Google Scholar] [CrossRef]
- Habraken, G.J.M.; Peeters, M.; Thornton, P.D.; Koning, C.E.; Heise, A. Selective enzymatic degradation of self-assembled particles from amphiphilic block copolymers obtained by the combination of N-carboxyanhydride and nitroxide-mediated polymerization. Biomacromolecules 2011, 12, 3761–3769. [Google Scholar] [CrossRef]
- Knoop, R.J.I.; de Geus, M.; Habraken, G.J.M.; Koning, C.E.; Menzel, H.; Heise, A. Stimuli responsive peptide conjugated polymer nanoparticles. Macromolecules 2010, 43, 4126–4132. [Google Scholar] [CrossRef]
- Merrifield, R.B. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Merrifield, R.B. Solid phase synthesis. Angew. Chem. Int. Ed. Engl. 1985, 10, 799–810. [Google Scholar] [CrossRef]
- White, P.D.; Chan, W.C. Basic principles. In Fmoc Solid Phase Peptide Synthesis: A Practical Approach; Chan, W.C., White, P.D., Eds.; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Becker, H.; Lucas, H.W.; Maul, J.; Pillai, V.N.R.; Anzinger, H.; Mutter, M. Polyethyleneglycols onto crosslinked polystyrenes: A new class of hydrophilic polymeric supports for peptide synthesis. Makromol. Chem. Rapid Commun. 1982, 3, 217–223. [Google Scholar] [CrossRef]
- Burkoth, T.S.; Benzinger, T.L.S.; Jones, D.N.S.; Hallenga, K.; Meredith, S.C.; Lynn, D.G. C-terminal PEG blocks the irreversible step in β-amyloid(10-35) fibrillogenesis. J. Am. Chem. Soc. 1998, 120, 7655–7656. [Google Scholar]
- Rosler, A.; Klok, H.A.; Hamley, I.W.; Castelletto, V.; Mykhaylyk, O.O. Nanoscale structure of poly(ethylene glycol) Hybrid block copolymers containing amphiphilic β-strand peptide sequences. Biomacromolecules 2003, 4, 859–863. [Google Scholar] [CrossRef]
- Hamley, I.W.; Krysmann, M.J. Effect of PEG crystallization on the self-assembly of PEG/peptide copolymers containing amyloid peptide fragments. Langmuir 2008, 24, 8210–8214. [Google Scholar] [CrossRef]
- Eckhardt, D.; Groenewolt, M.; Krause, E.; Börner, H.G. Rational design of oligopeptide organizers for the formation of poly(ethylene oxide) nanofibers. Chem. Commun. 2005, 2814–2816. [Google Scholar]
- Castelletto, V.; McKendrick, J.E.; Hamley, I.W.; Olsson, U.; Cenker, C. PEGylated amyloid peptide nanocontainer delivery and release system. Langmuir 2010, 26, 11624–11627. [Google Scholar] [CrossRef]
- Trimaille, T.; Mabrouk, K.; Monnier, V.; Charles, L.; Bertin, D.; Gigmes, D. SG1-Functionalized peptides as precursors for polymer-peptide conjugates: A straightforward approach. Macromolecules 2010, 43, 4864–4870. [Google Scholar] [CrossRef]
- de Graaf, A.J.; Mastrobattista, E.; Vermonden, T.; van Nostrum, C.F.; Rijkers, D.T.S.; Liskamp, R.M.J.; Hennink, W.E. Thermosensitive peptide-hybrid ABC block copolymers obtained by ATRP: Synthesis, self-assembly, and enzymatic degradation. Macromolecules 2012, 45, 842–851. [Google Scholar] [CrossRef]
- Paira, T.K.; Banerjee, S.; Mandal, T.K. Peptide-poly(ε-caprolactone) biohybrids by grafting-from ring-opening polymerization: synthesis, aggregation and crystalline properties. J. Polym. Sci. A Polym. Chem. 2012, 50, 2130–2141. [Google Scholar] [CrossRef]
- Paira, T.K.; Banerjee, S.; Raula, M.; Kotal, A.; Si, S.; Mandal, T.K. Peptide-polymer bioconjugates via atom transfer radical polymerization and their solution aggregation into hybrid micro/nanospheres for dye uptake. Macromolecules 2010, 43, 4050–4061. [Google Scholar] [CrossRef]
- Fuks, G.; Talom, R.M.; Gauffre, F. Biohybrid block copolymers: Towards functional micelles and vesicles. Chem. Soc. Rev. 2011, 40, 2475–2493. [Google Scholar] [CrossRef]
- Shi, H.; Liu, L.; Wang, X.; Li, J. Glycopolymer-peptide bioconjugates with antioxidant activity via RAFT polymerization. Polym. Chem. 2012, 3, 1182–1188. [Google Scholar] [CrossRef]
- Koga, T.; Iimura, M.; Higashi, N. Novel peptide-shelled dendrimer with dramatically changeable thermo-responsive character. Macromol. Biosci. 2012, 12, 1043–1047. [Google Scholar] [CrossRef]
- Singha, N.K.; Gibson, M.I.; Koiry, B.P.; Dainal, M.; Klok, H.A. Side-chain peptide-synthetic polymer conjugates via tandem “Ester-Amide/Thiol-Ene” post-polymerization modification of poly(pentafluorophenyl methacrylate) obtained using ATRP. Biomacromolecules 2011, 12, 2908–2913. [Google Scholar] [CrossRef]
- Tsurkan, M.V.; Chwalek, K.; Levental, K.R.; Freudenberg, U.; Werner, C. Modular starPEG-heparin gels with bifunctional peptide linkers. Macromol. Rapid Commun. 2010, 31, 1529–1533. [Google Scholar] [CrossRef]
- Börner, H.G.; Schlaad, H. Bioinspired functional block copolymers. Soft Matter 2007, 3, 394–408. [Google Scholar] [CrossRef]
- Hamley, I. Block copolymers in solution. In Fundamentals and Applications; Wiley: Chichester, UK, 2007. [Google Scholar]
- Yesodha, S.K.; Pillai, C.K.S.; Tsutsumi, N. Stable polymeric materials for nonlinear optics: A review based on azobenzene systems. Prog. Polym. Sci. 2004, 29, 45–74. [Google Scholar] [CrossRef]
- Cho, M.J.; Choi, D.H.; Sullivan, P.A.; Akelaitis, A.J.P.; Dalton, L.R. Recent progress in second-order nonlinear optical polymers and dendrimers. Prog. Polym. Sci. 2008, 33, 1013–1058. [Google Scholar] [CrossRef]
- Cornelissen, J.; Fischer, M.; Sommerdijk, N.; Nolte, R.J.M. Helical superstructures from charged poly(styrene)-poly(isocyanodipeptide) block copolymers. Science 1998, 280, 1427–1430. [Google Scholar]
- Klok, H.A.; Langenwalter, J.F.; Lecommandoux, S. Self-assembly of peptide-based diblock oligomers. Macromolecules 2000, 33, 7819–7826. [Google Scholar] [CrossRef]
- Hung, J.H.; Lin, Y.L.; Sheng, Y.J.; Tsao, H.K. Structure-photophysical property relationship of conjugated rod-coil block copolymers in solutions. Macromolecules 2012, 45, 2166–2170. [Google Scholar] [CrossRef]
- Antonietti, M.; Förster, S. Vesicles and liposomes: A self-assembly principle beyond lipids. Adv. Mater. 2003, 15, 1323–1333. [Google Scholar] [CrossRef]
- Chécot, F.; Lecommandoux, S.; Gnanou, Y.; Klok, H.A. Water-soluble stimuli-responsive vesicles from peptide-based diblock copolymers. Angew. Chem. Int. Ed. 2002, 41, 1339–1343. [Google Scholar] [CrossRef]
- Kukula, H.; Schlaad, A.; Antonietti, M.; Förster, S. The formation of polymer vesicles or “peptosomes” by polybutadiene-block-poly(L-glutamate)s in dilute aqueous solution. J. Am. Chem. Soc. 2002, 124, 1658–1663. [Google Scholar] [CrossRef]
- Holowka, E.P.; Sun, V.Z.; Kamei, D.T.; Deming, T.J. Polyarginine segments in block copolypeptides drive both vesicular assembly and intracellular delivery. Nat. Mater. 2007, 6, 52–57. [Google Scholar] [CrossRef]
- Lecommandoux, S.; Rodríguez-Hernández, J. Reversible inside-out micellization of pH-responsive and water-soluble vesicles based on polypeptide diblock copolymers. J. Am. Chem. Soc. 2005, 127, 2026–2027. [Google Scholar] [CrossRef]
- Zhou, Q.H.; Zheng, J.K.; Shen, Z.H.; Fan, X.H.; Chen, X.F.; Zhou, Q.F. Synthesis and hierarchical self-assembly of rod rod block copolymers via click chemistry between mesogen-jacketed liquid crystalline polymers and helical polypeptides. Macromolecules 2010, 43, 5637–5646. [Google Scholar]
- Hermes, F.; Otte, K.; Brandt, J.; Grawert, M.; Borner, H.G.; Schlaad, H. Polypeptide-based organogelators: Effects of secondary structure. Macromolecules 2011, 44, 7489–7492. [Google Scholar] [CrossRef]
- Kakwere, H.; Payne, R.J.; Jolliffe, K.A.; Perrier, S. Self-assembling macromolecular chimeras: Controlling fibrillization of a beta-sheet forming peptide by polymer. Soft Matter 2011, 7, 3754–3757. [Google Scholar] [CrossRef]
- Klok, H.A.; Langenwalter, J.F.; Lecommandoux, S. Self-assembly of peptide-based diblock oligomers. Macromolecules 2000, 33, 7819–7826. [Google Scholar] [CrossRef]
- Losik, M.; Kubowicz, S.; Smarsly, B.; Schlaad, H. Solid-state structure of polypeptide-based rod-coil block copolymers: Folding of helices. Eur. Phys. J. E. 2004, 15, 407–411. [Google Scholar] [CrossRef]
- Schlaad, H.; Smarsly, B.; Losik, M. The role of chain-length distribution in the formation of solid-state structures of polypeptide-based rod-coil block copolymers. Macromolecules 2004, 37, 2210–2214. [Google Scholar] [CrossRef]
- Sanchez-Ferrer, A.; Mezzenga, R. Secondary structure-induced micro- and macrophase separation in rod-coil polypeptide diblock, triblock, and star-block copolymer. Macromolecules 2010, 43, 1093–1100. [Google Scholar] [CrossRef]
- Junnila, S.; Houbenov, N.; Hanski, S.; Iatrou, H.; Hirao, A.; Hadjichristidis, N.; Ikkala, O. Hierarchical smectic self-assembly of an ABC miktoarm star terpolymer with a helical polypeptide arm. Macromolecules 2010, 43, 9071–9076. [Google Scholar]
- Junnila, S.; Houbenov, N.; Karatzas, A.; Hadjichristidis, N.; Hirao, A.; Iatrou, H.; Ikkala, O. Side-chain-controlled self-assembly of polystyrene-polypeptide miktoarm star copolymers. Macromolecules 2012, 45, 2850–2856. [Google Scholar] [CrossRef]
- Nuhn, H.; Klok, H.A. Aqueous solution self-assembly of polystyrene-b-poly(L-lysine) diblock oligomers. Eur. Polym. J. 2011, 47, 782–791. [Google Scholar] [CrossRef]
- Itoh, T.; Hatanaka, T.; Ihara, E.; Inoue, K. Helix-coil transformation of poly(gamma-benzyl-L-glutamate) with polystyrene attached to the N or C terminus in trifluoroacetic acid-chloroform mixtures. Polym. J. 2012, 44, 189–194. [Google Scholar] [CrossRef]
- Gkikas, M.; Iatrou, H.; Thomaidis, N.S.; Alexandridis, P.; Hadjichristidis, N. Well-defined homopolypeptides, copolypeptides, and hybrids of poly(L-proline). Biomacromolecules 2011, 12, 2396–2406. [Google Scholar] [CrossRef]
- Rapoport, N. Physical stimuli-responsive polymeric micelles for anti-cancer drug delivery. Prog. Polym. Sci. 2007, 32, 962–990. [Google Scholar] [CrossRef]
- Kotharangannagari, V.K.; Sánchez-Ferrer, A.; Ruokolainen, J.; Mezzenga, R. Photoresponsive reversible aggregation and dissolution of rod-coil polypeptide diblock copolymers. Macromolecules 2011, 44, 4569–4573. [Google Scholar] [CrossRef]
- Oh, H.J.; Joo, M.K.; Sohn, Y.S.; Jeong, B. Secondary structure effect of polypeptide on reverse thermal gelation and degradation of L/DL-poly(alanine)-poloxamer-L/DL-poly(alanine) copolymers. Macromolecules 2008, 41, 8204–8209. [Google Scholar] [CrossRef]
- Kotharangannagari, V.K.; Sanchez-Ferrer, A.; Ruokolainen, J.; Mezzenga, R. Thermoreversible gel-sol behavior of rod-coil-rod peptide-based triblock copolymers. Macromolecules 2012, 45, 1982–1990. [Google Scholar] [CrossRef]
- Chen, C.Y.; Wang, Z.H.; Li, Z.B. Thermoresponsive polypeptides from pegylated poly-L-glutamates. Biomacromolecules 2011, 12, 2859–2863. [Google Scholar] [CrossRef]
- Krishna, O.D.; Wiss, K.T.; Luo, T.Z.; Pochan, D.J.; Theato, P.; Kiick, K.L. Morphological transformations in a dually thermoresponsive coil-rod-coil bioconjugate. Soft Matter 2012, 8, 3832–3840. [Google Scholar]
- Kühnle, R.I.; Gebauer, D.; Borner, H.G. Calcium ions as bioinspired triggers to reversibly control the coil-to-helix transition in peptide-polymer conjugates. Soft Matter 2011, 7, 9616–9619. [Google Scholar] [CrossRef]
- Tangbunsuk, S.; Whittell, G.R.; Ryadnov, M.G.; Vandermeulen, G.W.M.; Woolfson, D.N.; Manners, I. Metallopolymer-peptide hybrid materials: synthesis and self-assembly of functional, polyferrocenylsilane-tetrapeptide conjugates. Chem. Eur. J. 2012, 18, 2524–2535. [Google Scholar]
- Börner, H.G. Precision polymers—modern tools to understand and program macromolecular interactions. Macromol. Rapid Commun. 2011, 32, 115–126. [Google Scholar] [CrossRef]
- Rabotyagova, O.S.; Cebe, P.; Kaplan, D.L. Protein-based block copolymers. Biomacromolecules 2011, 12, 269–289. [Google Scholar] [CrossRef]
- Börner, H.G.; Kuhnle, H.; Hentschel, J. Making “smart polymers” smarter: Modern concepts to regulate functions in polymer science. J. Polym. Sci. A Polym. Chem. 2010, 48, 1–14. [Google Scholar]
- Oh, J.K. Polylactide (PLA-based) amphiphilic block copolymers: Synthesis, self-assembly, and biomedical applications. Soft Matter 2011, 7, 5096–5108. [Google Scholar] [CrossRef]
- Harada, A.; Kataoka, K. Supramolecular assemblies of block copolymers in aqueous media as nanocontainers relevant to biological applications. Prog. Polym. Sci. 2006, 31, 949–982. [Google Scholar] [CrossRef]
- Tyrrell, Z.L.; Shen, Y.Q.; Radosz, M. Fabrication of micellar nanoparticles for drug delivery through the self-assembly of block copolymers. Prog. Polym. Sci. 2010, 35, 1128–1143. [Google Scholar] [CrossRef]
- Andrieu, J.; Kotman, N.; Maier, M.; Mailander, V.; Strauss, W.S.L.; Weiss, C.K.; Landfester, K. Live monitoring of cargo release from peptide-based hybrid nanocapsules induced by enzyme cleavage. Macromol. Rapid Commun. 2012, 33, 248–253. [Google Scholar] [CrossRef]
- Maier, M.; Kotman, N.; Friedrichs, C.; Andrieu, J.; Wagner, M.; Graf, R.; Strauss, W.S.L.; Mailander, V.; Weiss, C.K.; Landfester, K. Highly site specific, protease cleavable, hydrophobic peptide-polymer nanoparticles. Macromolecules 2011, 44, 6258–6267. [Google Scholar]
- Apostolovic, B.; Deacon, S.P.E.; Duncan, R.; Klok, H.A. Hybrid polymer therapeutics incorporating bioresponsive, coiled coil peptide linkers. Biomacromolecules 2010, 11, 1187–1195. [Google Scholar] [CrossRef]
- Ramos, J.; Imaz, A.; Callejas-Fernandez, J.; Barbosa-Barros, L.; Estelrich, J.; Quesada-Perez, M.; Forcada, J. Soft nanoparticles (thermo-responsive nanogels and bicelles) with biotechnological applications: from synthesis to simulation through colloidal characterization. Soft Matter 2011, 7, 5067–5082. [Google Scholar]
- Kopecek, J.; Yang, J.Y. Smart self-assembled hybrid hydrogel biomaterials. Angew. Chem. Int. Ed. 2012, 51, 7396–7417. [Google Scholar] [CrossRef]
- Kang, E.Y.; Moon, H.J.; Joo, M.K.; Jeong, B. Thermogelling chitosan-g-(PAF-PEG) aqueous solution as an injectable scaffold. Biomacromolecules 2012, 13, 1750–1757. [Google Scholar] [CrossRef]
- Kang, E.Y.; Yeon, B.; Moon, H.J.; Jeong, B. PEG-l-PAF and PEG-d-PAF: Comparative study on thermogellation and biodegradation. Macromolecules 2012, 45, 2007–2013. [Google Scholar] [CrossRef]
- Choi, B.G.; Park, M.H.; Cho, S.H.; Joo, M.K.; Oh, H.J.; Kim, E.H.; Kwideok Park, K.; Han, D.K.; Jeong, B. In situ thermal gelling polypeptide for chondrocytes 3D culture. Biomaterials 2010, 31, 9266–9272. [Google Scholar] [CrossRef]
- Min Hee Park, M.H.; Bo Gyu Choi, B.G.; Jeong, B. Complexation-induced biomimetic long range fibrous orientation in a rigid-flexible block copolymer thermogel. Adv. Funct. Mater. 2012, 22, 5118–5125. [Google Scholar] [CrossRef]
- Yun, E.J.; Yon, B.; Joo, M.K.; Jeong, B. Cell Therapy for skin wound using fibroblast encapsulated poly(ethylene glycol)-poly(L-alanine) thermogel. Biomacromolecules 2012, 13, 1106–1111. [Google Scholar] [CrossRef]
- Hamley, I.W.; Cheng, G.; Castelletto, V. A thermoresponsive hydrogel based on telechelic PEG end-capped with hydrophobic dipeptides. Macromol. Biosci. 2011, 11, 1068–1078. [Google Scholar] [CrossRef]
- Grieshaber, S.E.; Nie, T.; Yan, C.Q.; Zhong, S.; Teller, S.S.; Clifton, R.J.; Pochan, D.J.; Kiick, K.L.; Jia, X.Q. Assembly properties of an alanine-rich, lysine-containing peptide and the formation of peptide/polymer hybrid hydrogels. Macromol. Chem. Phys. 2011, 212, 229–239. [Google Scholar] [CrossRef]
- Grieshaber, S.E.; Farran, A.J.E.; Bai, S.; Kiick, K.L.; Jia, X.Q. Tuning the properties of elastin mimetic hybrid copolymers via a modular polymerization method. Biomacromolecules 2012, 13, 1774–1786. [Google Scholar] [CrossRef]
- Chawla, K.; Yu, T.B.; Liao, S.W.; Guan, Z.B. Biodegradable and biocompatible synthetic saccharide-peptide hydrogels for three-dimensional stem cell culture. Biomacromolecules 2011, 12, 560–567. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).