Functionalized Polymers from Lignocellulosic Biomass: State of the Art

Abstract
:
{kind=link}
{kind=link}
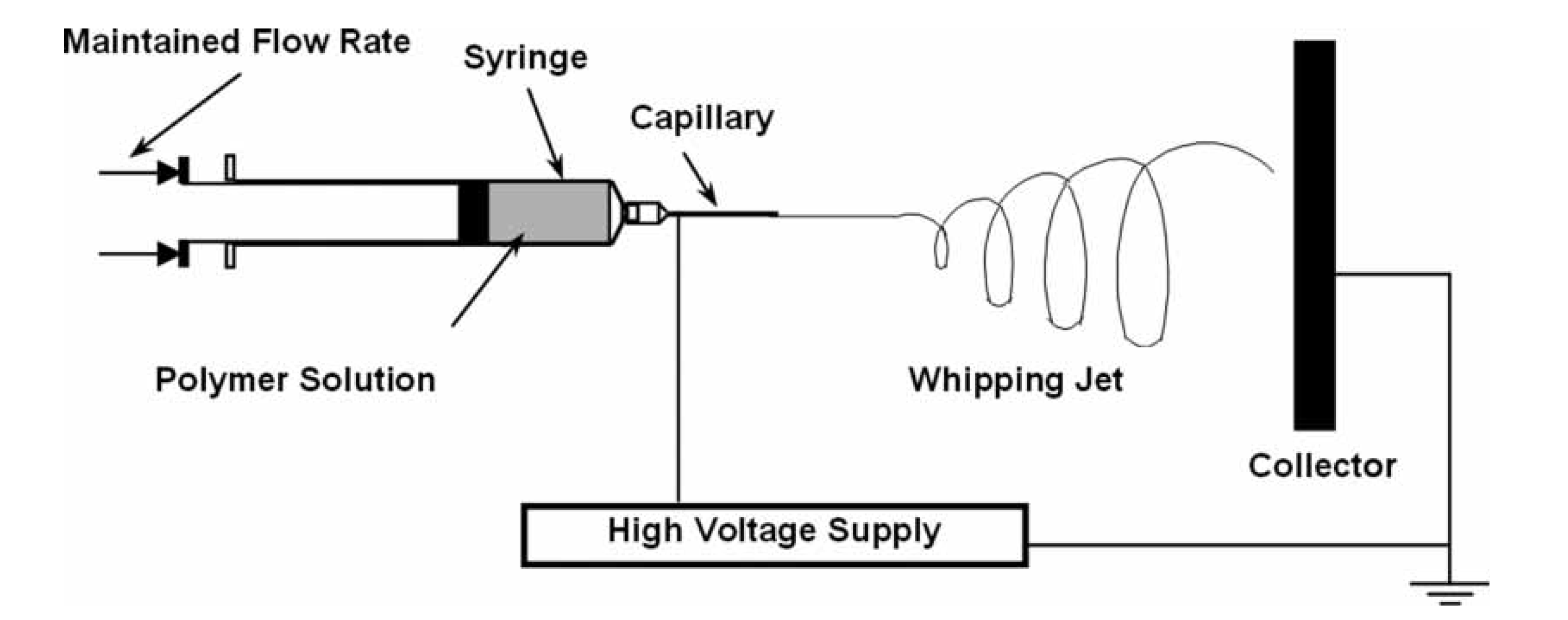{kind=link}
{kind=link}
{kind=link}
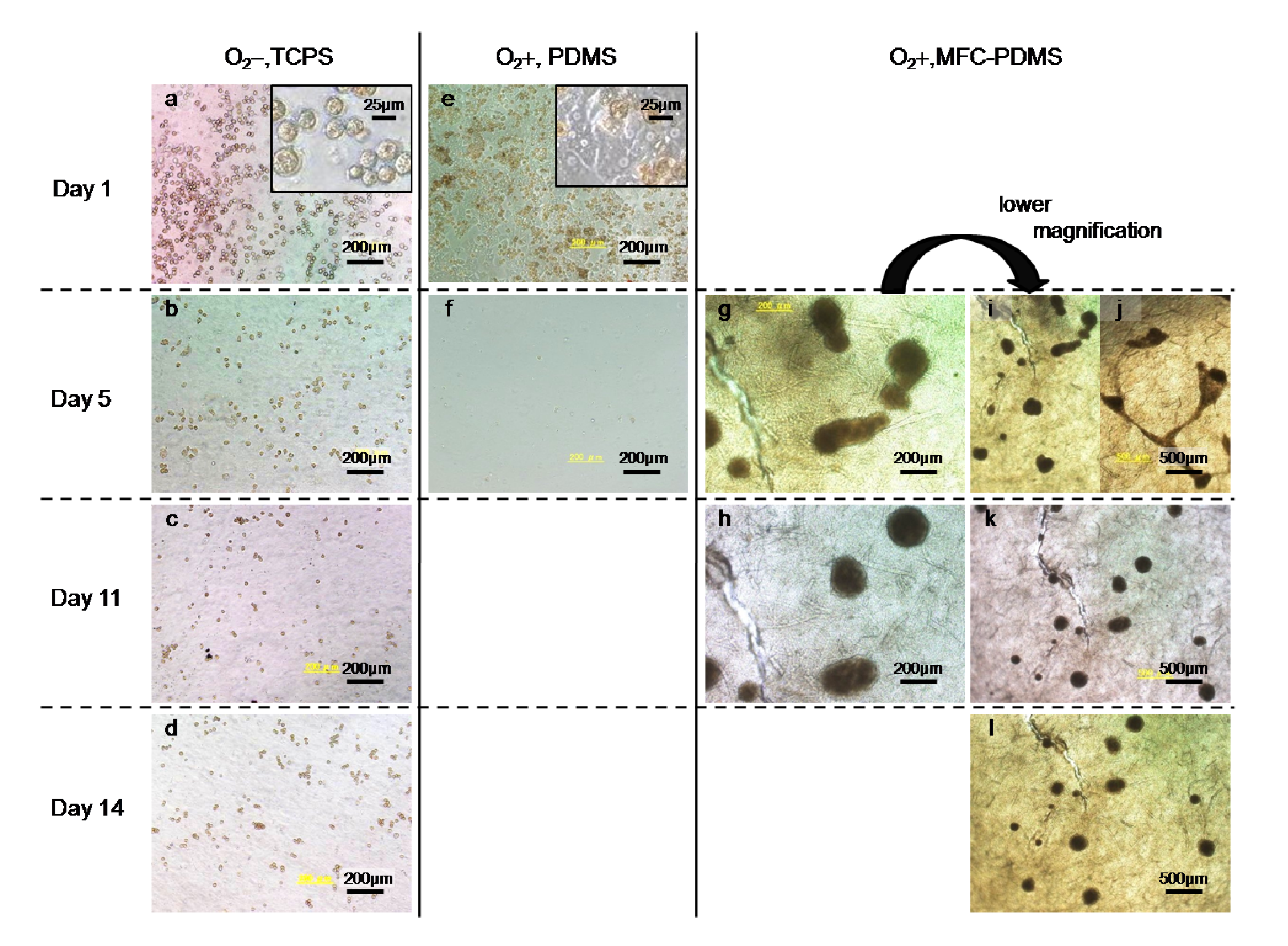{kind=link}
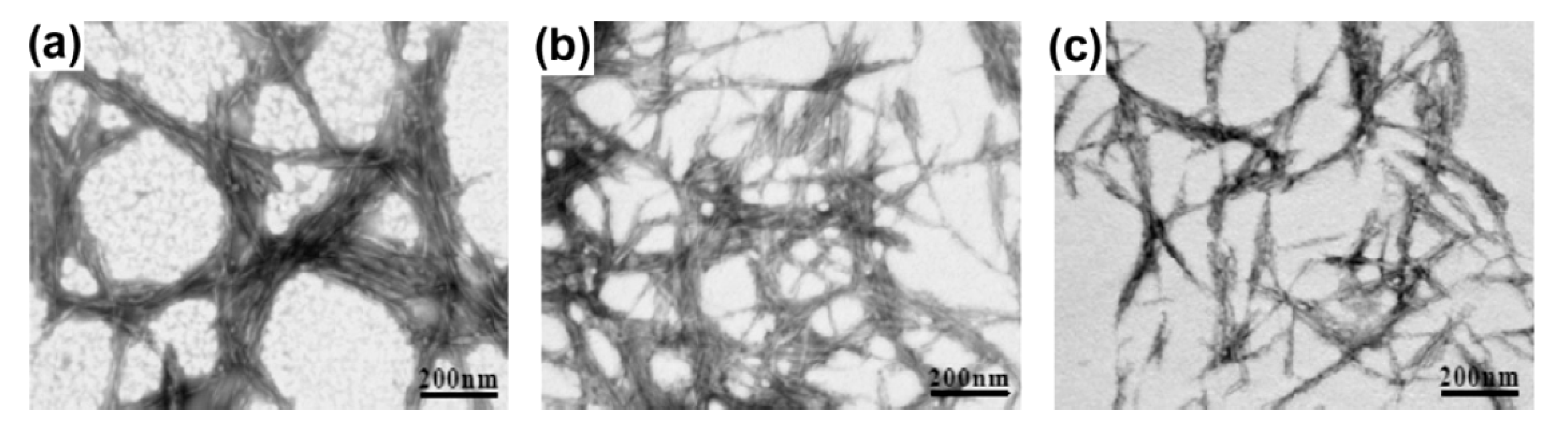{kind=link}
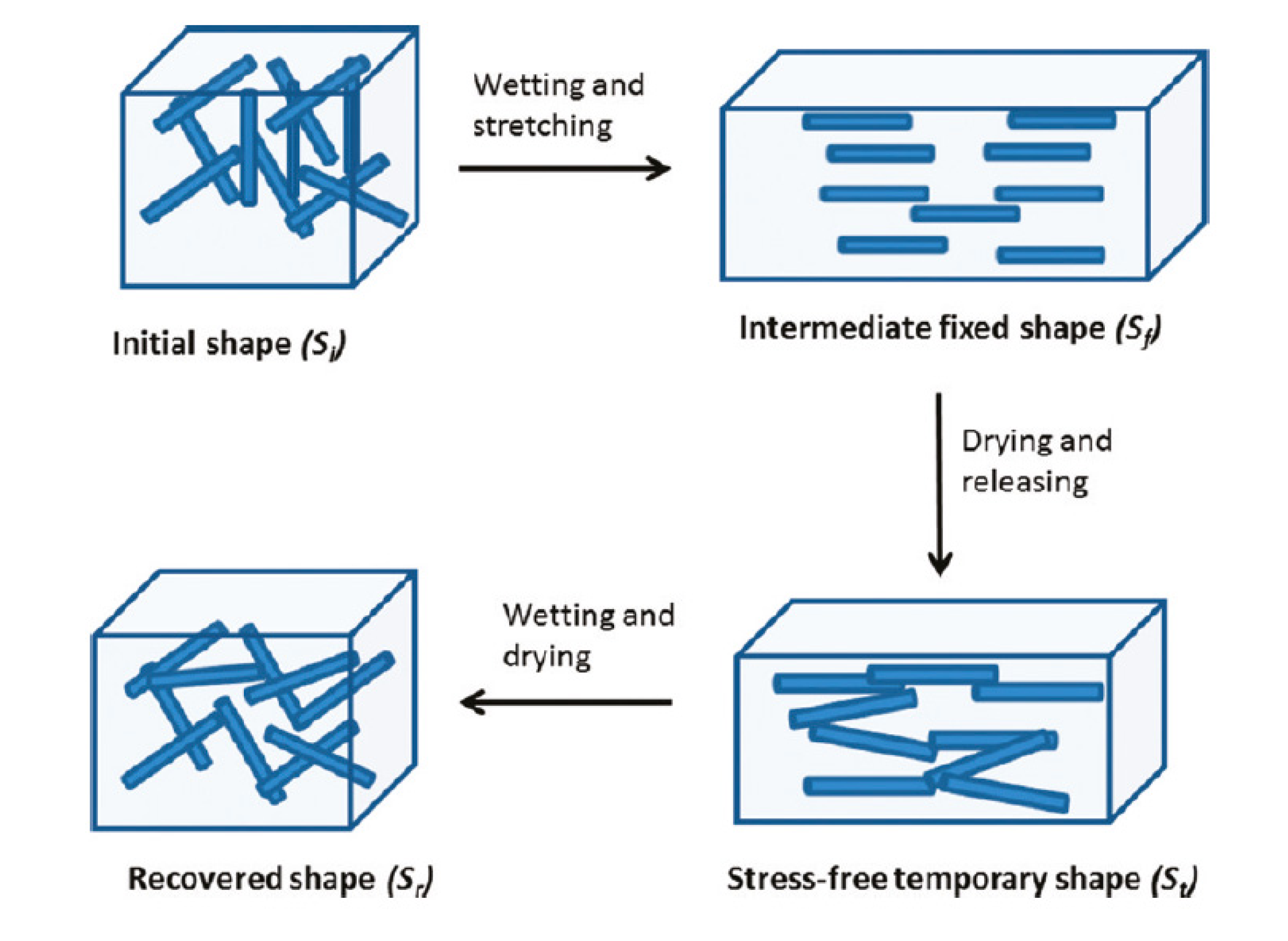{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Bio-Based Polymers Can Lead the Way to a Future with Reduced Reliance on Fossil Fuels
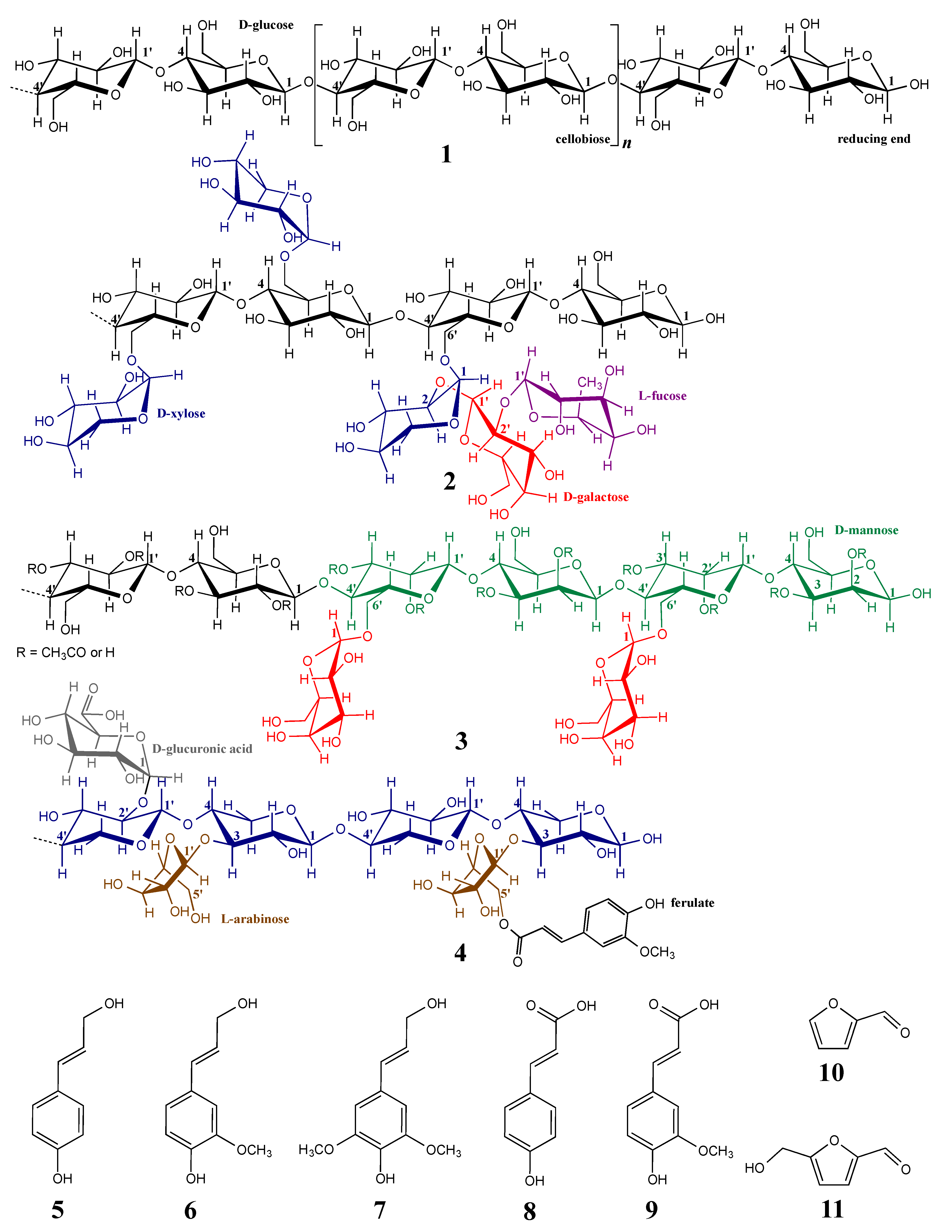
1.2. Lignocellulosic Biomass Consists of Different Cell Wall Polymers
1.3. The Biorefinery
1.4. Bio-Based Polymers from Plant Cell Walls
- (1)
- The cell wall polymers themselves. In this case cellulose, hemicellulosic polysaccharides or lignin are isolated, processed, and converted to end products.
- (2)
- Fermentable sugars generated in a biorefinery. The production of second-generation cellulosic biofuels relies on the deconstruction of the cell wall polysaccharides into monomeric hexose and pentose sugars that are then converted to fuels such as ethanol or butanol via microbial fermentation [21]. The use of different microbial strains enables the production of chemical feedstocks that can be used for the production of bio-based products, including polymers, with poly-lactic acid (PLA) used for bio-degradable plastics currently representing the main application [28,29].
- (3)
- The waste stream of the biorefinery or paper mill. In the former, these residues include most of the lignin, and a portion (~5%) of the cellulose that is resistant to deconstruction, along with monomeric sugars that cannot be converted microbially, and compounds formed from the monomeric sugars during processing (e.g., furfural, HMF), as well as various extractives. In the case of paper mills, the waste stream consists primarily of lignin and extractives. Both the lignin and cellulose can be used for the production of biopolymers and composites. The remaining compounds are often not present in high enough concentrations to enable cost-effective recovery.
2. Cellulose Fibers and Composites
2.1. Cellulose Acetate
2.2. Microfibrillated Cellulose and Cellulose Nanocrystals for Use in Composites

2.2.1. Polyelectrolyte Multilayers

2.2.2. Superoleophobic Surfaces
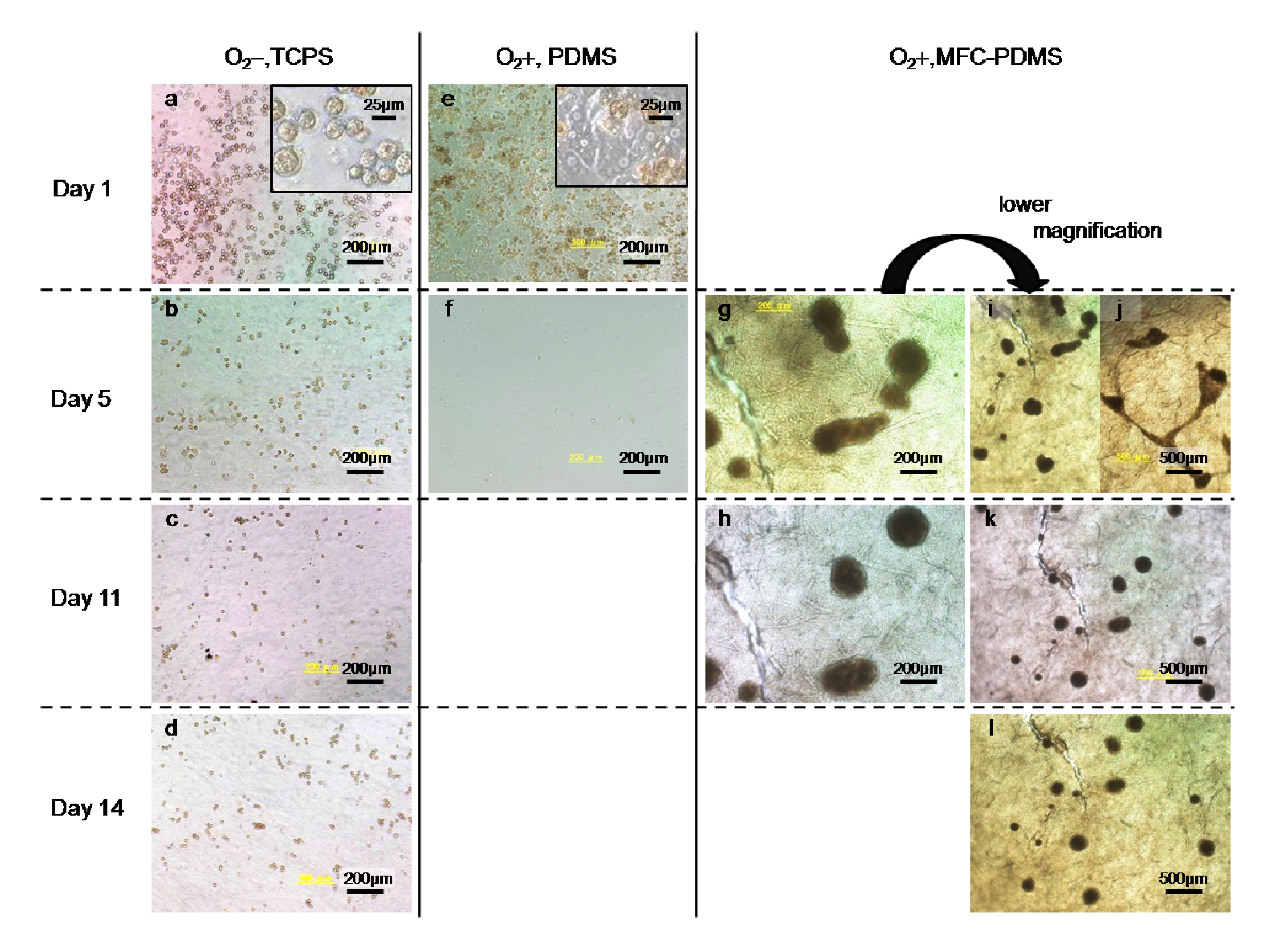
2.2.3. MFC as a Matrix for Hepatocyte Cell Cultures
2.2.4. Micro-Electro-Mechanical Systems
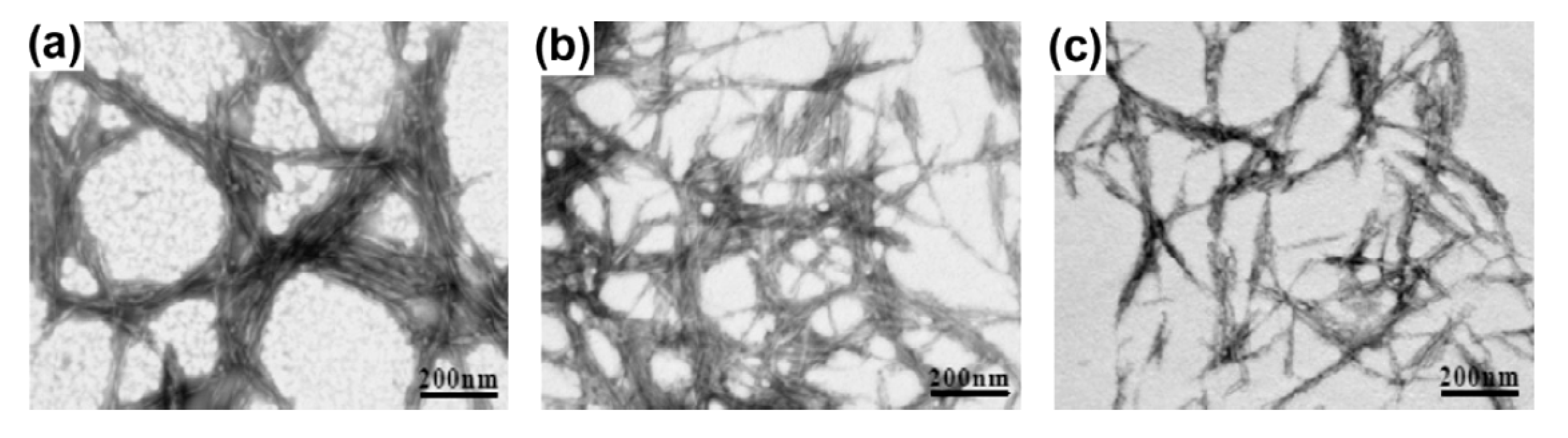
2.3. Cellulose Nanocrystals/Nanowhiskers

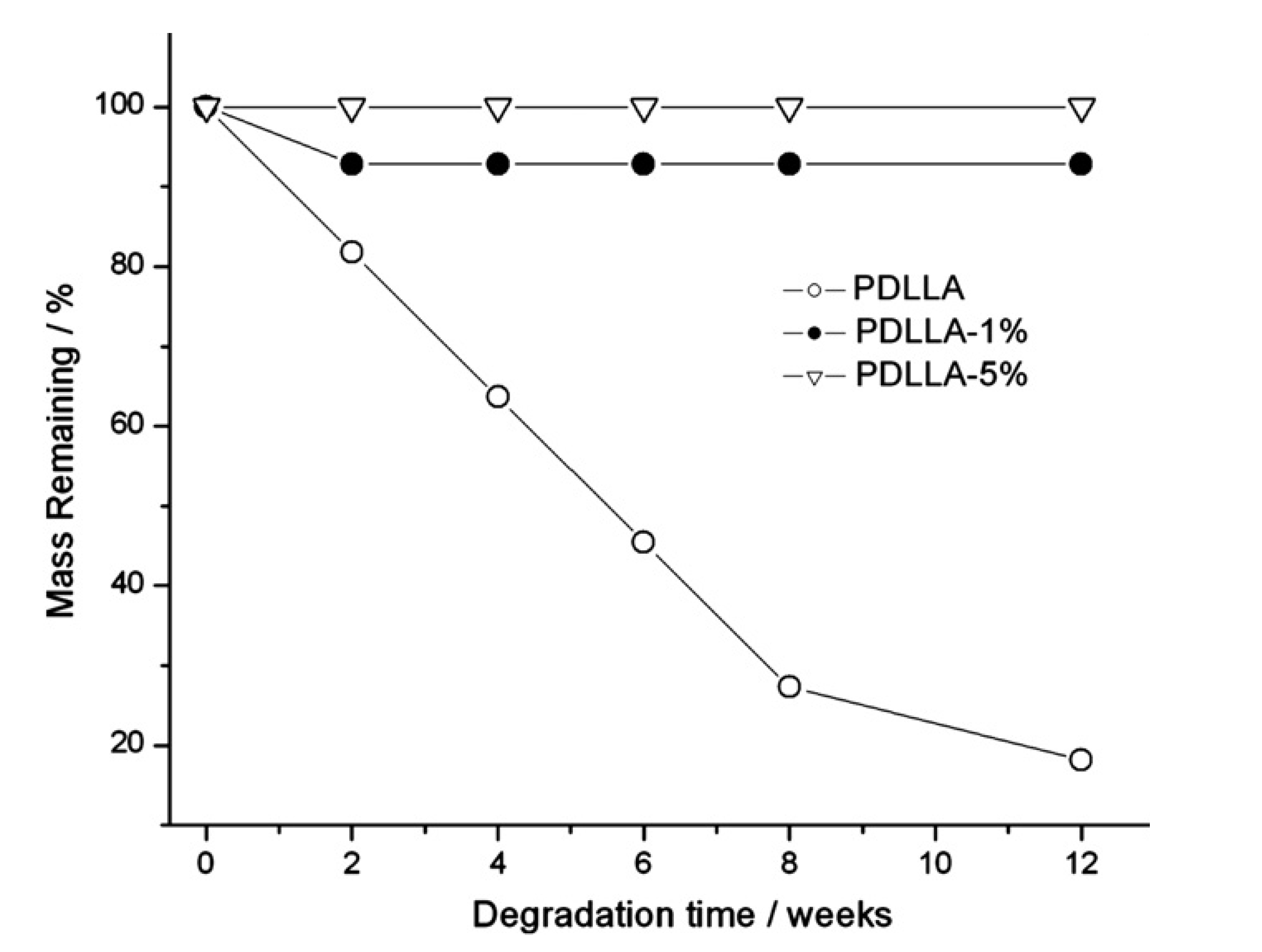
2.3.1. CNW Biodegradability
3. Hemicellulosic Polysaccharides
3.1. Packaging Films
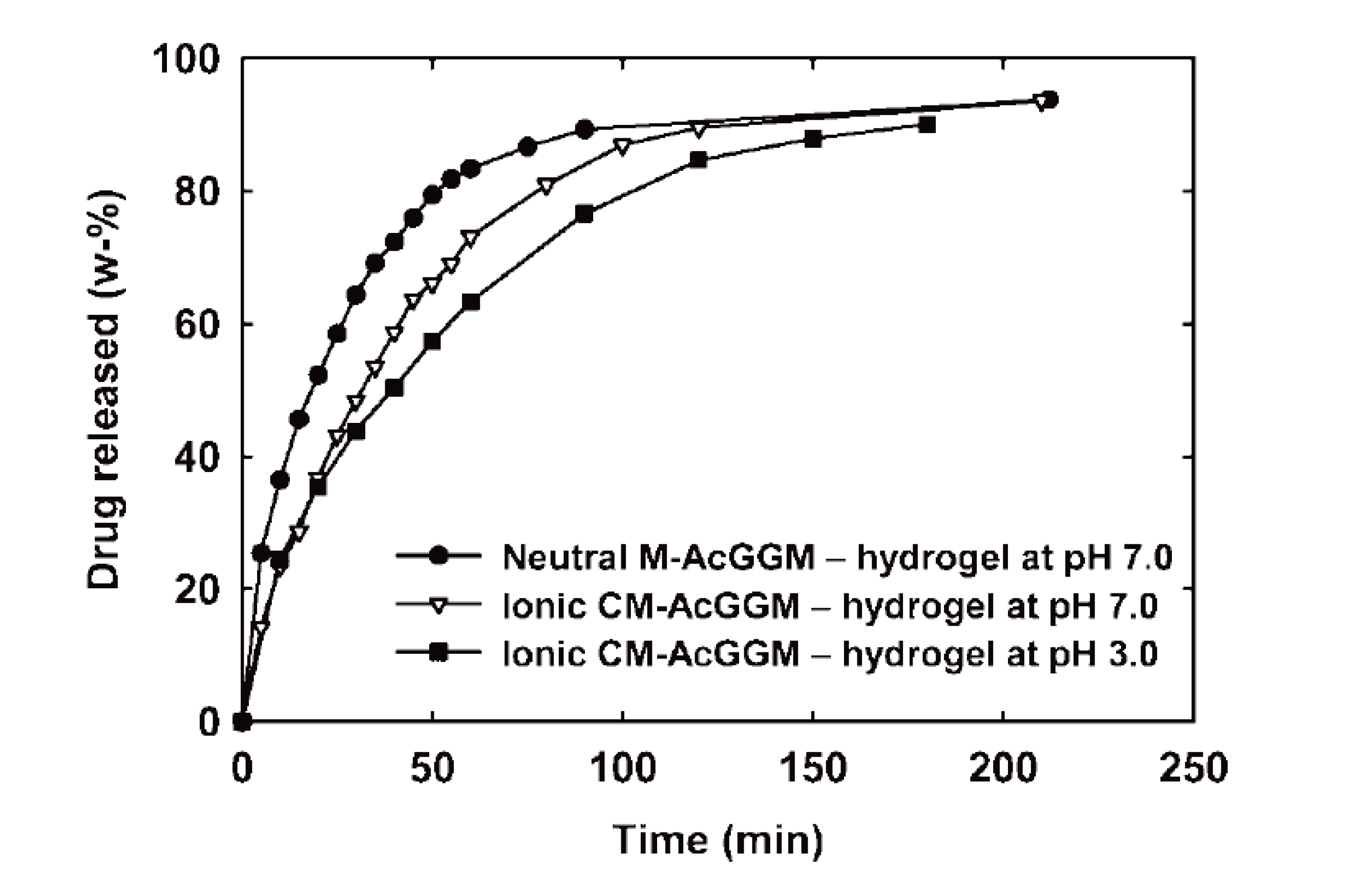
3.2. Hydrogels
4. Lignin
4.1. Lignin as an Additive to Enhance Composites
4.2. Lignosulfonates in Active Packaging
4.3. Lignin-Coated Electrodes in Sensors and Batteries
4.4. Lignin Nanotubes

5. Genetic Modification of Biomass Composition: Tailoring Plants for Specific Applications

6. Conclusion and Prospects
Acknowledgements
References
- Vermerris, W. Why Bioenergy Makes Sense. In Genetic Improvement of Bioenergy Crops; Vermerris, W., Ed.; Springer: New York, NY, USA, 2008; pp. 3–42. [Google Scholar]
- Independent Statistics & Analysis U.S. Energy Information Administration Web Page. Frequently Asked Questions. Available online: http://www.eia.gov/tools/faqs/faq.cfm?id=34&t=6 (accessed on 22 February 2013).
- Loock, M. Going beyond best technology and lowest price: On renewable energy investors’ preference for service-driven business models. Energy Policy 2012, 40, 21–27. [Google Scholar] [CrossRef]
- Masini, A.; Menichetti, E. The impact of behavioral factors in the renewable energy investment decision making process: Conceptual framework and empirical findings. Energy Policy 2012, 40, 28–38. [Google Scholar]
- Chakravorty, U.; Hubert, M.-H.; Nøstbakken, L. Fuel versus food. Annu. Rev. Resour. Econ. 2009, 1, 645–663. [Google Scholar] [CrossRef]
- Thompson, P.B. The agricultural ethics of biofuels: Climate ethics and mitigation arguments. Agriculture 2012, 8, 169–189. [Google Scholar]
- Carpita, N.C.; Gibeaut, D.M. Structural models of primary cell walls in flowering plants: Consistency of molecular structure with the physical properties of the walls during growth. Plant J. 1993, 3, 1–30. [Google Scholar] [CrossRef]
- Crowell, E.F.; Gonneau, M.; Stierhof, Y.-D.; Höfte, H.; Vernhettes, S. Regulated trafficking of cellulose synthases. Curr. Opin. Plant Biol. 2010, 13, 700–705. [Google Scholar]
- Guerriero, G.; Fugelstad, J.; Bulone, V. What do we really know about cellulose biosynthesis in higher plants? J. Integr. Plant Biol. 2010, 52, 161–175. [Google Scholar]
- Mizrachi, E.; Mansfield, S.; Myburg, A. Cellulose factories: Advancing bioenergy production from forest trees. New Phytol. 2012, 194, 54–62. [Google Scholar] [CrossRef]
- Dahlgren, G. An updated angiosperm classification. Bot. J. Linn. Soc. 1989, 100, 197–203. [Google Scholar]
- Burton, R.A.; Wilson, S.M.; Hrmova, M.; Harvey, A.J.; Shirley, N.J.; Medhurst, A.; Stone, B.A.; Newbigin, E.J.; Bacic, A.; Fincher, G.B. Cellulose Synthase-like CSLF genes mediate the synthesis of cell wall (1,3;1,4)-β-d-glucans. Science 2006, 311, 1940–1942. [Google Scholar] [CrossRef]
- Cocuron, J.; Lerouxel, O.; Drakakaki, G.; Alonso, A.; Liepman, A.; Keegstra, K.; Raikhel, N.; Wilkerson, C. A gene from the cellulose synthase-like C family encodes a beta-1,4 glucan synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 8550–8555. [Google Scholar] [CrossRef]
- Scheller, H.V.; Ulvskov, P. Hemicelluloses. Annu. Rev. Plant Biol. 2010, 61, 263–289. [Google Scholar] [CrossRef]
- Zabotina, O.A. Xyloglucan and its biosynthesis. Front. Plant Sci. 2012, 3, 134. [Google Scholar] [CrossRef]
- Vermerris, W.; Nicholson, R. Phenolic Compound Biochemistry; Springer: Dordrecht, The Netherlands, 2006; p. 276. [Google Scholar]
- Ralph, J.; Peng, J.; Lu, F.; Hatfield, R.; Helm, R. Are lignins optically active? J. Agric. Food Chem. 1999, 47, 2991–2996. [Google Scholar] [CrossRef]
- Hatfield, R.; Vermerris, W. Lignin formation in plants. The dilemma of linkage specificity. Plant Physiol. 2001, 126, 1351–1357. [Google Scholar] [CrossRef]
- Ralph, J.; Lundquist, K.; Brunow, G.; Lu, F.; Kim, H.; Schatz, P.F.; Marita, J.M.; Hatfield, R.D.; Ralph, S.A.; Christensen, J.H.; Boerjan, W. Lignins: Natural polymers from oxidative coupling of 4-hydroxyphenylpropanoids. Phytochem. Rev. 2004, 3, 29–60. [Google Scholar] [CrossRef]
- Vanholme, R.; Demedts, B.; Morreel, K.; Ralph, J.; Boerjan, W. Lignin biosynthesis and structure. Plant Physiol. 2010, 153, 895–905. [Google Scholar] [CrossRef]
- Ragauskas, A.; Williams, C.; Davison, B.; Britovsek, G.; Cairney, J.; Eckert, C.; Frederick, W.; Hallett, J.; Leak, D.; Liotta, C.; et al. The path forward for biofuels and biomaterials. Science 2006, 311, 484–489. [Google Scholar] [CrossRef]
- Agbor, V.B.; Cicek, N.; Sparling, R.; Berlin, A.; Levin, D.B. Biomass pretreatment: Fundamentals toward application. Biotechnol. Adv. 2011, 29, 675–685. [Google Scholar] [CrossRef]
- Sun, Y.; Cheng, J. Hydrolysis of lignocellulosic materials for ethanol production: A review. Bioresour. Technol. 2002, 83, 1–11. [Google Scholar] [CrossRef]
- Saritha, M.; Arora, A.; Lata. Biological pretreatment of lignocellulosic substrates for enhanced delignification and enzymatic digestibility. Indian J. Microbiol. 2012, 52, 122–130. [Google Scholar] [CrossRef]
- Singh, P.; Sulaiman, O.; Hashim, R.; Rupani, P.F.; Peng, L.C. Biopulping of lignocellulosic material using different fungal species: A review. Rev. Environ. Sci. Biotechnol. 2010, 9, 141–151. [Google Scholar] [CrossRef]
- Pu, Y.; Kosa, M.; Kalluri, U.C.; Tuskan, G.A.; Ragauskas, A.J. Challenges of the utilization of wood polymers: How can they be overcome? Appl. Microbiol. Biotechnol. 2011, 91, 1525–1536. [Google Scholar] [CrossRef]
- Gullón, P.; Romaní, A.; Vila, C.; Garrote, G.; Parajó, J.C. Potential of hydrothermal treatments in lignocellulose biorefineries. Biofuels Bioprod. Bioref. 2012, 6, 219–232. [Google Scholar] [CrossRef]
- Nampoothiri, K.; Nair, N.; John, R. An overview of the recent developments in polylactide (PLA) research. Bioresour. Technol. 2010, 101, 8493–8501. [Google Scholar]
- Inkinen, S.; Hakkarainen, M.; Albertsson, A.; Sodergard, A. From lactic acid to poly(lactic acid) (PLA): Characterization and analysis of PLA and its precursors. Biomacromolecules 2011, 12, 523–532. [Google Scholar] [CrossRef]
- Klemm, D.; Heinze, T.; Wagenknecht, W. Comprehensive Cellulose Chemistry; Wiley-VCG: Weinheim, Germany, 1998; pp. 115–133. [Google Scholar]
- Schutzenberger, P. Action de l'acide acétique anhydre sur la cellulose, l'amidon, les sucres, la mannite et ses congénères, les glucosides et certaines matières colorantes végétales. [in French]. Compt. Rend. Hebd. Séances Acad. Sci. 1865, 61, 484–487. [Google Scholar]
- Ghaemi, N.; Madaeni, S.S.; Alizadeh, A.; Daraei, P.; Zinatizadeh, A.A.; Rahimpour, F. Separation of nitrophenols using cellulose acetate nanofiltration membrane: Influence of surfactant additives. Sep. Purif. Technol. 2012, 85, 147–156. [Google Scholar] [CrossRef]
- Zaky, A.; Escobar, I.; Motlagh, A.M.; Gruden, C. Determining the influence of active cells and conditioning layer on early stage biofilm formation using cellulose acetate ultrafiltration membranes. Desalination 2012, 286, 296–303. [Google Scholar]
- Koseoglu-Imer, D.Y.; Nadir Dizge, N.; Koyuncu, I. Enzymatic activation of cellulose acetate membrane for reducing of protein fouling. Colloids Surf. B 2012, 92, 334–339. [Google Scholar] [CrossRef]
- Jayalakshmi, A.; Rajesh, S.; Senthilkumar, S.; Mohan, D. Epoxy functionalized poly(ether-sulfone) incorporated cellulose acetate ultrafiltration membrane for the removal of Chromium ions. Sep. Purif. Technol. 2012, 90, 120–132. [Google Scholar]
- Kumari, A.; Sarkhel, G.; Choudhury, A. Preparation and characterization of polyvinylpyrollidone incorporated cellulose acetate membranes for ultrafiltration of metal ion. J. Appl. Polym. Sci. 2012, 124, E300–E308. [Google Scholar] [CrossRef]
- Harper, M.; Ashley, K. Preliminary studies on the use of acid-soluble cellulose acetate internal capsules for workplace metals sampling and analysis. J. Occup. Environ. Hyg. 2012, 9, D125–D129. [Google Scholar] [CrossRef]
- Magosso, H.A.; Fattori, N.; Arenas, L.T.; Gushikem, Y. New promising composite materials useful in the adsorption of Cu(II) in ethanol based on cellulose and cellulose acetate. Cellulose 2012, 19, 913–923. [Google Scholar] [CrossRef]
- Ghaemi, N.; Madaeni, S.S.; Alizadeh, A.; Rajabi, H.; Daraei, P.; Falsafi, M. Effect of fatty acids on the structure and performance of cellulose acetate nanofiltration membranes in retention of nitroaromatic pesticides. Desalination 2012, 301, 26–41. [Google Scholar]
- Rana, D.; Scheier, B.; Narbaitz, R.M.; Matsuura, T.; Tabe, S.; Jasimd, S.Y.; Khulbe, K.C. Comparison of cellulose acetate (CA) membrane and novel CA membranes containing surface modifying macromolecules to remove pharmaceutical and personal care product micropollutants from drinking water. J. Membr. Sci. 2012, 409–410, 346–354. [Google Scholar] [CrossRef]
- Kiran; Rana, D.S.; Balokhra, R.L.; Umar, A.; Chauhan, S. A thermodynamic study of 1,4-dioxane across cellulose acetate membrane under different conditions. Fluid Phase Equilib. 2012, 322–323, 148–158. [Google Scholar] [CrossRef]
- Nista, S.V.G.; Peres, L.; D’Ávila, M.A.; Schmidt, F.L.; Mei, L.H.I. Nanostructured membranes based on cellulose acetate obtained by electrospinning, part 1: Study of the best solvents and conditions by designof experiments. J. Appl. Polym. Sci. 2012, 126, E70–E78. [Google Scholar]
- De Lima, J.A.; Pinotti, C.A.; Felisberti, M.I.; do Carmo Gonçalves, M. Blends and clay nanocomposites of cellulose acetate and poly(epichlorohydrin). Compos. B 2012, 43, 2375–2381. [Google Scholar] [CrossRef]
- De Lima, J.A.; Pinotti, C.A.; Felisberti, M.I.; do Carmo Gonçalves, M. Morphology and mechanical properties of nanocomposites of cellulose acetate and organic montmorillonite prepared with different plasticizers. J. Appl. Polym. Sci. 2012, 124, 4628–4635. [Google Scholar]
- Rodriguez, F.J.; Galotto, M.J.; Guarda, A.; Bruna, J.E. Modification of cellulose acetate films using nanofillers based on organoclays. J. Food Eng. 2012, 110, 262–268. [Google Scholar] [CrossRef]
- Liu, X.; Lin, T.; Gao, Y.; Xu, Z.; Huang, C.; Yao, G.; Jiang, L.; Tang, Y.; Wang, X. Antimicrobial electrospun nanofibers of cellulose acetate and polyester urethane composite for wound dressing. J. Biomed. Mater. Res. B 2012, 100, 1556–1565. [Google Scholar]
- Gouma, P.; Xue, R.; Goldbeck, C.P.; Perrotta, P.; Balázsi, C. Nano-hydroxyapatite-cellulose acetate composites for growing of bone cells. Mater. Sci. Eng. C 2012, 32, 607–612. [Google Scholar] [CrossRef]
- Kanyong, P.; Pemberton, R.M.; Jackson, S.K.; Hart, J.P. Development of a sandwich format, amperometric screen-printed uric acid biosensor for urine analysis. Anal. Biochem. 2012, 428, 39–43. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, T.; Chen, L.; Ren, M.; Zhang, B.; Wang, Z.; Wang, Y. Bifunctional polyethersulfone hollow fiber with a porous, single-layer skin for use as a bioartificial liver bioreactor. J. Mater. Sci. 2012, 23, 2001–2011. [Google Scholar]
- Harun, N.I.; Ali, R.M.; Ali, A.M.M.; Yahya, M.Z.A. Dielectric behaviour of cellulose acetate-based polymer electrolytes. Ionics 2012, 18, 599–606. [Google Scholar] [CrossRef]
- Ramesh, S.; Shanti, R.; Morris, E. Plasticizing effect of 1-allyl-3-methylimidazolium chloride in cellulose acetate based polymer electrolytes. Carbohydr. Polym. 2012, 87, 2624–2629. [Google Scholar] [CrossRef]
- Shamsipur, M.; Pourmortazavi, S.M.; Hajimirsadeghi, S.S.; Atifeh, S.M. Effect of functional group on thermal stability of cellulose derivative energetic polymers. Fuel 2012, 95, 394–399. [Google Scholar] [CrossRef]
- Gutiérrez, M.C.; de Paoli, M.-A.; Felisberti, M.I. Biocomposites based on cellulose acetate and short curauá fibers: Effect of plasticizers and chemical treatments of the fibers. Compos. A 2012, 43, 1338–1346. [Google Scholar] [CrossRef]
- Stiubianu, G.; Nicolescu, A.; Nistor, A.; Cazacu, M.; Varganici, C.; Simionescu, B.C. Chemical modification of cellulose acetate by allylation and crosslinking with siloxane derivatives. Polym. Int. 2012, 61, 1115–1126. [Google Scholar]
- Wan, S.; Sun, Y.; Qi, X.; Tan, F. Improved bioavailability of poorly water-soluble drug curcumin in cellulose acetate solid dispersion. AAPS PharmSciTech 2012, 13, 159–166. [Google Scholar] [CrossRef]
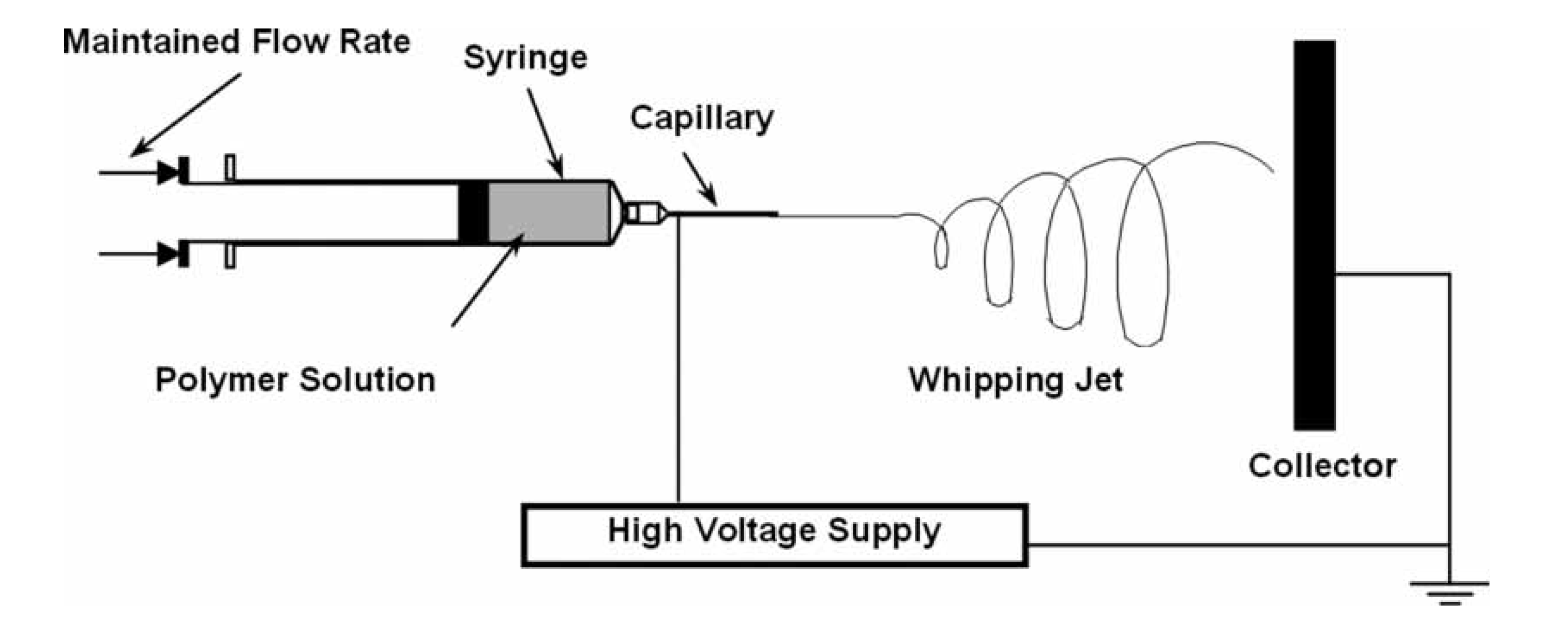
- Díaz, J.E.; Barrero, A.; Márquez, M.; Loscertales, I.G. Controlled encapsulation of hydrophobic liquids in hydrophilic polymer nanofibers by co-electrospinning. Adv. Funct. Mater. 2006, 16, 2110–2116. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Wang, X.; Feng, Y.; Li, J.; Lim, C.T.; Ramakrishna, S. Coaxial electrospinning of (fluorescein isothiocyanate-conjugated bovine serum albumin)-encapsulated poly(ε-caprolactone) nanofibers for sustained release. Biomacromolecules 2006, 7, 1049–1057. [Google Scholar] [CrossRef]
- Moghe, A.K.; Gupta, B.S. Co-axial electrospinning for nanofiber structures: Preparation and applications. Polym. Rev. 2008, 48, 353–377. [Google Scholar] [CrossRef]
- Yan, J.; Yu, D.-G. Smoothening electrospinning and obtaining high-quality cellulose acetate nanofibers using a modified coaxial process. J. Mater. Sci. 2012, 47, 7138–7147. [Google Scholar] [CrossRef]
- Yu, D.-G.; Yu, J.-H.; Chen, L.; Williams, G.R.; Wang, X. Modified coaxial electrospinning for the preparation of high-quality ketoprofen-loaded cellulose acetate nanofibers. Carbohydr. Polym. 2012, 90, 1016–1023. [Google Scholar] [CrossRef]
- Hornig, S.; Heinze, T. Efficient approach to design stable water-dispersible nanoparticles of hydrophobic cellulose esters. Biomacromolecules 2008, 9, 1487–1492. [Google Scholar] [CrossRef]
- Kulterer, M.R.; Reichel, V.E.; Hribernik, S.; Wu, M.; Kostlerd, S.; Kargl, R.; Ribitsch, V. Nanoprecipitation of cellulose acetate using solvent/nonsolvent mixtures as dispersive media. Colloids Surf. A Eng. Asp. 2011, 375, 23–29. [Google Scholar] [CrossRef]
- Kulterer, M.R.; Reichel, V.E.; Kargl, R.; Köstler, S.; Sarbova, V.; Heinze, T.; Stana-Kleinschek, K.; Ribitsch, V. Functional polysaccharide composite nanoparticles from cellulose acetate and potential applications. Adv. Funct. Mater. 2012, 22, 1749–1758. [Google Scholar] [CrossRef]
- Zhang, L.; Ma, H.; Cao, F.; Gong, J.; Su, Z. Nonaqueous synthesis of uniform polyaniline nanospheres via cellulose acetate template. J. Polym. Sci. A Polym. Chem. 2012, 50, 912–917. [Google Scholar] [CrossRef]
- Yoo, S.; Hsieh, J.S. Enzyme-assisted preparation of fibrillated cellulose fibers and its effect on physical and mechanical properties of paper sheet composites. Ind. Eng. Chem. Res. 2010, 49, 2161–2168. [Google Scholar] [CrossRef]
- Saito, T.; Kimura, S.; Nishiyama, Y.; Isogai, A. Cellulose nanofibers prepared by TEMPO-mediated oxidation of native cellulose. Biomacromolecules 2007, 8, 2485–2491. [Google Scholar] [CrossRef]
- Soykeabkaew, N.; Nishino, T.; Peijs, T. All-cellulose composites of regenerated cellulose fibres by surface selective dissolution. Compos. A Appl. Sci. Manuf. 2009, 40, 321–328. [Google Scholar] [CrossRef]
- Duchemin, B.J.C.; Mathew, A.P.; Oksman, K. All-cellulose composites by partial dissolution in the ionic liquid 1-butyl-3-methylimidazolium chloride. Compos. A 2009, 40, 2031–2037. [Google Scholar] [CrossRef]
- Shakeri, A.; Mathew, A.P.; Oksman, K. Self-reinforced nanocomposite by partial dissolution of cellulose microfibrils in ionic liquid. J. Compos. Mater. 2012, 46, 1305–1311. [Google Scholar] [CrossRef]
- Zuluaga, R.; Putaux, J.L.; Cruz, J.; Vélez, J.; Mondragon, I.; Gañán, P. Cellulose microfibrils from banana rachis: Effect of alkaline treatments on structural and morphological features. Carbohydr. Polym. 2009, 76, 51–59. [Google Scholar] [CrossRef]
- Hassan, E.A.; Hassan, M.L.; Oksman, K. Improving bagasse pulp paper sheet properties with microfibrillated cellulose isolated from xylanase-treated bagasse. Wood Fiber Sci. 2011, 43, 76–82. [Google Scholar]
- Henriksson, M.; Henriksson, G.; Berglund, L.A.; Lindstrom, T. An environmentally friendly method for enzyme-assisted preparation of microfibrillated cellulose (MFC) nanofibers. Eur. Polym. J. 2007, 43, 3434–3441. [Google Scholar] [CrossRef]
- Spence, K.L.; Venditti, R.A.; Rojas, O.J.; Habibi, Y.; Pawlak, J.J. A comparative study of energy consumption and physical properties of microfibrillated cellulose produced by different processing methods. Cellulose 2011, 18, 1097–1111. [Google Scholar] [CrossRef]
- Spence, K.L.; Venditti, R.A.; Rojas, O.J.; Habibi, Y.; Pawlak, J.J. The effect of chemical composition on microfibrillar cellulose films from wood pulps: Water interactions and physical properties for packaging applications. Cellulose 2010, 17, 835–848. [Google Scholar] [CrossRef]
- Agoda-Tandjawa, G.; Durand, S.; Berot, S.; Blassel, C.; Gaillard, C.; Garnier, C.; Doublier, J.-L. Rheological characterization of microfibrillated cellulose suspensions after freezing. Carbohydr. Polym. 2010, 80, 677–686. [Google Scholar] [CrossRef]
- Spence, K.L.; Venditti, R.A.; Habibi, Y.; Rojas, O.J.; Pawlak, J.J. The effect of chemical composition on microfibrillar cellulose films fromwood pulps: Mechanical processing and physical properties. Bioresour. Technol. 2010, 101, 5961–5968. [Google Scholar] [CrossRef]
- Taipale, T.; Österberg, M.; Nykänen, A.; Ruokolainen, J.; Laine, J. Effect of microfibrillated cellulose and fines on the drainage of kraft pulp suspension and paper strength. Cellulose 2010, 17, 1005–1020. [Google Scholar]
- Tingaut, P.; Zimmermann, T.; Lopez-Suevos, F. Synthesis and characterization of bionanocomposites with tunable properties from poly(lactic acid) and acetylated microfibrillated cellulose. Biomacromolecules 2010, 11, 454–464. [Google Scholar] [CrossRef]
- Mikkonen, K.S.; Pitkänen, L.; Liljeström, V.; Bergström, E.M.; Serimaa, R.; Salmén, L.; Tenkanen, M. Arabinoxylan structure affects the reinforcement of films by microfibrillated cellulose. Cellulose 2012, 19, 467–480. [Google Scholar] [CrossRef]
- Qiu, K.; Netravali, A.N. Fabrication and characterization of biodegradable composites based on microfibrillated cellulose and polyvinyl alcohol. Compos. Sci. Technol. 2012, 72, 1588–1594. [Google Scholar] [CrossRef]
- Fortunato, G.; Zimmermann, T.; Lübben, J.; Bordeanu, N.; Hufenus, R. Reinforcement of polymeric submicrometer-sized fibers by microfibrillated cellulose. Macromol. Mater. Eng. 2012, 297, 576–584. [Google Scholar] [CrossRef]
- Tanpichai, S.; Sampson, W.W.; Eichhorn, S.J. Stress-transfer in microfibrillated cellulose reinforced poly(lactic acid) composites using Raman spectroscopy. Compos. A 2012, 43, 1145–1152. [Google Scholar] [CrossRef]
- Ridgway, C.J.; Gane, P.A.C. Constructing NFC-pigment composite surface treatment for enhanced paper stiffness and surface properties. Cellulose 2012, 19, 547–560. [Google Scholar] [CrossRef]
- Syverud, K.; Stenius, P. Strength and barrier properties of MFC films. Cellulose 2009, 16, 75–85. [Google Scholar] [CrossRef]
- Bulota, M.; Jääskeläinen, A.S.; Paltakari, J.; Hughes, M. Properties of biocomposites: Influence of preparation method, testing environment and a comparison with theoretical models. J. Mater. Sci. 2011, 46, 3387–3398. [Google Scholar]
- Okubo, K.; Fujii, T.; Thostenson, E.T. Multi-scale hybrid biocomposite: Processing and mechanical characterization of bamboo fiber reinforced PLA with microfibrillated cellulose. Compos. A 2009, 40, 469–475. [Google Scholar] [CrossRef]
- Nakagaito, A.N.; Fujimura, A.; Sakai, T.; Hama, Y.; Yano, H. Production of microfibrillated cellulose (MFC)-reinforced polylactic acid (PLA) nanocomposites from sheets obtained by a papermaking-like process. Compos. Sci. Technol. 2009, 69, 1293–1297. [Google Scholar] [CrossRef]
- Bulota, M.; Kreitsmann, K.; Hughes, M.; Paltakari, J. Acetylated microfibrillated cellulose as a toughening agent in poly(lactic acid). J. Appl. Polym. Sci. 2012, 126, E448–E457. [Google Scholar]
- Bulota, M.; Hughes, M. Toughening mechanisms in poly(lactic) acid reinforced with TEMPO-oxidized cellulose. J. Mater. Sci. 2012, 47, 5517–5523. [Google Scholar] [CrossRef]
- Gabr, M.H.; Elrahman, M.A.; Okubo, K.; Fujii, T. Effect of microfibrillated cellulose on mechanical properties of plain-woven CFRP reinforced epoxy. Compos. Struct. 2010, 92, 1999–2006. [Google Scholar] [CrossRef]
- Gabr, M.H.; Elrahman, M.A.; Okubo, K.; Fujii, T. Interfacial adhesion improvement of plain woven carbon fiber reinforced epoxy filled with micro-fibrillated cellulose by addition liquid rubber. J. Mater. Sci. 2010, 45, 3841–3850. [Google Scholar]
- Spence, K.L.; Venditti, R.A.; Rojas, O.J.; Pawlak, J.J.; Hubbe, M.A. Water vapor barrier properties of coated and filled microfibrillated cellulose composite films. Bioresources 2011, 6, 4370–4388. [Google Scholar]
- Fernández, A.; Sánchez, M.D.; Ankerfors, M.; Lagaron, J.M. Effects of ionizing radiation in ethylene-vinyl alcohol copolymers and in composites containing microfibrillated cellulose. J. Appl. Polym. Sci. 2008, 109, 126–134. [Google Scholar] [CrossRef]
- Minelli, M.; Baschetti, M.G.; Doghieri, F.; Ankerfors, M.; Lindström, T.; Siró, I.; Plackett, D. Investigation of mass transport properties of microfibrillated cellulose (mfc) films. J. Membr. Sci. 2010, 358, 67–75. [Google Scholar] [CrossRef]
- Aulin, C.; Gällstedt, M.; Lindström, T. Oxygen and oil barrier properties of microfibrillated cellulose films and coatings. Cellulose 2010, 17, 559–574. [Google Scholar] [CrossRef]
- Rodionova, G.; Lenes, M.; Eriksen, Ø.; Gregersen, Ø. Surface chemical modification of microfibrillated cellulose: Improvement of barrier properties for packaging applications. Cellulose 2011, 18, 127–134. [Google Scholar] [CrossRef]
- Siró, I.; Plackett, D. Microfibrillated cellulose and new nanocomposite materials: A review. Cellulose 2010, 17, 459–494. [Google Scholar] [CrossRef]
- Lavoine, N.; Desloges, I.; Dufresne, A.; Bras, J. Microfibrillated cellulose—Its barrier properties and applications in cellulosic materials: A review. Carbohydr. Polym. 2012, 90, 735–764. [Google Scholar] [CrossRef]
- Johansson, C.; Bras, J.; Mondragon, I.; Nechita, P.; Plackett, D.; Šimon, P.; Svetec, D.G.; Virtanen, S.; Baschetti, M.G.; Breen, C.; et al. Renewable fibers and bio-based materials for packaging applications—A review of recent developments. Bioresources 2012, 7, 2506–2552. [Google Scholar]
- Wågberg, L.; Decher, G.; Norgren, M.; Lindström, T.; Ankerfors, M.; Axnäs, K. The build-up of polyelectrolyte multilayers of microfibrillated cellulose and cationic polyelectrolytes. Langmuir 2008, 24, 784–795. [Google Scholar] [CrossRef]
- Larsson, P.A.; Wågberg, L. Diffusion-Induced dimensional changes in papers and fibrillar films: Influence of hydrophobicity and fibre-wall cross-linking. Cellulose 2010, 17, 891–901. [Google Scholar] [CrossRef]
- Aulin, C.; Varga, I.; Claesson, P.M.; Wågberg, L.; Lindström, T. Buildup of polyelectrolyte multilayers of polyethyleneimine and microfibrillated cellulose studied by in situ dual-polarization interferometry and quartz crystal microbalance with dissipation. Langmuir 2008, 24, 2509–2518. [Google Scholar]
- Enarsson, L.-E.; Wågberg, L. Polyelectrolyte adsorption on thin cellulose films studied with reflectometry and quartz crystal microgravimetry with dissipation. Biomacromolecules 2009, 10, 134–141. [Google Scholar] [CrossRef]
- Nyström, G.; Mihranyan, A.; Razaq, A.; Lindstro¨m, T.; Nyholm, L.; Strømme, M. A nanocellulose polypyrrole composite based on microfibrillated cellulose from wood. J. Phys. Chem. B 2010, 114, 4178–4182. [Google Scholar] [CrossRef]
- Jabbour, L.; Gerbaldi, C.; Chaussy, D.; Zeno, E.; Bodoardo, S.; Beneventi, D. Microfibrillated cellulose-graphite nanocomposites for highly flexible paper-like Li-ion battery electrodes. J. Mater. Chem. 2010, 20, 7344–7347. [Google Scholar]
- Chiappone, A.; Nair, J.R.; Gerbaldi, C.; Jabbour, L.; Bongiovanni, R.; Zeno, E.; Beneventi, D.; Penazzi, N. Microfibrillated cellulose as reinforcement for li-ion battery polymer electrolytes with excellent mechanical stability. J. Power Sources 2011, 196, 10280–10288. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, X.; Lu, C.; Wang, Y.; Deng, Y. Flexible and transparent paper-based ionic diode fabricated from oppositely charged microfibrillated cellulose. J. Phys. Chem. C 2012, 116, 9227–9234. [Google Scholar]
- Aulin, C.; Netrval, J.; Wågberg, L.; Lindström, T. Aerogels from nanofibrillated cellulose with tunable oleophobicity. Soft Matter 2010, 6, 3298–3305. [Google Scholar]
- Larsson, M.; Zhou, Q.; Larsson, A. Different types of microfibrillated cellulose as filler materials in polysodium acrylate suprasorbents. Chin. J. Polym. Sci. 2011, 29, 407–413. [Google Scholar] [CrossRef]
- Xhanari, K.; Syverud, K.; Stenius, P. Emulsions stabilized by microfibrillated cellulose: The effect of hydrophobization, concentration and O/W ratio. J. Disper. Sci. Technol. 2011, 32, 447–452. [Google Scholar] [CrossRef]
- Lif, A.; Stenstad, P.; Syverud, K.; Nydén, M.; Holmberg, K. Fischer–Tropsch diesel emulsions stabilised by microfibrillated cellulose and nonionic surfactants. J. Colloid Interface Sci. 2010, 352, 585–592. [Google Scholar] [CrossRef]
- Boissard, C.I.R.; Bourban, P.-E.; Tingaut, P.; Zimmermann, T.; Månson, J.-A.E. Water of functionalized microfibrillated cellulose as foaming agent for the elaboration of poly(lactic acid) biocomposites. J. Reinf. Plast. Compos. 2011, 30, 709–719. [Google Scholar] [CrossRef]
- Qu, P.; Tang, H.; Gao, Y.; Zhang, L.; Wang, S. Polyethersulfone composite membrane blended with cellulose fibrils. Bioresources 2010, 5, 2323–2336. [Google Scholar]
- Evenou, F.; Couderc, S.; Kim, B.; Fujii, T.; Sakai, Y. Microfibrillated cellulose sheets coating oxygen-permeable PDMS membranes induce rat hepatocytes 3D aggregation into stably-attached 3D hemispheroids. J. Biomater. Sci. 2011, 22, 1509–1522. [Google Scholar] [CrossRef]
- Couderc, S.; Ducloux, O.; Kim, B.J.; Someya, T. A mechanical switch device made of a polyimide-coated microfibrillated cellulose sheet. J. Micromech. Microeng. 2009, 19, 055006:1–055006:11. [Google Scholar]
- Tuteja, A.; Choi, W.; Ma, M.; Mabry, J.M.; Mazzella, S.A.; Rutledge, G.C.; McKinley, G.H.; Cohen, R.E. Designing superoleophobic surfaces. Science 2007, 318, 1618–1622. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, C.; Chang, P.R.; Cao, X.; Anderson, D.P. Bionanocomposites based on pea starch and cellulose nanowhiskers hydrolyzed from pea hull fibre: Effect of hydrolysis time. Carbohydr. Polym. 2009, 76, 607–615. [Google Scholar] [CrossRef]
- Rosa, M.F.; Medeiros, E.S.; Malmonge, J.A.; Gregorski, K.S.; Wood, D.F.; Mattoso, L.H.C.; Glenn, G.; Orts, W.J.; Imam, S.H. Cellulose nanowhiskers from coconut husk fibers: Effect of preparation conditions on their thermal and morphological behavior. Carbohydr. Polym. 2010, 81, 83–92. [Google Scholar] [CrossRef]
- Kargarzadeh, H.; Ahmad, I.; Abdullah, I.; Dufresne, A.; Zainudin, S.Y.; Sheltami, R.M. Effects of hydrolysis conditions on the morphology, crystallinity, and thermal stability of cellulose nanocrystals extracted from kenaf bast fibers. Cellulose 2012, 19, 855–866. [Google Scholar] [CrossRef]
- Pandey, J.K.; Kim, C.-S.; Chu, W.-S.; Lee, C.S.; Jang, D.-Y.; Ahn, S.-H. Evaluation of morphological architecture of cellulose chains in grass during conversion from macro to nano dimensions. E-Polymers 2009, 102, 1–15. [Google Scholar]
- De Oliveira Taipina, M.; Ferrarezi, M.M.F.; do Carmo Goncalves, M. Morphological evolution of curauá fibers under acid hydrolysis. Cellulose 2012, 19, 1199–1207. [Google Scholar] [CrossRef]
- Lu, P.; Hsieh, Y.-L. Preparation and characterization of cellulose nanocrystals from rice straw. Carbohydr. Polym. 2012, 87, 564–573. [Google Scholar] [CrossRef]
- Sèbe, G.; Ham-Pichavant, F.; Ibarboure, E.; Koffi, A.L.C.; Tingaut, P. Supramolecular structure characterization of cellulose II nanowhiskers produced by acid hydrolysis of cellulose I substrates. Biomacromolecules 2012, 13, 570–578. [Google Scholar] [CrossRef]
- Li, W.; Yue, J.; Liu, S. Preparation of nanocrystalline cellulose via ultrasound and its reinforcement capability for poly(vinyl alcohol) composites. Ultrason. Sonochem. 2012, 19, 479–485. [Google Scholar] [CrossRef]
- Sturcova, A.; Davies, G.R.; Eichhorn, S.J. The elastic modulus and stress-transfer properties of tunicate cellulose whiskers. Biomacromolecules 2005, 6, 1055–1061. [Google Scholar] [CrossRef]
- Ahola, S.; Österberg, M.; Laine, J. Cellulose nanofibrils-adsorption with poly(amideamine) epichlorohydrin studied by QCM-D and application as a paper strength additive. Cellulose 2008, 15, 303–314. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, C.; Chang, P.R.; Anderson, D.P.; Huneault, M.A. Pea starch-based composite films with pea hull fibers and pea hull fiber-derived nanowhiskers. Polym. Eng. Sci. 2009, 49, 369–378. [Google Scholar] [CrossRef]
- Da Silva, J.B.A.; Pereira, F.V.; Druzian, J.I. Cassava starch-based films plasticized with sucrose and inverted sugar and reinforced with cellulose nanocrystals. J. Food Sci. 2012, 77, N14–N19. [Google Scholar]
- Yang, H.-S.; Gardner, D.J.; Nader, J.W. Characteristic impact resistance model analysis of cellulose nanofibril-filled polypropylene composites. Compos. A 2011, 42, 2028–2035. [Google Scholar]
- Ten, E.; Jiang, L.; Wolcott, M.P. Preparation and properties of aligned poly (3-hydroxybutyrate-co-3-hydroxyvalerate)/cellulose nanowhiskers composites. Carbohydr. Polym. 2013, 92, 206–213. [Google Scholar] [CrossRef]
- Ten, E.; Jiang, L.; Bahr, D.F.; Li, B.; Wolcott, M.P. Effects of cellulose nanowhiskers on mechanical, dielectric and rheological properties of poly(3-hydroxybutyrate-co-3-hydroxyvalerate)(PHBV)/cellulose nanowhiskers (CNW) composites. Ind. Eng. Chem. Res. 2012, 51, 2941–2951. [Google Scholar] [CrossRef]
- Ten, E.; Turtle, J.; Bahr, D.; Jiang, L.; Wolcott, M.P. Thermal and mechanical properties of poly(3-hydroxybutyrate-co-3-hydroxyvalerate)/cellulose nanowhiskers composites. Polymer 2010, 51, 2652–2660. [Google Scholar] [CrossRef]
- Yu, H.-Y.; Qin, Z.-Y.; Liu, Y.-N.; Chen, L.; Liu, N.; Zhou, Z. Simultaneous improvement of mechanical properties and thermal stability of bacterial polyester by cellulose nanocrystals. Carbohydr. Polym. 2012, 89, 971–978. [Google Scholar]
- Petersson, L.; Mathew, A.P.; Oksman, K. Dispersion and properties of cellulose nanowhiskers and layered silicates in cellulose acetate butyrate nanocomposites. J. Appl. Polym. Sci. 2009, 112, 2001–2009. [Google Scholar] [CrossRef]
- Siqueira, G.; Mathew, A.P.; Oksman, K. Processing of cellulose nanowhiskers/cellulose acetate butyrate nanocomposites using sol-gel process to facilitate dispersion. Compos. Sci. Technol. 2011, 71, 1886–1892. [Google Scholar] [CrossRef]
- Siqueira, G.; Bras, J.; Dufresne, A. Cellulose whiskers versus microfibrils: Influence of the nature of the nanoparticle and its surface functionalization on the thermal and mechanical properties of nanocomposites. Biomacromolecules 2009, 10, 425–432. [Google Scholar] [CrossRef]
- Mabrouk, A.B.; Kaddami, H.; Boufi, S.; Erchiqui, F.; Dufresne, A. Cellulosic nanoparticles from alfa fibers (Stipa tenacissima): Extraction procedures and reinforcement potential in polymer nanocomposites. Cellulose 2012, 19, 843–853. [Google Scholar] [CrossRef]
- Bendahou, A.; Kaddami, H.; Dufresne, A. Investigation on the effect of cellulosic nanoparticles’ morphology on the properties of natural rubber based nanocomposites. Eur. Polym. J. 2010, 46, 609–620. [Google Scholar] [CrossRef]
- Visakh, P.M.; Thomas, S.; Oksman, K.; Mathew, A.P. Cellulose nanofibres and cellulose nanowhiskers based natural rubber composites: Diffusion, sorption, and permeation of aromatic organic solvents. J. Appl. Polym. Sci. 2012, 124, 1614–1623. [Google Scholar] [CrossRef]
- Fahma, F.; Iwamoto, S.; Hori, N.; Iwata, T.; Takemura, A. Effect of pre-acid-hydrolysis treatment on morphology and properties of cellulose nanowhiskers from coconut husk. Cellulose 2011, 18, 443–450. [Google Scholar] [CrossRef]
- Cao, X.; Ding, B.; Yu, J.; Al-Deyab, S.S. Cellulose nanowhiskers extracted from TEMPO-oxidized jute fibers. Carbohydr. Polym. 2012, 90, 1075–1080. [Google Scholar] [CrossRef]
- Çetin, N.M.; Tingaut, P.; Özmen, N.; Henry, N.; Harper, D.; Dadmun, M.; Sébe, G. Acetylation of cellulose nanowhiskers with vinyl acetate under moderate conditions. Macromol. Biosci. 2009, 9, 997–1003. [Google Scholar] [CrossRef]
- Siqueira, G.; Bras, J.; Dufresne, A. New process of chemical grafting of cellulose nanoparticles with a long chain isocyanate. Langmuir 2010, 26, 402–411. [Google Scholar] [CrossRef]
- Dufresne, A. Processing of polymer nanocomposites reinforced with polysaccharide nanocrystals. Molecules 2010, 15, 4111–4128. [Google Scholar] [CrossRef]
- Zoppe, J.O.; Habibi, Y.; Rojas, O.J.; Venditti, R.A.; Johansson, L.-S.; Efimenko, K.; Österberg, M.; Laine, J. Poly(N-isopropylacrylamide) brushes grafted from cellulose nanocrystals via surface-initiated single-electron transfer living radical polymerization. Biomacromolecules 2010, 11, 2683–2691. [Google Scholar] [CrossRef]
- Goffin, A.-L.; Raquez, J.-M.; Duquesne, E.; Siqueira, G.; Habibi, Y.; Dufresne, A.; Dubois, P. From interfacial ring-opening polymerization to melt processing of cellulose nanowhisker-filled polylactide-based nanocomposites. Biomacromolecules 2011, 12, 2456–2465. [Google Scholar] [CrossRef]
- Goffin, A.-L.; Raquez, J.-M.; Duquesne, E.; Siqueira, G.; Habibi, Y.; Dufresne, A.; Dubois, P. Poly(ε-caprolactone) based nanocomposites reinforced by surface-grafted cellulose nanowhiskers via extrusion processing: Morphology, rheology, and thermo-mechanical properties. Polymer 2011, 52, 1532–1538. [Google Scholar] [CrossRef]
- Labet, M.; Thielemans, W. Citric acid as a benign alternative to metal catalysts for the production of cellulose-grafted-polycaprolactone copolymers. Polym. Chem. 2012, 3, 679–684. [Google Scholar] [CrossRef]
- Goffin, A.-L.; Habibi, Y.; Raquez, J.-M.; Dubois, P. Polyester-grafted cellulose nanowhiskers: A new approach for tuning the microstructure of immiscible polyester blends. ACS Appl. Mater. Interfaces 2012, 4, 3364–3371. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Kuga, S.; Wada, M.; Okano, T. Cellulose microcrystal film of high uniaxial orientation. Macromolecules 1997, 30, 6395–6397. [Google Scholar] [CrossRef]
- Edgar, C.D.; Gray, D.G. Smooth model cellulose I surfaces from nanocrystal suspensions. Cellulose 2003, 10, 299–306. [Google Scholar] [CrossRef]
- Cranston, E.D.; Gray, D.G. Morphological and optical characterization of polyelectrolyte multilayers incorporating nanocrystalline cellulose. Biomacromolecules 2006, 7, 2522–2530. [Google Scholar] [CrossRef]
- Cranston, E.D.; Gray, D.G. Birefringence in spin-coated films containing cellulose nanocrystals. Colloids Surf. A 2008, 325, 44–51. [Google Scholar] [CrossRef]
- Cranston, E.D.; Gray, D.G.; Rutland, M.W. Direct surface force measurements of polyelectrolyte multilayer films containing nanocrystalline cellulose. Langmuir 2010, 26, 17190–17197. [Google Scholar] [CrossRef]
- Hoeger, I.; Rojas, O.J.; Efimenko, K.; Velev, O.D.; Kelley, S.S. Ultrathin film coatings of aligned cellulosenanocrystalsfrom a convective-shearassemblysystem and their surface mechanical properties. Soft Matter 2011, 7, 1957–1967. [Google Scholar] [CrossRef]
- Sugiyama, J.; Chanzy, H.; Maretg, G. Orientation of cellulose microcrystals by strong magnetic fields. Macromolecules 1992, 25, 4232–4234. [Google Scholar] [CrossRef]
- Cranston, E.D.; Gray, D.G. Formation of cellulose-based electrostatic layer-by-layer films in a magnetic field. Sci. Technol. Adv. Mater. 2006, 7, 319–321. [Google Scholar] [CrossRef]
- Kimura, F.; Kimura, T.; Tamura, M.; Hirai, A.; Ikuno, M.; Horii, F. Three-dimensional crystal alignment using a time-dependent elliptic magnetic field. Langmuir 2005, 21, 2034–2037. [Google Scholar] [CrossRef]
- Kvien, I.; Oksman, K. Orientiation of cellulose nanowhiskers in polyvinyl alcohol. Appl. Phys. A 2007, 87, 641–643. [Google Scholar] [CrossRef]
- Li, D.; Liu, Z.; Al-Haik, M.; Tehrani, M.; Murray, F.; Tannenbaum, R.; Garmestani, H. Magnetic alignment of cellulose nanowhiskers in an all-cellulose composite. Polym. Bull. 2010, 65, 635–642. [Google Scholar] [CrossRef]
- Li, D.; Sun, X.; Khaleel, M.A. Materials design of all-cellulose composite using microstructure based finite element analysis. J. Eng. Mater. Technol. 2012, 134, 010911:1–010911:9. [Google Scholar]
- Bordel, D.; Putaux, J.-L.; Heux, L. Orientation of native cellulose in an electric field. Langmuir 2006, 22, 4899–4901. [Google Scholar] [CrossRef]
- Habibi, Y.; Heim, T.; Douillard, R. AC Electric field-assisted assembly and alignment of cellulose nanocrystals. J. Polym. Sci. B Polym. Phys. 2008, 46, 1430–1436. [Google Scholar] [CrossRef]
- Huang, J.; Liu, L.; Yao, J. Electrospinning of Bombyx mori silk fibroin nanofiber mats reinforced by cellulose nanowhiskers. Fiber Polym. 2011, 12, 1002–1006. [Google Scholar] [CrossRef]
- Changsarn, S.; Mendez, J.D.; Shanmuganathan, K.; Foster, E.J.; Weder, C.; Supaphol, P. Biologically inspired hierarchical design of nanocomposites based on poly(ethylene oxide) and cellulose nanofibers. Macromol. Rapid Commun. 2011, 32, 1367–1372. [Google Scholar] [CrossRef]
- Herrera, N.V.; Mathew, A.P.; Wang, L.Y.; Oksman, K. Randomly oriented and aligned cellulose fibres reinforced with cellulose nanowhiskers, prepared by electrospinning. Plast. Rubber Compos. 2011, 40, 57–64. [Google Scholar] [CrossRef]
- Lee, J.; Deng, Y. Increased Mechanical properties of aligned and isotropic electrospun PVA nanofiber webs by cellulose nanowhisker reinforcement. Macromol. Res. 2012, 20, 76–83. [Google Scholar] [CrossRef]
- Sanchez-García, M.D.; Hilliou, L.; Lagarón, J.M. Morphology and water barrier properties of nanobiocomposites of κ/ι-Hybrid carrageenan and cellulose nanowhiskers. J. Agric. Food Chem. 2010, 58, 12847–12857. [Google Scholar] [CrossRef]
- Belbekhouche, S.; Bras, J.; Siqueira, G.; Chappey, C.; Lebrun, L.; Khelifi, B.; Marais, S.; Dufresne, A. Water sorption behavior and gas barrier properties of cellulose whiskers and microfibrils films. Carbohydr. Polym. 2011, 83, 1740–1748. [Google Scholar] [CrossRef]
- Fortunati, E.; Peltzer, M.; Armentano, I.; Torre, L.; Jiménez, A.; Kenny, J.M. Effects of modified cellulose nanocrystals on the barrier and migration propertiesof PLA nano-biocomposites. Carbohydr. Polym. 2012, 90, 948–956. [Google Scholar] [CrossRef]
- Savadekar, N.R.; Mhaske, S.T. Synthesis of nano cellulose fibers and effect on thermoplastics starch based films. Carbohydr. Polym. 2012, 89, 146–151. [Google Scholar] [CrossRef]
- Eyley, S.; Shariki, S.; Dale, S.E.C.; Bending, S.; Marken, F.; Thielemans, W. Ferrocene-decorated nanocrystalline cellulose with charge carrier mobility. Langmuir 2012, 28, 6514–6519. [Google Scholar]
- Eyley, S.; Thielemans, W. Imidazolium grafted cellulose nanocrystals for ion exchange applications. Chem. Commun. 2011, 47, 4177–4179. [Google Scholar] [CrossRef]
- Moreau, C.; Beury, N.; Delorme, N.; Cathala, B. Tuning the architecture of cellulose nanocrystal−poly(allylamine hydrochloride) multilayered thin films: Influence of dipping parameters. Langmuir 2012, 28, 10425–10436. [Google Scholar] [CrossRef]
- Mendez, J.; Annamalai, P.K.; Eichhorn, S.J.; Rusli, R.; Rowan, S.J.; Foster, E.J.; Weder, C. Bioinspired mechanically adaptive polymer nanocomposites with water-activated shape-memory effect. Macromolecules 2011, 44, 6827–6835. [Google Scholar] [CrossRef]
- Kümmerer, K.; Menz, J.; Schubert, T.; Thielemans, W. Biodegradability of organic nanoparticles in the aqueous environment. Chemosphere 2011, 82, 1387–1392. [Google Scholar] [CrossRef]
- Ni, H.; Zeng, S.; Wu, J.; Cheng, X.; Luo, T.; Wang, W.; Zeng, W.; Chen, Y. Cellulose nanowhiskers: Preparation, characterization and cytotoxicity evaluation. Biomed. Mater. Eng. 2012, 22, 121–127. [Google Scholar]
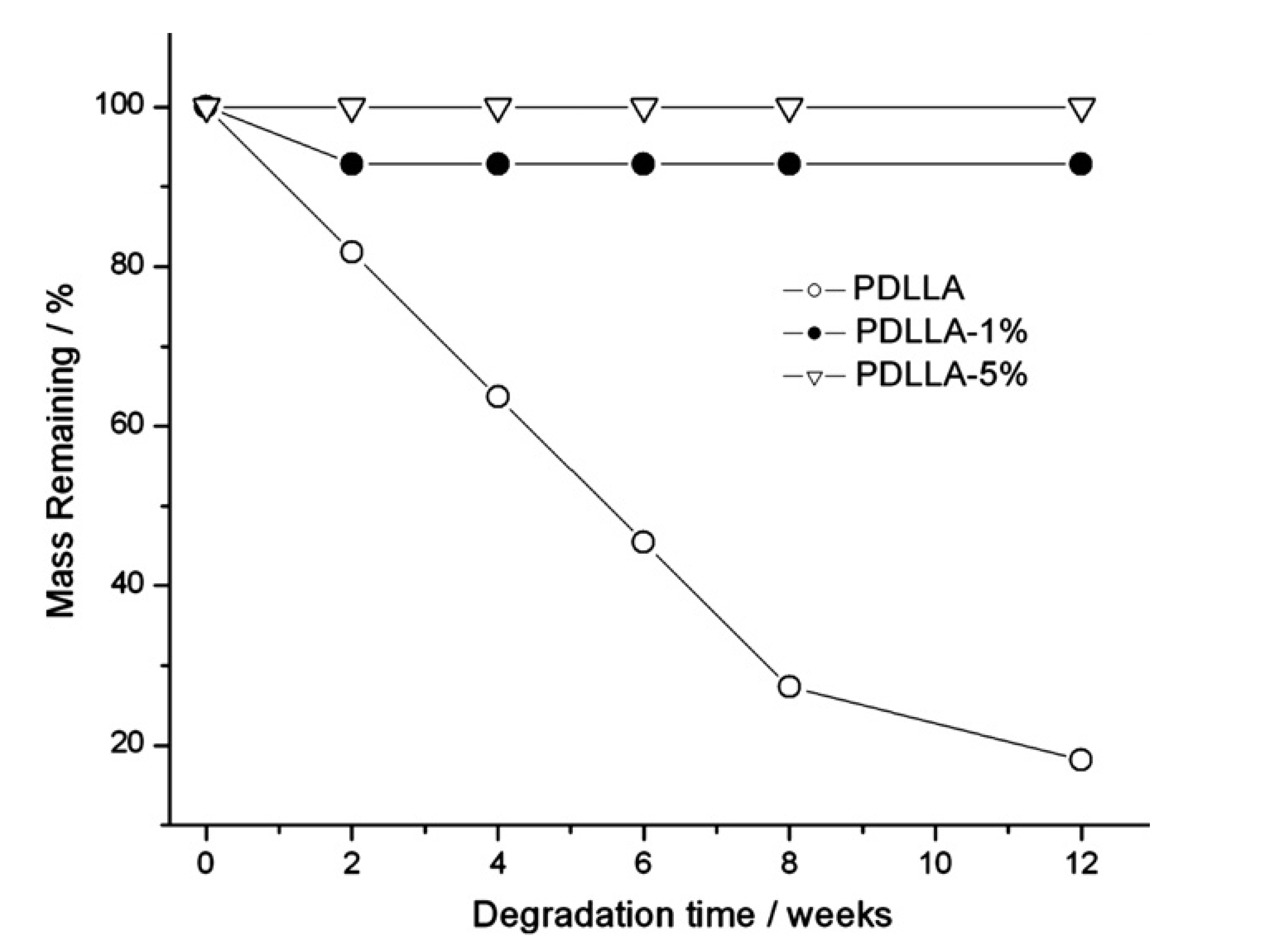
- De Paula, E.L.; Mano, V.; Pereira, F.V. Influence of cellulose nanowhiskers on the hydrolytic degradation behavior of poly(d,l-lactide). Polym. Degrad. Stabil. 2011, 96, 1631–1638. [Google Scholar] [CrossRef]
- Dash, R.; Ragauskas, A.J. Synthesis of a novel cellulose nanowhisker-based drug delivery system. RSC Adv. 2012, 2, 3403–3409. [Google Scholar] [CrossRef]
- Chirat, C.; Lachenal, D.; Dufresne, A. Biorefinery in a kraft pulp mill: From bioethanol to cellulose nanocrystals. Cellul. Chem. Technol. 2010, 44, 59–64. [Google Scholar]
- Oksman, K.; Etang, J.A.; Mathew, A.P.; Jonoobi, M. Cellulose nanowhiskers separated from a bio-residue from wood bioethanol production. Biomass Bioenergy 2011, 35, 146–152. [Google Scholar]
- Peng, F.; Peng, P.; Xu, F.; Sun, R.-C. Fractional purification and bioconversion of hemicelluloses. Biotechnol. Adv. 2012, 30, 879–903. [Google Scholar] [CrossRef]
- Peng, P.; Peng, F.; Bian, J.; Xu, F.; Sun, R.-C.; Kennedy, J.F. Isolation and structural characterization of hemicelluloses from the bamboo species Phyllostachys incarnata Wen. Carbohydr. Polym. 2011, 86, 883–890. [Google Scholar] [CrossRef]
- Haimer, E.; Wendland, M.; Potthast, A.; Rosenau, T.; Liebner, F. Precipitation of hemicelluloses from DMSO/water mixtures using carbon dioxide as an antisolvent. J. Nanomater. 2008, 2008, 1–5. [Google Scholar]
- Haimer, E.; Wendlana, M.; Potthast, A.; Henniges, U.; Rosenau, T.; Liebner, F. Controlled precipitation and purification of hemicellulose from DMSO and DMSO/water mixtures by carbon dioxide as anti-solvent. J. Supercrit. Fluids 2010, 53, 121–130. [Google Scholar] [CrossRef]
- Kaczmar, J.W.; Pach, J.; Burgstaller, C. The chemically treated hemp fibres to reinforce polymers. Polimery 2011, 56, 11–12. [Google Scholar]
- Reddy, K.O.; Guduri, B.R.; Rajulu, A.V. Structural characterization and tensile properties of Borassus fruit fibers. J. Appl. Polym. Sci. 2009, 114, 603–611. [Google Scholar] [CrossRef]
- Zhang, Y.; Hosseinaei, O.; Wang, S.; Zhou, Z. Influence of hemicellulose extraction on water uptake behavior of wood strands. Wood Fiber Sci. 2011, 43, 244–250. [Google Scholar]
- Hosseinaei, O.; Wang, S.; Rials, T.G.; Xing, C.; Taylor, A.M.; Kelley, S.S. Effect of hemicellulose extraction on physical and mechanical properties and mold susceptibility of flakeboard. For. Prod. J. 2011, 61, 31–37. [Google Scholar]
- Chang, W.-P.; Kim, K.-J.; Gupta, R.K. Ultrasound-assisted surface-modification of wood particulates for improved wood/plastic composites. Compos. Interfaces 2009, 16, 687–709. [Google Scholar] [CrossRef]
- Cordeiro, N.; Gouveia, C.; Moraes, A.G.O.; Amico, S.C. Natural fibers characterization by inverse gas chromatography. Carbohydr. Polym. 2011, 84, 110–117. [Google Scholar] [CrossRef]
- Rao, R.S.; Jyothi, C.P.; Prakasham, R.S.; Rao, C.S.; Sarma, P.N.; Rao, L.V. Xylitol production from corn fiber and sugarcane bagasse hydrolysates by Candida tropicalis. Bioresour. Technol. 2006, 97, 1974–1978. [Google Scholar] [CrossRef]
- Li, M.; Meng, X.; Diao, E.; Du, F. Xylitol production by Candida tropicalis from corn cob hemicellulose hydrolysate in a two-stage fed-batch fermentation process. J. Chem. Technol. Biotechnol. 2012, 87, 387–392. [Google Scholar] [CrossRef]
- Silva, C.J.S.M.; Mussatto, S.S.; Roberto, I.C. Study of xylitol production by Candida guilliermondii on a bench bioreactor. J Food Eng. 2006, 75, 115–119. [Google Scholar] [CrossRef]
- Mayerhoff, Z.D.V.L.; Roberto, I.C.; Silva, S.S. Xylitol production from rice straw hemicellulose using different yeast strains. Biotechnol. Lett. 1997, 19, 407–409. [Google Scholar] [CrossRef]
- Cruz, J.M.; Domínguez, J.M.; Domínguez, H.; Parajó, J.C. Solvent extraction of hemicellulosic wood hydrolysates: A procedure useful for obtaining both detoxified fermentation media and polyphenols with antioxidant activity. Food Chem. 1999, 67, 147–153. [Google Scholar] [CrossRef]
- Stepan, A.M.; Höije, A.; Schols, H.A.; de Waard, P.; Gatenholm, P. Arabinose content of arabinoxylans contributes to flexibility of acetylated arabinoxylan films. J. Appl. Polym. Sci. 2012, 125, 2348–2355. [Google Scholar] [CrossRef]
- Stevanic, J.S.; Bergström, E.M.; Gatenholm, P.; Berglund, L.; Salmén, L. Arabinoxylan/nanofibrillated cellulose composite films. J. Mater. Sci. 2012, 47, 6724–6732. [Google Scholar] [CrossRef]
- Edlund, U.; Yu, Y.; Zhu Ryberg, Y.; Krause-Rehberg, R.; Albertsson, A.-C. Positron lifetime reveals the nano level packing in complex polysaccharide-rich hydrolysate matrixes. Anal. Chem. 2012, 84, 3676–3681. [Google Scholar] [CrossRef]
- Voepel, J.; Edlund, U.; Albertsson, A.-C. Alkenyl-functionalized precursors for renewable hydrogels design. J. Polym. Sci. Polym. Chem. 2009, 47, 3595–3606. [Google Scholar] [CrossRef]
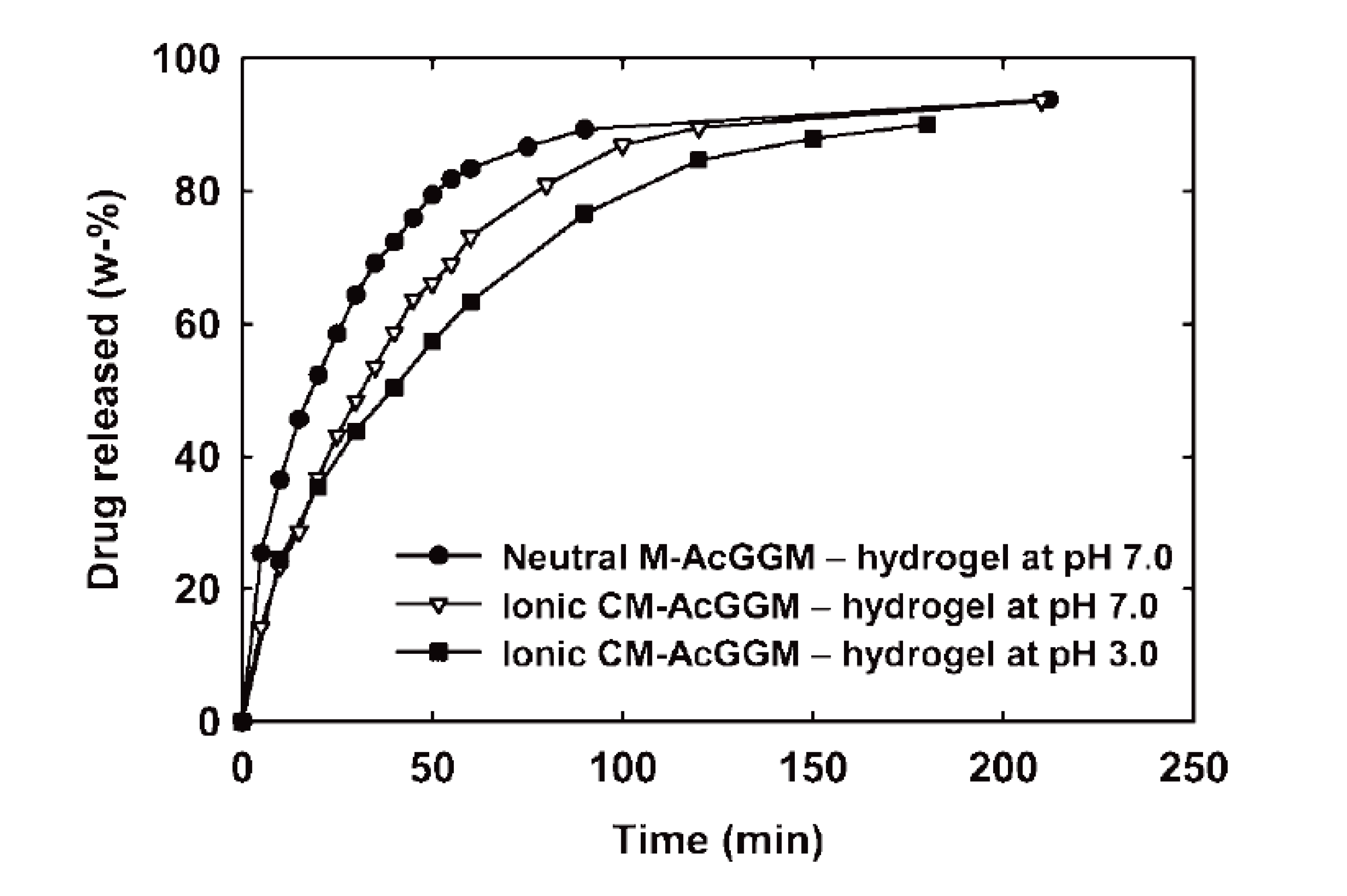
- Voepel, J.; Sjöberg, J.; Reif, M.; Albertsson, A.-C.; Hultin, U.-K.; Gasslander, U. Drug diffusion in neutral and ionic hydrogels assembled from acetylated galactoglucomannan. J. Appl. Polym. Sci. 2009, 112, 2401–2412. [Google Scholar] [CrossRef]
- Silva, T.C.F.; Habibi, Y.; Colodette, J.L.; Lucia, L.A. The influence of the chemical and structural features of xylan on the physical properties of its derived hydrogels. Soft Matter 2011, 7, 1090–1099. [Google Scholar] [CrossRef]
- Karaaslan, M.A.; Tshabalala, M.A.; Buschle-Diller, G. Wood Hemicellulose/chitosan-based semi-interpenetrating network hydrogels: Mechanical swelling and controlled drug release properties. Bioresources 2010, 5, 1036–1054. [Google Scholar]
- Karaaslan, M.A.; Tshabalala, M.A.; Yelle, D.J.; Buschle-Diller, G. Nanoreinforced biocompatible hydrogels from wood hemicelluloses and cellulose whiskers. Carbohydr. Polym. 2011, 86, 192–201. [Google Scholar] [CrossRef]
- Paës, G.; Chabbert, B. Characterization of arabinoxylan/cellulose nanocrystals gels to investigate fluorescent probes mobility in bioinspired models of plant secondary cell wall. Biomacromolecules 2012, 13, 206–214. [Google Scholar] [CrossRef]
- Peng, X.-W.; Ren, J.-L.; Zhong, L.-X.; Peng, F.; Sun, R.-C. Xylan-rich hemicelluloses-graft-acrylic acid ionic hydrogels with rapid responses to pH, salt, and organic solvents. J. Agric. Food Chem. 2011, 59, 8208–8215. [Google Scholar] [CrossRef]
- Peng, X.-W.; Zhong, L.-X.; Ren, J.-L.; Sun, R.-C. Highly effective adsorption of heavy metal ions from aqueous solutions by macroporous xylan-rich hemicelluloses-based hydrogel. J. Agric. Food Chem. 2012, 60, 3909–3916. [Google Scholar] [CrossRef]
- Pohjanlehto, H.; Setälä, H.; Kammiovirta, K.; Harlin, A. The use of N,N′-diallylaldardiamides as cross-linkers in xylan derivatives-based hydrogels. Carbohydr. Res. 2011, 346, 2736–2745. [Google Scholar]
- Oliveira, E.E.; Silva, A.E.; Nagashima, T.; Gomes, M.C.S.; Aguiar, L.M.; Marcelino, H.R.; Araújo, I.B.; Bayer, M.P.; Ricardo, N.M.P.S.; Oliveira, A.O.; et al. Xylan from corn cobs, a promising polymer for drug delivery: Production and characterization. Bioresour. Technol. 2010, 101, 5402–5406. [Google Scholar] [CrossRef]
- Olsson, A.; Salmen, L. Viscoelasticity of in situ lignin as affected by structure softwood vs. hardwood. ACS Symp. Ser. 1992, 489, 133–143. [Google Scholar] [CrossRef]
- Szczesniak, L.; Rachocki, A.; Tritt-Goc, J. Glass transition temperature and thermal decomposition of cellulose powder. Cellulose 2008, 15, 445–451. [Google Scholar] [CrossRef]
- Naqvi, M.; Yan, J.; Dahlquist, E. Black liquor gasification integrated in pulp and paper mills: A critical review. Bioresour. Technol. 2010, 101, 8001–8015. [Google Scholar] [CrossRef]
- Kang, S.; Li, X.; Fan, J.; Chang, J. Solid fuel production by hydrothermal carbonization of black liquor. Bioresour. Technol. 2012, 110, 715–718. [Google Scholar] [CrossRef]
- Braaten, S.; Christensen, B.; Fredheim, G. Comparison of molecular weight and molecular weight distributions of softwood and hardwood lignosulfonates. J. Wood Chem. Technol. 2003, 23, 197–215. [Google Scholar] [CrossRef]
- Yu, G.; Li, B.; Wang, H.; Liu, C.; Mu, X. Preparation of concrete superplasticizer by oxidation sulfomethylation of sodium lignosulfonate. Bioresources 2013, 8, 1055–1063. [Google Scholar]
- Mittal, M.; Sharma, C. Studies on lignin-based adhesives for plywood panels. Polym. Int. 2007, 29, 7–8. [Google Scholar] [CrossRef]
- Nadji, H.; Rodrigue, D.; Benaboura, A.; Bedard, Y.; Stevanovic, T.; Riedl, B. Value-added derivatives of soda lignin alfa grass (Stipa tenacissima). II. Uses as lubricants in plastic processing. J. Appl. Polym. Sci. 2009, 114, 3003–3007. [Google Scholar] [CrossRef]
- Ibrahim, M.; Azreena, M.; Ismail, M. Lignin graft copolymer as a drilling mud thinner for high temperature well. J. Appl. Sci. 2006, 6, 1808–1813. [Google Scholar] [CrossRef]
- Windeisen, E.; Wegener, G. Lignin as building unit for polymers. Polym. Sci. 2012, 10, 255–265. [Google Scholar]
- Kumar, M.; Mohanty, A.; Erickson, L.; Misra, M. Lignin and its applications with polymers. J. Biobased Mater. Bioenergy 2009, 3, 1–24. [Google Scholar] [CrossRef]
- Doherty, W.; Mousavioun, P.; Fellows, C. Value-adding to cellulosic ethanol: Lignin polymers. Ind. Crop Prod. 2011, 33, 259–276. [Google Scholar] [CrossRef] [Green Version]
- Johansson, K.; Winestrand, S.; Johansson, C.; Jarnstrom, L.; Jonsson, L. Oxygen-scavenging coatings and films based on lignosulfonates and laccase. J. Biotechnol. 2012, 161, 14–18. [Google Scholar]
- Milczarek, G. Kraft lignin as dispersing agent for carbon nanotubes. J. Electroanal. Chem. 2010, 638, 178–181. [Google Scholar] [CrossRef]
- Faria, F.; Evtuguin, D.; Rudnitskaya, A.; Gomes, M.; Oliveira, J.; Graça, M.; Costa, L. Lignin-basedpolyurethane doped with carbon nanotubes for sensor applications. Polym. Int. 2012, 61, 788–794. [Google Scholar] [CrossRef]
- Rudnitskaya, A.; Evtuguin, D.; Costa, L.; Graça, M.; Fernandes, A.; Correia, M.; Gomes, M.; Oliveira, J. Potentiometric chemical sensors from lignin-poly(propylene oxide) copolymers doped by carbon nanotubes. Analyst 2013, 138, 501–508. [Google Scholar] [CrossRef]
- Milczarek, G.; Inganäs, O. Renewable cathode materials from biopolymer/conjugated polymer interpenetrating networks. Science 2012, 335, 1468–1471. [Google Scholar] [CrossRef]
- Bhirde, A.; Sousa, A.A.; Patel, V.; Azari, A.; Gutkind, J.S.; Leapman, R.D.; Rusling, J.F. Imaging the distribution of individual platinum-based anticancer drug molecules attached to single-wall carbon nanotubes. Nanomedicine 2009, 4, 763–772. [Google Scholar] [CrossRef]
- Li, R.; Wu, R.; Zhao, L.; Wu, M.; Yang, L.; Zou, H. P-glycoprotein antibody functionalized carbon nanotube overcomes the multidrug resistance of human leukemia cells. ACS Nano 2010, 4, 1399–1408. [Google Scholar] [CrossRef]
- Thostenson, E.; Ren, Z.; Chou, T. Advances in the science and technology of carbon nanotubes and their composites: A review. Compos. Sci. Technol. 2001, 61, 1899–1912. [Google Scholar] [CrossRef]
- Kang, Z.; Wang, E.; Mao, B.; Su, Z.; Chen, L.; Xu, L. Obtaining carbon nanotubes from grass. Nanotechnology 2005, 16, 1192–1195. [Google Scholar] [CrossRef]
- Shin, Y.; Wang, C.; Li, X.; Exarhos, G. Synthesis of supportedcarbon nanotubes in mineralized silica-wood composites. Carbon 2005, 43, 1096–1098. [Google Scholar] [CrossRef]
- Xie, X.; Goodell, B.; Qian, Y.; Daniel, G.; Zhang, D.; Nagle, D.; Peterson, M.; Jellison, J. A Method of producing carbon nanotubes directly from plant materials. For. Prod. J. 2009, 59, 26–28. [Google Scholar]
- Magrez, A.; Kasas, S.; Salicio, V.; Pasquier, N.; Seo, J.; Celio, M.; Catsicas, S.; Schwaller, B.; Forro, L. Cellular toxicity of carbon-based nanomaterials. Nano Lett. 2006, 6, 1121–1125. [Google Scholar] [CrossRef]
- Poland, C.; Duffin, R.; Kinloch, I.; Maynard, A.; Wallace, W.; Seaton, A.; Stone, V.; Brown, S.; MacNee, W.; Donaldson, K. Carbon Nanotubes introduced into the abdominal cavity of mice show asbestos-like pathogenicity in a pilot study. Nat. Nanotechnol. 2008, 3, 423–428. [Google Scholar] [CrossRef]
- Caicedo, H.M.; Dempere, A.L.; Vermerris, W. Template-mediated synthesis and bio-functionalization of flexible lignin-based nanotubes and nanowires. Nanotechnology 2012, 23, 105605. [Google Scholar] [CrossRef]
- Vermerris, W. Composition and biosynthesis of lignocellulosic biomass. In Genetic Improvement of Bioenergy Crops; Vermerris, W., Ed.; Springer: New York, NY, USA, 2008; pp. 89–142. [Google Scholar]
- Vanholme, R.; Van Acker, R.; Boerjan, W. Potential of Arabidopsis systems biology to advance the biofuel field. Trends Biotechnol. 2010, 28, 543–547. [Google Scholar] [CrossRef]
- Vermerris, W. Survey of genomics approaches to improve bioenergy traits in maize, sorghum and sugarcane. J. Integr. Plant Biol. 2011, 53, 105–119. [Google Scholar] [CrossRef]
- Pilate, G.; Guiney, E.; Holt, K.; Petit-Conil, M.; Lapierre, C.; Leplé, J.; Pollet, B.; Mila, I.; Webster, E.; Marstorp, H.; et al. Field and pulping performances of transgenic trees with altered lignification. Nat. Biotechnol. 2002, 20, 607–612. [Google Scholar] [CrossRef]
- Herrera, S. Struggling to see the forest through the trees. Nat. Biotechnol. 2005, 23, 165–167. [Google Scholar] [CrossRef]
- Chen, F.; Dixon, R.A. Lignin modification improves fermentable sugar yields for biofuel production. Nat. Biotechnol. 2007, 25, 759–761. [Google Scholar] [CrossRef]
- Vermerris, W.; Saballos, A.; Ejeta, G.; Mosier, N.S.; Ladisch, M.R.; Carpita, N.C. Molecular breeding to enhance ethanol production from corn and sorghum stover. Crop Sci. 2007, 47, S142–S153. [Google Scholar]
- Vanholme, R.; Morreel, K.; Darrah, C.; Oyarce, P.; Grabber, J.; Ralph, J.; Boerjan, W. Metabolic engineering of novel lignin in biomass crops. New Phytol. 2012, 196, 978–1000. [Google Scholar] [CrossRef]
- Davison, B.H.; Drescher, S.R.; Tuskan, G.A.; Davis, M.F.; Nghiem, N.P. Variation of S/G ratio and lignin content in a Populus family influences the release of xylose by dilute acid hydrolysis. Appl. Biochem. Biotechnol. 2006, 129-132, 427–435. [Google Scholar]
- Fu, C.; Mielenz, J.; Xiao, X.; Ge, Y.; Hamilton, C.; Rodriguez, M.; Chen, F.; Foston, M.; Ragauskas, A.; Bouton, J.; Dixon, R.; Wang, Z. Genetic manipulation of lignin reduces recalcitrance and improves ethanol production from switchgrass. Proc. Natl. Acad. Sci. USA. 2011, 108, 3803–3808. [Google Scholar] [CrossRef]
- Jung, J.H.; Fouad, W.M.; Vermerris, W.; Gallo, M.; Altpeter, F. RNAi suppression of lignin biosynthesis in sugarcane reduces recalcitrance for biofuel production from lignocellulosic biomass. Plant Biotechnol. J. 2012, 10, 1067–1076. [Google Scholar] [CrossRef]
- Sattler, S.E.; Palmer, N.A.; Saballos, A.; Greene, A.M.; Xin, Z.; Sarath, G.; Vermerris, W.; Pedersen, J.F. Identification and characterization of 4 missense mutations in Brown midrib 12 (Bmr12); the caffeic O-methyltranferase (COMT) of sorghum. Bioenerg. Res. 2012, 5, 855–865. [Google Scholar] [CrossRef]
- Sattler, S.E.; Funnell-Harris, D.L.; Pedersen, J.F. Brown midrib mutations and their importance to the utilization of maize, sorghum, and pearl millet lignocellulosic tissues. Plant Sci. 2010, 178, 229–238. [Google Scholar] [CrossRef]
- Sindhu, A.; Langewisch, T.; Olek, A.; Multani, D.S.; McCann, M.C.; Vermerris, W.; Carpita, N.C.; Johal, G. Maize Brittle stalk2 encodes a COBRA-Like protein expressed in early organ development but required for tissue flexibility at maturity. Plant Physiol. 2007, 145, 1444–1459. [Google Scholar] [CrossRef]
- 250. Penning, B.W.; Hunter, C.T., III; Tayengwa, R.; Eveland, A.L.; Dugard, C.K.; Olek, A.T.; Vermerris, W.; Koch, K.E.; McCarty, D.R.; Davis, M.F.; Thomas, S.R.; McCann, M.C.; Carpita, N.C. Genetic resources for maize cell wall biology. Plant Physiol. 2009, 151, 1703–1728. [Google Scholar] [CrossRef]
- Xin, Z.; Wang, M.L.; Barkley, N.A.; Burow, G.; Franks, C.; Pederson, G.; Burke, J. Applying genotyping (TILLING) and phenotyping analyses to elucidate gene function in a chemically induced sorghum mutant population. BMC Plant Biol. 2008, 8, 103. [Google Scholar] [CrossRef]
- Vermerris, W.; Saballos, A. Genetic enhancement of sorghum for biomass utilization. In Genetics and Genomics of the Saccharinae; Paterson, A., Ed.; Springer: New York, NY, USA, 2012; pp. 391–428. [Google Scholar]
- Vandenbrink, J.; Hilten, R.; Das, K.; Paterson, A.; Feltus, F. Analysis of crystallinity index and hydrolysis rates in the bioenergy crop Sorghum bicolor. Bioenerg. Res. 2012, 5, 387–397. [Google Scholar] [CrossRef]
- Studer, M.; Brethauer, S.; DeMartini, J.; McKenzie, H.; Wyman, C. Co-hydrolysis of hydrothermal and dilute acid pretreated Populus slurries to support development of a high-throughput pretreatment system. Biotechnol. Biofuels 2011, 4, 19–29. [Google Scholar] [CrossRef]
- Vermerris, W.; Sherman, D.M.; McIntyre, L.M. Phenotypic plasticity in cell walls of maize brown midrib mutants is limited by lignin composition. J. Exp. Bot. 2010, 61, 2479–2490. [Google Scholar] [CrossRef]
- Funnell-Harris, D.; Pedersen, J.; Sattler, S. Alteration in lignin biosynthesis restricts growth of Fusarium spp. in brown midrib sorghum. Phytopathology 2010, 100, 671–681. [Google Scholar] [CrossRef]
- Jackson, L.A.; Shadle, G.L.; Zhou, R.; Nakashima, J.; Chen, F.; Dixon, R.A. Improving Saccharification Efficiency of alfalfa stems through modification of the terminal stages of monolignol biosynthesis. Bioenerg. Res. 2008, 1, 180–192. [Google Scholar] [CrossRef]
- Lee, Y.; Chen, F.; Gallego-Giraldo, L.; Dixon, R.A.; Voit, E.O. Integrative analysis of transgenic alfalfa (Medicago sativa L.) suggests new metabolic control mechanisms for monolignol biosynthesis. PLoS Comput. Biol. 2011, 7, 1–13. [Google Scholar]
- Vanholme, R.; Storme, V.; Vanholme, B.; Sundin, L.; Christensen, J.; Goeminne, G.; Halpin, C.; Rohde, A.; Morreel, K.; Boerjan, W. A Systems biology view of responses to lignin biosynthesis perturbations in Arabidopsis. Plant Cell. 2012, 24, 3506–3529. [Google Scholar] [CrossRef] [Green Version]
- Eranki, P.L.; Bals, B.D.; Dale, B.E. Advanced regional biomass processing depots: A key to the logistical challenges of the cellulosic biofuel industry. Biofuels Bioprod. Bioref. 2011, 5, 621–630. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ten, E.; Vermerris, W. Functionalized Polymers from Lignocellulosic Biomass: State of the Art. Polymers 2013, 5, 600-642. https://doi.org/10.3390/polym5020600
Ten E, Vermerris W. Functionalized Polymers from Lignocellulosic Biomass: State of the Art. Polymers. 2013; 5(2):600-642. https://doi.org/10.3390/polym5020600
Chicago/Turabian StyleTen, Elena, and Wilfred Vermerris. 2013. "Functionalized Polymers from Lignocellulosic Biomass: State of the Art" Polymers 5, no. 2: 600-642. https://doi.org/10.3390/polym5020600