Surface Initiated Polymerizations via e-ATRP in Pure Water


Abstract
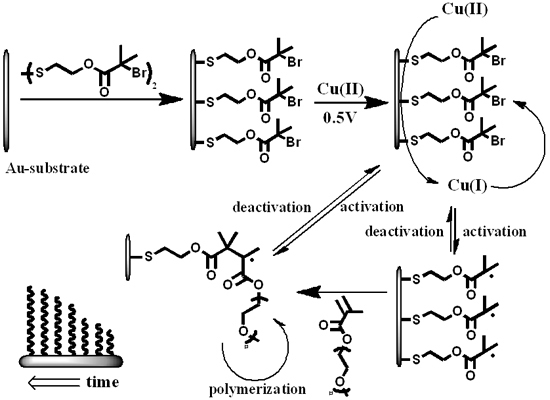
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

2. Experimental Section
2.1. Materials
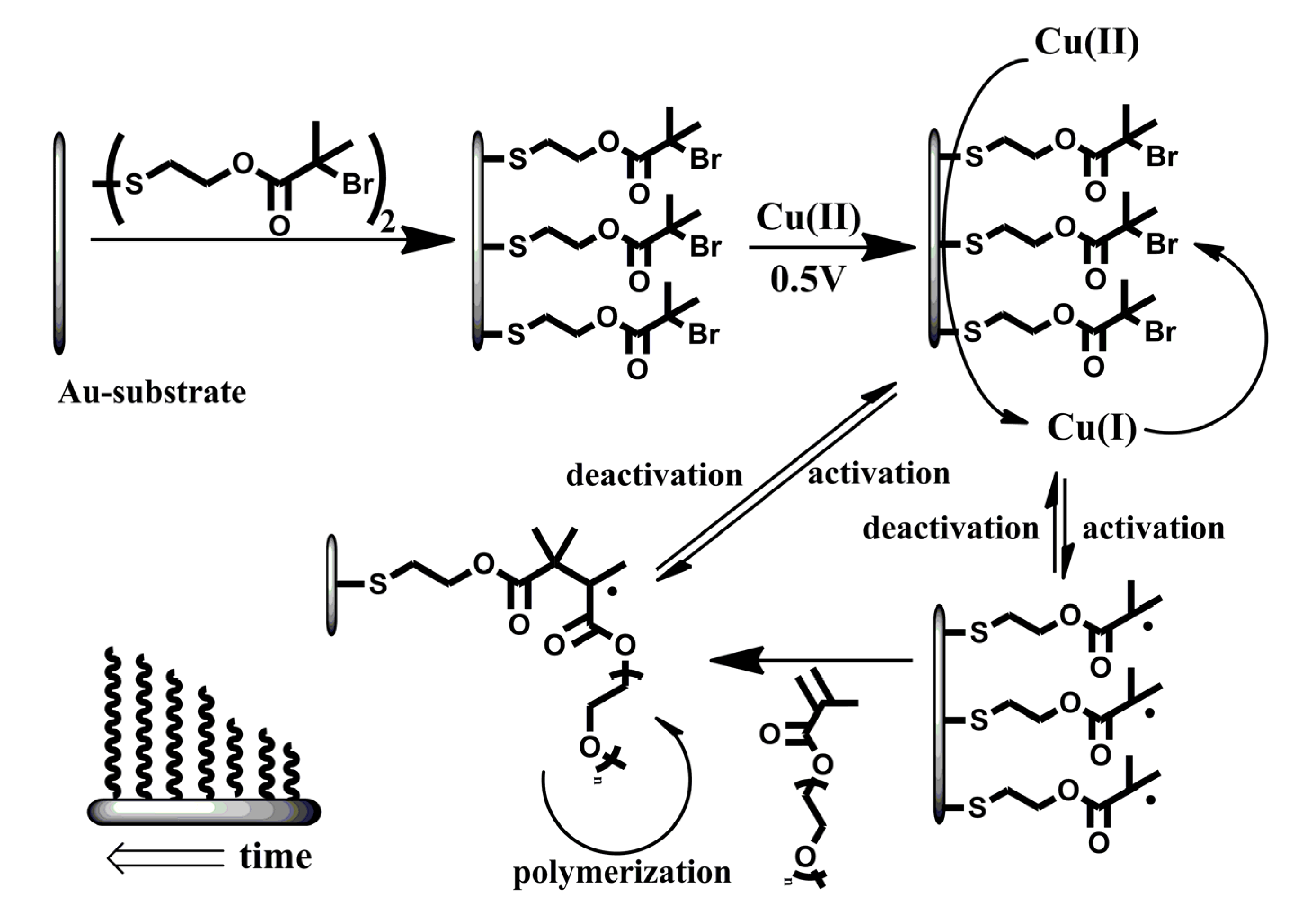
2.2. Electrochemical Polymerization
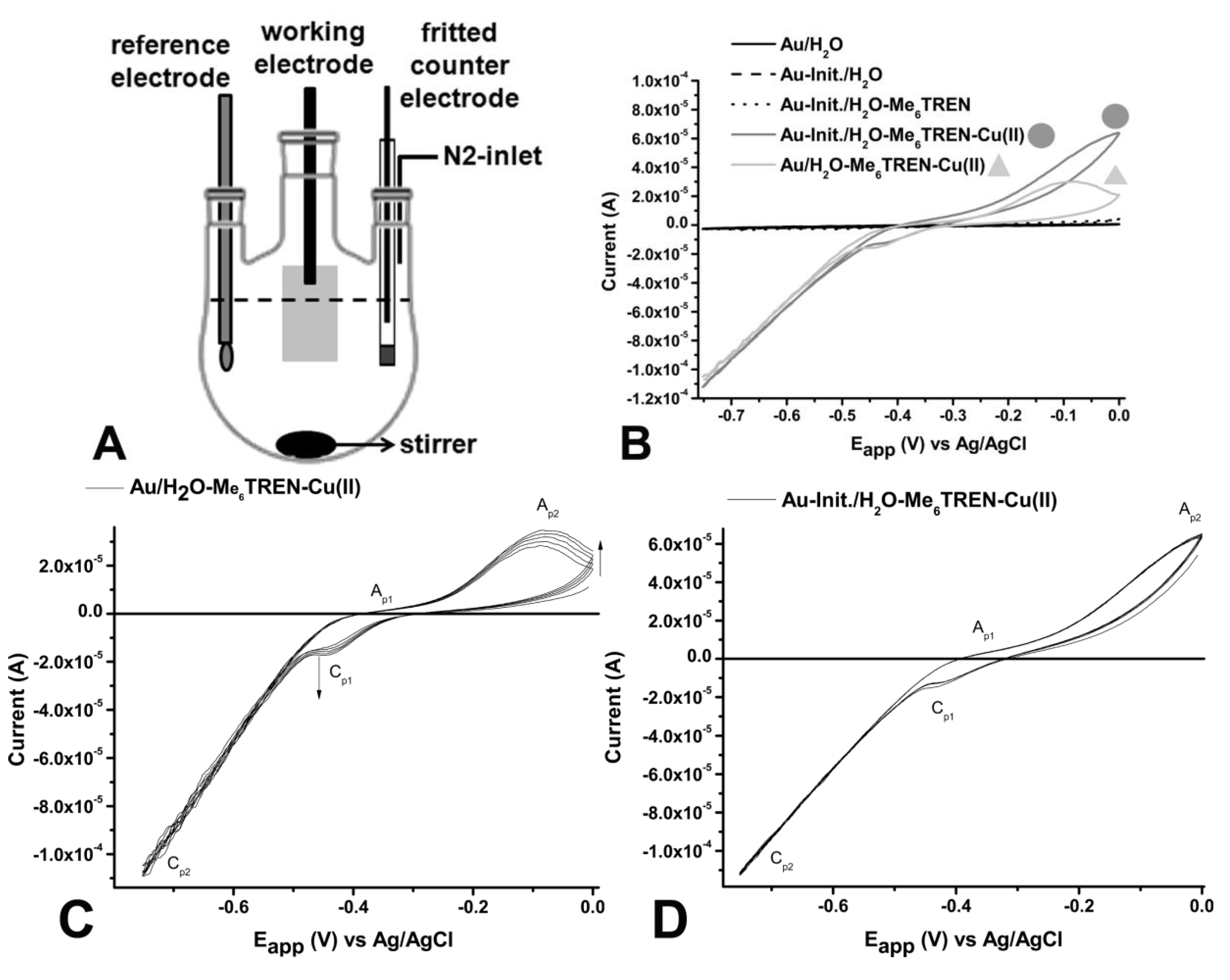
 ) and Cu(II)Br2/Me6TREN (1:1) on unmodified gold-electrode (—,
) and Cu(II)Br2/Me6TREN (1:1) on unmodified gold-electrode (—,  ) recorded at scan speed (v) v = 0.02 V∙s−1; (C) Cyclic voltammograms of flat unmodified gold-electrodes in degassed MilliQ-water in the presence of 1mM Cu(II)Br2/Me6TREN (1:1) recorded at v = 0.02 V∙s−1. Arrows indicate the direction to how changes in signals progress with each cycle; and (D) Cyclic voltammograms of flat with initiator modified gold-electrodes in degassed MilliQ-water in the presence of 1 mM Cu(II)Br2/Me6TREN (1:1) recorded at v = 0.02 V∙s−1. The cyclic voltammogram does not display any changes over multiple runs.
) and Cu(II)Br2/Me6TREN (1:1) on unmodified gold-electrode (—, ) recorded at scan speed (v) v = 0.02 V∙s−1; (C) Cyclic voltammograms of flat unmodified gold-electrodes in degassed MilliQ-water in the presence of 1mM Cu(II)Br2/Me6TREN (1:1) recorded at v = 0.02 V∙s−1. Arrows indicate the direction to how changes in signals progress with each cycle; and (D) Cyclic voltammograms of flat with initiator modified gold-electrodes in degassed MilliQ-water in the presence of 1 mM Cu(II)Br2/Me6TREN (1:1) recorded at v = 0.02 V∙s−1. The cyclic voltammogram does not display any changes over multiple runs.
) recorded at scan speed (v) v = 0.02 V∙s−1; (C) Cyclic voltammograms of flat unmodified gold-electrodes in degassed MilliQ-water in the presence of 1mM Cu(II)Br2/Me6TREN (1:1) recorded at v = 0.02 V∙s−1. Arrows indicate the direction to how changes in signals progress with each cycle; and (D) Cyclic voltammograms of flat with initiator modified gold-electrodes in degassed MilliQ-water in the presence of 1 mM Cu(II)Br2/Me6TREN (1:1) recorded at v = 0.02 V∙s−1. The cyclic voltammogram does not display any changes over multiple runs.
) and Cu(II)Br2/Me6TREN (1:1) on unmodified gold-electrode (—, ) recorded at scan speed (v) v = 0.02 V∙s−1; (C) Cyclic voltammograms of flat unmodified gold-electrodes in degassed MilliQ-water in the presence of 1mM Cu(II)Br2/Me6TREN (1:1) recorded at v = 0.02 V∙s−1. Arrows indicate the direction to how changes in signals progress with each cycle; and (D) Cyclic voltammograms of flat with initiator modified gold-electrodes in degassed MilliQ-water in the presence of 1 mM Cu(II)Br2/Me6TREN (1:1) recorded at v = 0.02 V∙s−1. The cyclic voltammogram does not display any changes over multiple runs.
3. Results and Discussion
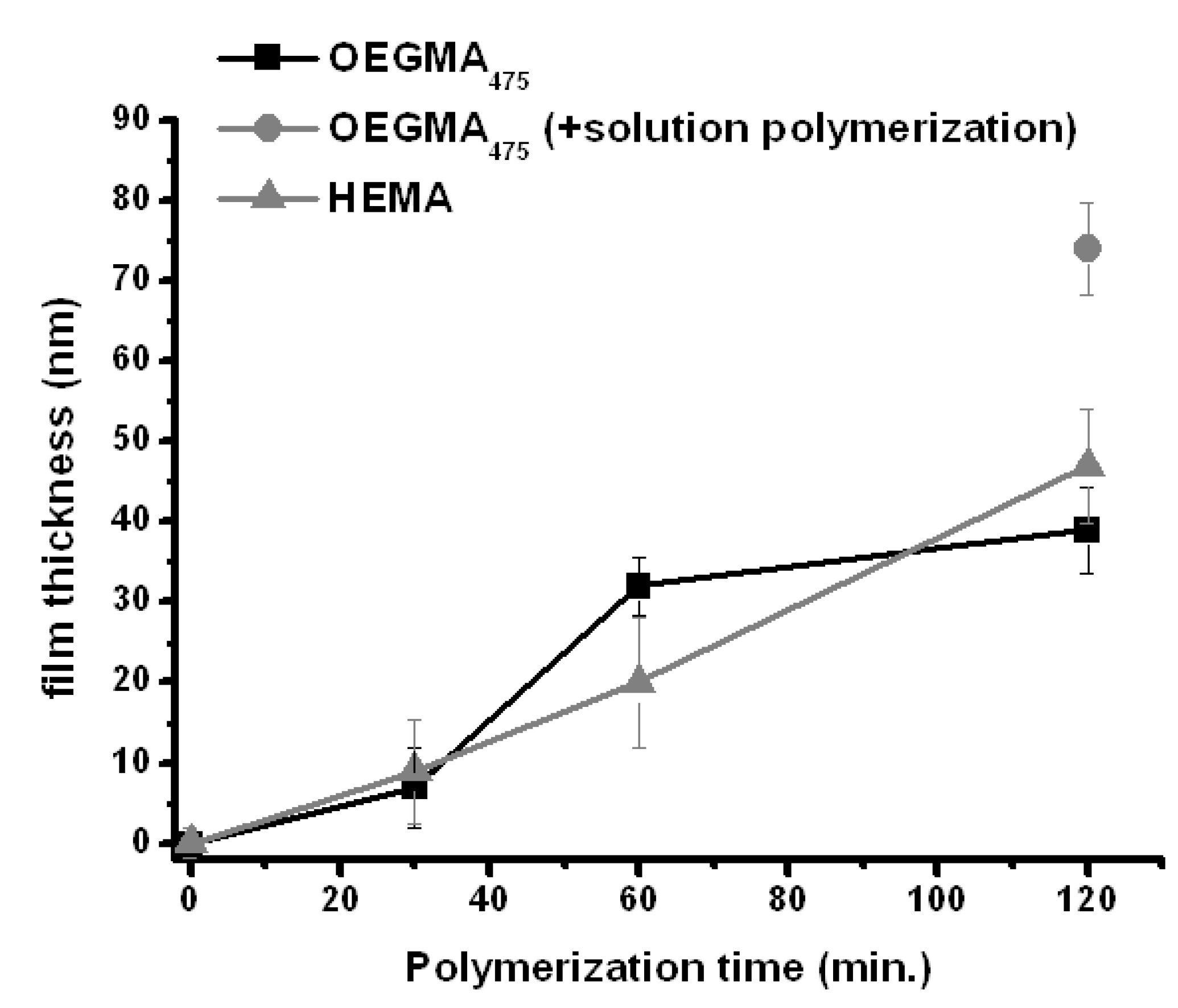
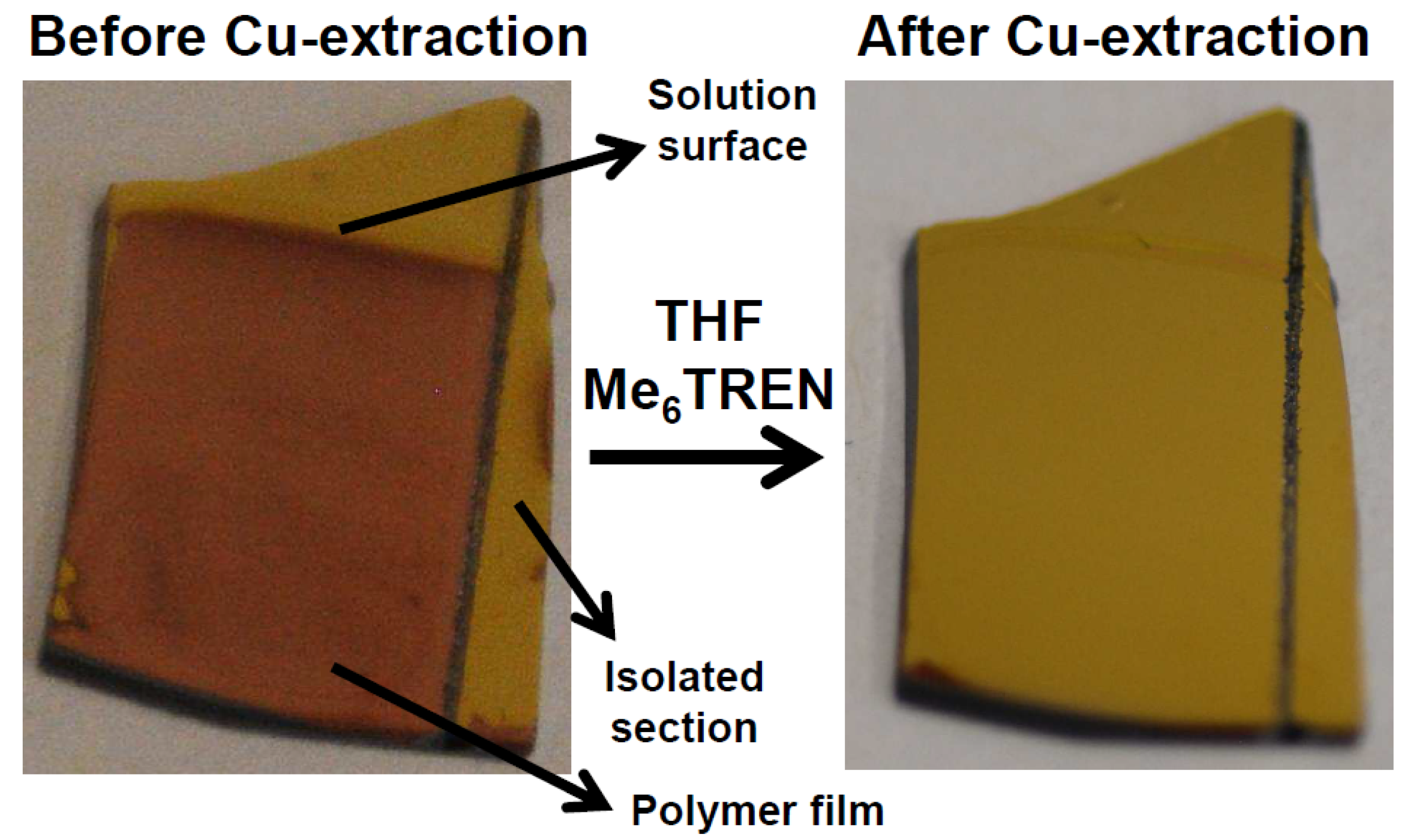
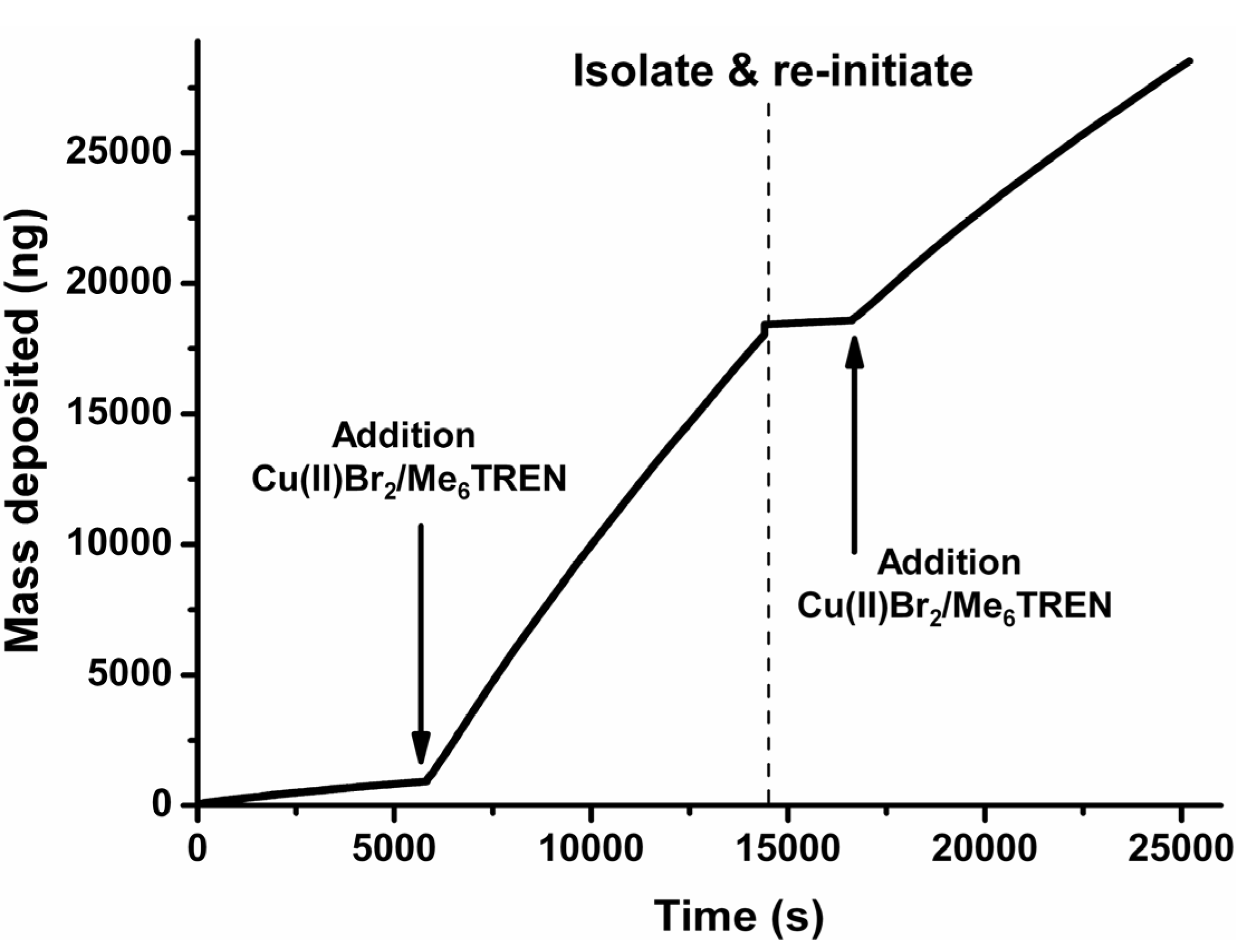
 ). Additionally OEGMA475 was polymerized with the connecting electrode positioned also in solution to see the effect on film thickness of additional solution-based Cu(I)-formation (
). Additionally OEGMA475 was polymerized with the connecting electrode positioned also in solution to see the effect on film thickness of additional solution-based Cu(I)-formation (  ). Error bars depict the standard deviation for the analyzed sample which is obtained from large area analysis.
). Additionally OEGMA475 was polymerized with the connecting electrode positioned also in solution to see the effect on film thickness of additional solution-based Cu(I)-formation ( ). Error bars depict the standard deviation for the analyzed sample which is obtained from large area analysis.
). Error bars depict the standard deviation for the analyzed sample which is obtained from large area analysis.
). Additionally OEGMA475 was polymerized with the connecting electrode positioned also in solution to see the effect on film thickness of additional solution-based Cu(I)-formation ( ). Error bars depict the standard deviation for the analyzed sample which is obtained from large area analysis.


4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Cunningham, M.F. Controlled/living radical polymerization in aqueous dispersed systems. Prog. Polym. Sci. 2008, 33, 365–398. [Google Scholar] [CrossRef]
- Qiu, J.; Charleux, B.; Matyjaszewski, K.; Pierre, Â.; Curie, M. Controlled/living radical polymerization in aqueous media: Homogeneous and heterogeneous systems. Prog. Polym. Sci. 2001, 26, 2083–2134. [Google Scholar]
- Tsarevsky, N.V.; Matyjaszewski, K. “Green” atom transfer radical polymerization: From process design to preparation of well-defined environmentally friendly polymeric materials. Chem. Rev. 2007, 107, 2270–2299. [Google Scholar]
- Matyjaszewski, K.; Tsarevsky, N.V. Nanostructured functional materials prepared by atom transfer radical polymerization. Nat. Chem. 2009, 1, 276–288. [Google Scholar] [CrossRef]
- Edmondson, S.; Osborne, V.L.; Huck, W.T.S. Polymer brushes via surface-initiated polymerizations. Chem. Soc. Rev. 2004, 33, 14–22. [Google Scholar]
- Stuart, M.A.C.; Huck, W.T.; Genzer, J.; Müller, M.; Ober, C.; Stamm, M.; Sukhorukov, G.B.; Szleifer, I.; Tsukruk, V.V.; Urban, M.; et al. Emerging applications of stimuli-responsive polymer materials. Nat. Mater. 2010, 9, 101–113. [Google Scholar] [CrossRef]
- Minko, S. Responsive polymer brushes. J. Macromol. Sci. Polym. Rev. 2006, 46, 397–420. [Google Scholar] [CrossRef]
- Ramstedt, M.; Cheng, N.; Azzaroni, O.; Mossialos, D.; Mathieu, H.J.; Huck, W.T. Synthesis and characterization of poly(3-sulfopropylmethacrylate) brushes for potential antibacterial applications. Langmuir 2007, 23, 3314–3321. [Google Scholar]
- Kenawy, E.-R.; Worley, S.D.; Broughton, R. The chemistry and applications of antimicrobial polymers: A state-of-the-art review. Biomacromolecules 2007, 8, 1359–1384. [Google Scholar]
- Ma, H.; Hyun, J.; Stiller, P.; Chilkoti, A. “Non-fouling” oligo(ethylene glycol)-functionalized polymer brushes synthesized by surface-initiated atom transfer radical polymerization. Adv. Mater. 2004, 16, 338–341. [Google Scholar] [CrossRef]
- Feng, W.; Gao, X.; McClung, G.; Zhu, S.; Ishihara, K.; Brash, J.L. Methacrylate polymer layers bearing poly(ethylene oxide) and phosphorylcholine side chains as non-fouling surfaces: In vitro interactions with plasma proteins and platelets. Acta Biomater. 2011, 7, 3692–3699. [Google Scholar]
- Zhang, Z.; Chen, S.; Jiang, S. Dual-functional biomimetic materials: Nonfouling poly(carboxybetaine) with active functional groups for protein immobilization. Biomacromolecules 2006, 7, 3311–3315. [Google Scholar]
- Ladd, J.; Zhang, Z.; Chen, S.; Hower, J.C.; Jiang, S. Zwitterionic polymers exhibiting high resistance to nonspecific protein adsorption from human serum and plasma. Biomacromolecules 2008, 9, 1357–1361. [Google Scholar] [CrossRef]
- Sun, L.; Baker, G.L.; Bruening, M.L. Polymer brush membranes for pervaporation of organic solvents from water. Macromolecules 2005, 38, 2307–2314. [Google Scholar]
- Zhou, F.; Biesheuvel, P.M.; Choi, E.Y.; Shu, W.; Poetes, R.; Steiner, U.; Huck, W.T. Polyelectrolyte brush amplified electroactuation of microcantilevers. Nano Lett. 2008, 8, 725–730. [Google Scholar] [CrossRef]
- Ma, H.; He, J.; Liu, X.; Gan, J.; Jin, G.; Zhou, J. Surface initiated polymerization from substrates of low initiator density and its applications in biosensors. ACS Appl. Mater. Interfaces 2010, 2, 3223–3230. [Google Scholar]
- Braunecker, W.A.; Matyjaszewski, K. Controlled/living radical polymerization: Features, developments, and perspectives. Prog. Polym. Sci. 2007, 32, 93–146. [Google Scholar] [CrossRef]
- Grover, G.N.; Maynard, H.D. Protein-polymer conjugates: Synthetic approaches by controlled radical polymerizations and interesting applications. Curr. Opin. Chem. Biol. 2010, 14, 818–827. [Google Scholar] [CrossRef]
- Wang, X.-S.; Lascelles, S.F.; Jackson, R.A.; Armes, S.P. Facile synthesis of well-defined water-soluble polymers via atom transfer radical polymerization in aqueous media at ambient temperature. Chem. Commun. 1999, 1817–1818. [Google Scholar]
- Slavin, S.; Khoshdel, E.; Haddleton, D.M. Biological surface modification by “thiol-ene” addition of polymers synthesised by catalytic chain transfer polymerisation (CCTP). Polym. Chem. 2012, 3, 1461–1466. [Google Scholar] [CrossRef]
- Siegwart, D.J.; Oh, J.K.; Matyjaszewski, K. ATRP in the design of functional materials for biomedical applications. Prog. Polym. Sci. 2012, 37, 18–37. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.J.; Neoh, K.G.; Kang, E.T. Bioactive surfaces and biomaterials via atom transfer radical polymerization. Prog. Polym. Sci. 2009, 34, 719–761. [Google Scholar]
- Zeng, Q.; Li, T.; Cash, B.; Li, S.; Xie, F.; Wang, Q. Chemoselective derivatization of a bionanoparticle by click reaction and ATRP reaction. Chem. Commun. 2007, 1453–1455. [Google Scholar]
- Hu, Y.; Samanta, D.; Parelkar, S.S.; Hong, S.W.; Wang, Q.; Russell, T.P.; Emrick, T. Ferritin-polymer conjugates: Grafting chemistry and integration into nanoscale assemblies. Adv. Funct. Mater. 2010, 20, 3603–3612. [Google Scholar]
- Mougin, N.C.; van Rijn, P.; Park, H.; Müller, A.H.E.; Böker, A. Hybrid capsules via self-assembly of thermoresponsive and interfacially active bionanoparticle-polymer conjugates. Adv. Funct. Mater. 2011, 21, 2470–2476. [Google Scholar]
- Bünsow, J.; Mänz, M.; Vana, P.; Johannsmann, D. Electrochemically induced RAFT polymerization of thermoresponsive hydrogel films: Impact on film thickness and surface morphology. Macromol. Chem. Phys. 2010, 211, 761–767. [Google Scholar]
- Magenau, A.J.D.; Strandwitz, N.C.; Gennaro, A.; Matyjaszewski, K. Electrochemically mediated atom transfer radical polymerization. Science 2011, 332, 81–84. [Google Scholar]
- Matyjaszewski, K.; Dong, H.; Jakubowski, W.; Pietrasik, J.; Kusumo, A. Grafting from surfaces for “everyone”: ARGET ATRP in the presence of air. Langmuir 2007, 23, 4528–4531. [Google Scholar]
- Bortolamei, N.; Isse, A.A.; Magenau, A.J.D.; Gennaro, A.; Matyjaszewski, K. Controlled aqueous atom transfer radical polymerization with electrochemical generation of the active catalyst. Angew. Chem. Int. Ed. 2011, 50, 11391–11394. [Google Scholar]
- Gurunathan, K.; Murugan, A.V.; Marimuthu, R.; Mulik, U.P.; Amalnerkar, D.P. Electrochemically synthesised conducting polymeric materials for applications towards technology in electronics, optoelectronics and energy storage devices. Mater. Chem. Phys. 1999, 61, 173–191. [Google Scholar]
- Li, B.; Yu, B.; Huck, W.T.S.; Zhou, F.; Liu, W. Electrochemically induced surface-initiated atom-transfer radical polymerization. Angew. Chem. Int. Ed. 2012, 51, 5092–5095. [Google Scholar]
- Ibrahim, M.M.; Mersal, G.A.M. Solution studies of tris(2-aminoethyl)amine with metal ions of biological interest: The electroanalytical determination of the phosphate triester hydrolysis. J. Inorg. Organomet. Polym. Mater. 2009, 19, 549–557. [Google Scholar] [CrossRef]
- Li, B.; Yu, B.; Huck, W.T.S.; Liu, W.; Zhou, F. Electrochemically mediated atom transfer radical polymerization on nonconducting substrates: Controlled brush growth through catalyst diffusion. J. Am. Chem. Soc. 2013, 135, 1708–1710. [Google Scholar]
- Jones, D.M.; Brown, A.A.; Huck, W.T.S. Surface-initiated polymerizations in aqueous media: Effect of initiator density. Langmuir 2002, 18, 1265–1269. [Google Scholar]
- Ciampolini, M.; Nardi, N. Five-coordinated high-spin complexes of bivalent cobalt, nickel, and copper with tris(2-dimethylaminoethyl)amine. Inorg. Chem. 1966, 5, 41–44. [Google Scholar] [CrossRef]
- Cosnier, S.; Holzinger, M. Electrosynthesized polymers for biosensing. Chem. Soc. Rev. 2011, 40, 2146–2156. [Google Scholar] [CrossRef]
- Isse, A.A.; Gennaro, A.; Lin, C.Y.; Hodgson, J.L.; Coote, M.L.; Guliashvili, T. Mechanism of carbon-halogen bond reductive cleavage in activated alkyl halide initiators relevant to living radical polymerization: Theoretical and experimental study. J. Am. Chem. Soc. 2011, 133, 6254–6264. [Google Scholar]
- He, J.; Wu, Y.; Wu, J.; Mao, X.; Fu, L.; Qian, T.; Fang, J.; Xiong, C.; Xie, J.; Ma, H. Study and application of a linear frequency–thickness relation for surface-initiated atom transfer radical polymerization in a quartz crystal microbalance. Macromolecules 2007, 40, 3090–3096. [Google Scholar] [CrossRef]
- Moya, S.E.; Brown, A.A.; Azzaroni, O.; Huck, W.T.S. Following polymer brush growth using the quartz crystal microbalance technique. Macromol. Rapid Commun. 2005, 26, 1117–1121. [Google Scholar] [CrossRef]
- Buchmann, M.B.; Fyles, T.M.; Mischki, T.; Sutherland, T.; Tong, C.C.; Yip, V.L.Y. Intramolecular thiolysis of 4-mercaptobutyrate esters: Developing a “traceless” linker for alcohol release from self-assembled monolayers on gold. Arch. Org. Chem. 2011, 2011, 19–26. [Google Scholar]
- Broyer, R.M.; Grover, G.N.; Maynard, H.D. Emerging synthetic approaches for protein–polymer conjugations. Chem. Commun. 2011, 47, 2212–2226. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hosseiny, S.S.; Van Rijn, P. Surface Initiated Polymerizations via e-ATRP in Pure Water. Polymers 2013, 5, 1229-1240. https://doi.org/10.3390/polym5041229
Hosseiny SS, Van Rijn P. Surface Initiated Polymerizations via e-ATRP in Pure Water. Polymers. 2013; 5(4):1229-1240. https://doi.org/10.3390/polym5041229
Chicago/Turabian StyleHosseiny, Seyed Schwan, and Patrick Van Rijn. 2013. "Surface Initiated Polymerizations via e-ATRP in Pure Water" Polymers 5, no. 4: 1229-1240. https://doi.org/10.3390/polym5041229
APA StyleHosseiny, S. S., & Van Rijn, P. (2013). Surface Initiated Polymerizations via e-ATRP in Pure Water. Polymers, 5(4), 1229-1240. https://doi.org/10.3390/polym5041229