Addressing the Inflammatory Response to Clinically Relevant Polymers by Manipulating the Host Response Using ITIM Domain-Containing Receptors

Abstract
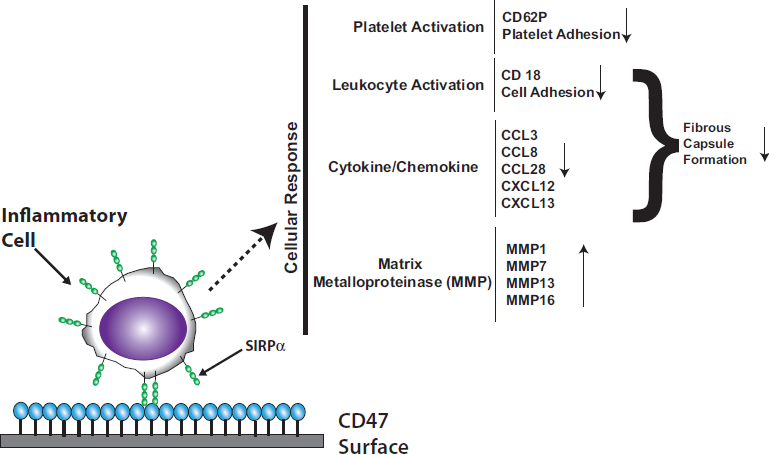
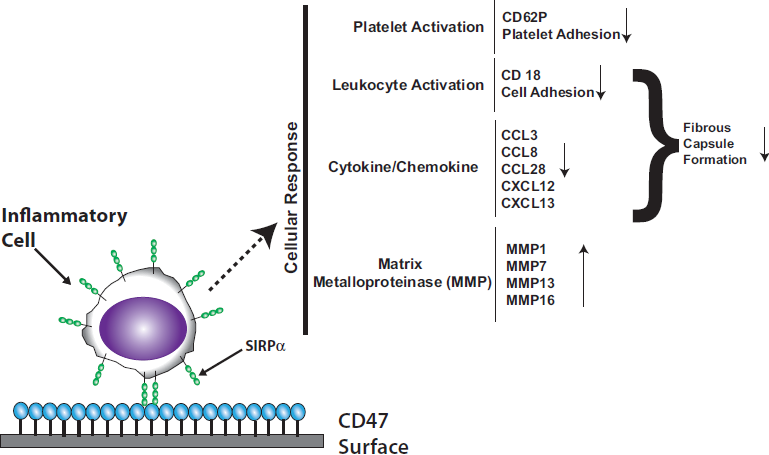
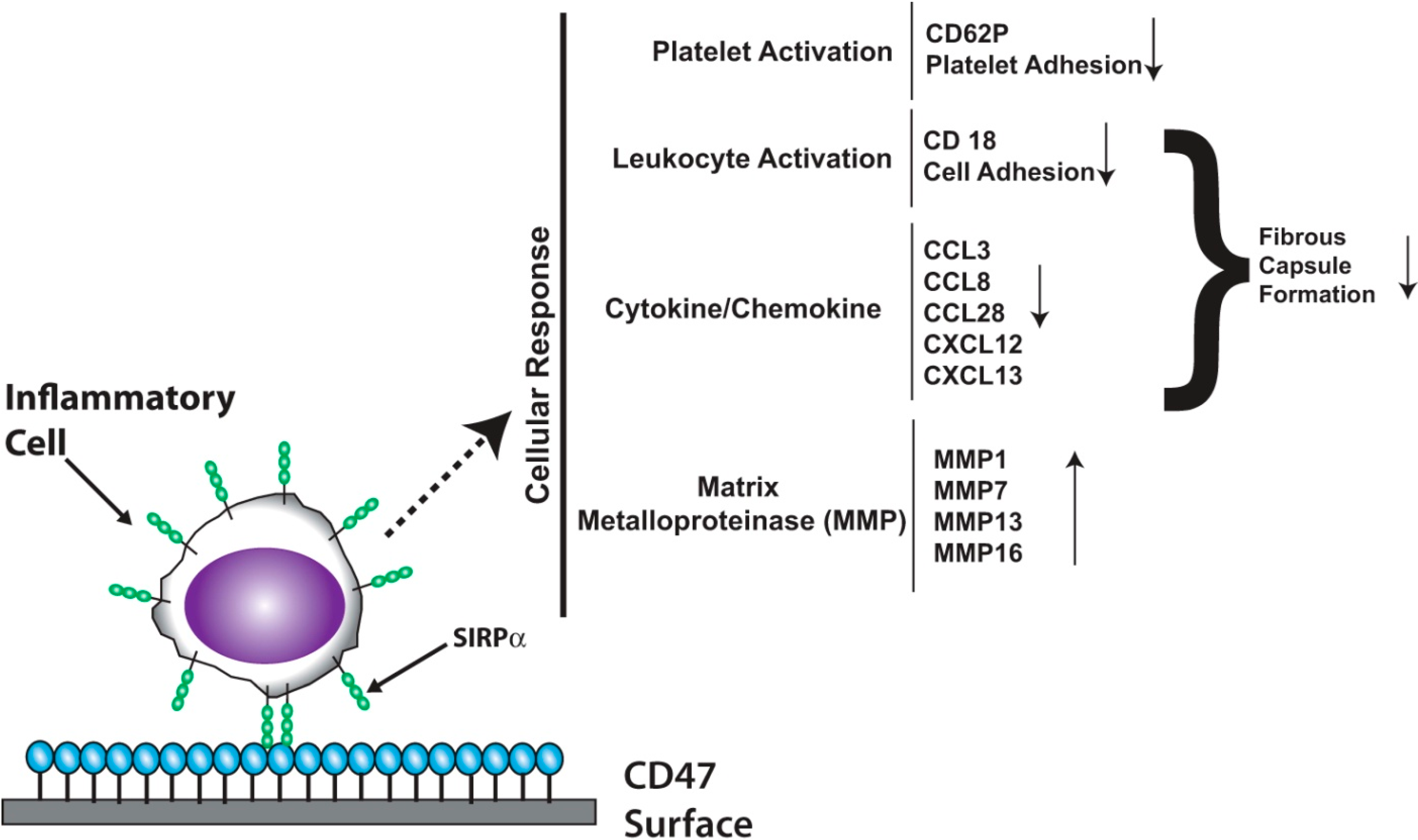
:
1. Introduction

2. Bioinert and Bioactive Surfaces
3. Immunoreceptor Tyrosine-Based Inhibitory Motif (ITIM)
3.1. Signal Regulatory Protein Alpha (SIRPα)


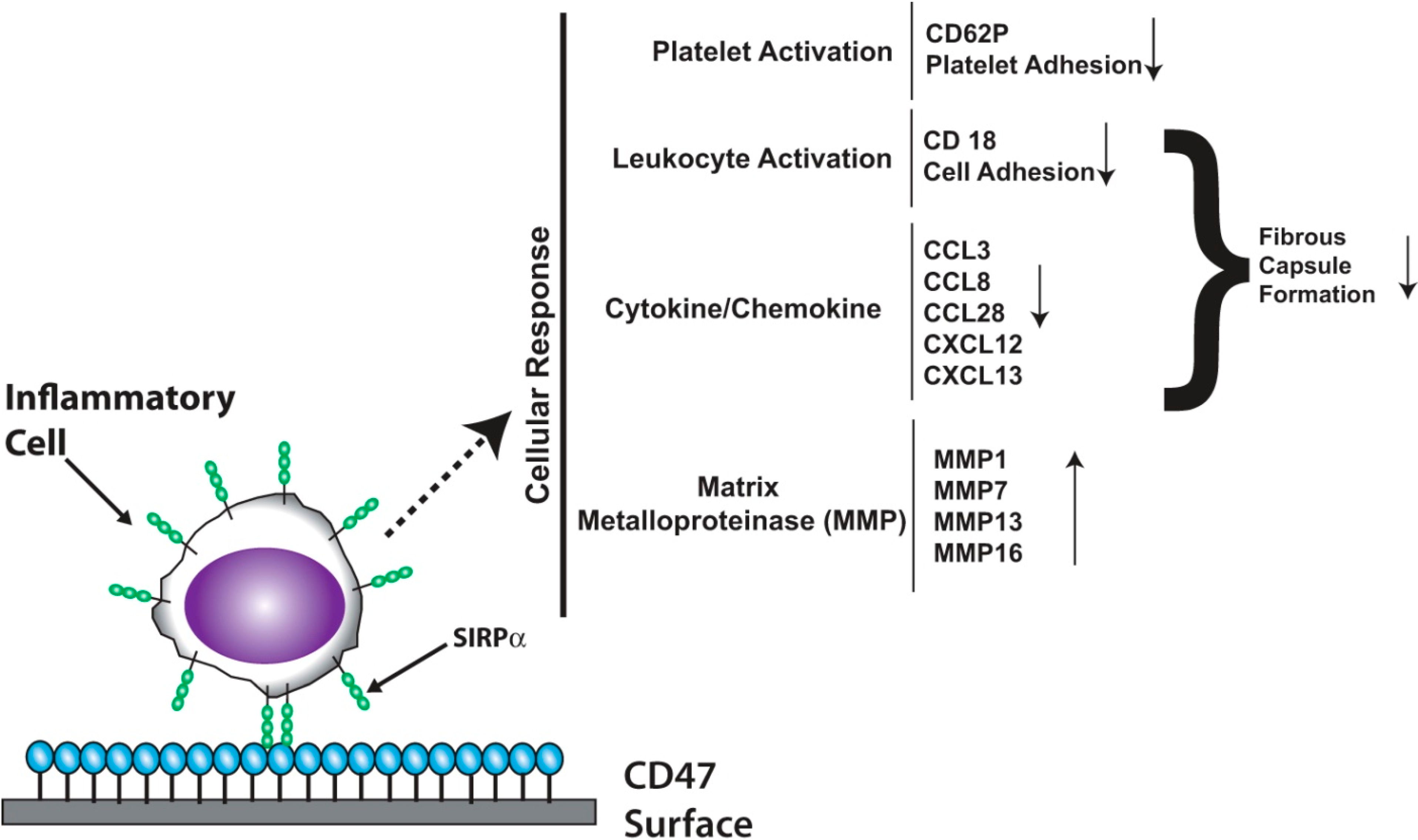
3.2. Platelet Endothelial Cell Adhesion Molecule-1 (PECAM-1)
3.3. CD200R
4. ITIM Receptors and the Adaptive Immune Response: Implications for Biomaterials

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | T or B Cell Distribution | Ligand |
|---|---|---|
| FcγRIIB | B | IgG |
| CTLA-4 | T | CD80, CD86 |
| PD-1 | T | PD-1 ligand 1 and 2 |
| CD72 | B | Unknown |
| CD22 | B | Sialic Acid |
| CD66a | T,B | CD66, CD62E |
5. Limitation of ITIM-Based Therapeutics
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Anderson, J.M.; Rodriguez, A.; Chang, D.T. Foreign body reaction to biomaterials. Semin. Immunol. 2008, 20, 86–100. [Google Scholar] [PubMed]
- Rossaint, J.; Berger, C.; van Aken, H.; Scheld, H.H.; Zahn, P.K.; Rukosujew, A.; Zarbock, A. Cardiopulmonary bypass during cardiac surgery modulates systemic inflammation by affecting different steps of the leukocyte recruitment cascade. PLoS One 2012, 7, e45738. [Google Scholar] [PubMed]
- Paparella, D.; Yau, T.M.; Young, E. Cardiopulmonary bypass induced inflammation: Pathophysiology and treatment. An update. Eur. J. Cardiothorac. Surg. 2002, 21, 232–244. [Google Scholar] [PubMed]
- Rodriguez, A.; Meyerson, H.; Anderson, J.M. Quantitative in vivo cytokine analysis at synthetic biomaterial implant sites. J. Biomed. Mater. Res. A 2009, 89, 152–159. [Google Scholar] [PubMed]
- Wiggins, M.J.; Wilkoff, B.; Anderson, J.M.; Hiltner, A. Biodegradation of polyether polyurethane inner insulation in bipolar pacemaker leads. J. Biomed. Mater. Res. 2001, 58, 302–307. [Google Scholar] [PubMed]
- Stokes, K.; McVenes, R.; Anderson, J.M. Polyurethane elastomer biostability. J. Biomater. Appl. 1995, 9, 321–354. [Google Scholar] [PubMed]
- Luttikhuizen, D.T.; Harmsen, M.C.; van Luyn, M.J.A. Cellular and molecular dynamics in the foreign body reaction. Tissue Eng. 2006, 12, 1955–1970. [Google Scholar] [PubMed]
- Vroman, L.; Adams, A.L.; Fischer, G.C.; Munoz, P.C. Interaction of high molecular weight kininogen, factor XII, and fibrinogen in plasma at interfaces. Blood 1980, 55, 156–159. [Google Scholar] [PubMed]
- Love, R.J.; Jones, K.S. The recognition of biomaterials: Pattern recognition of medical polymers and their adsorbed biomolecules. J. Biomed. Mater. Res. A 2013, 101, 2740–2752. [Google Scholar] [PubMed]
- Tang, L.; Eaton, J.W. Fibrin(ogen) mediates acute inflammatory responses to biomaterials. J. Exp. Med. 1993, 178, 2147–2156. [Google Scholar] [PubMed]
- Tang, L.; Lucas, A.H.; Eaton, J.W. Inflammatory responses to implanted polymeric biomaterials: Role of surface-adsorbed immunoglobulin G. J. Lab. Clin. Med. 1993, 122, 292–300. [Google Scholar] [PubMed]
- Gorbet, M.B.; Sefton, M.V. Biomaterial-associated thrombosis: Roles of coagulation factors, complement, platelets and leukocytes. Biomaterials 2004, 25, 5681–5703. [Google Scholar] [PubMed]
- Wilson, C.J.; Clegg, R.E.; Leavesley, D.I.; Pearcy, M.J. Mediation of biomaterial-cell interactions by adsorbed proteins: A review. Tissue Eng. 2005, 11, 1–18. [Google Scholar] [PubMed]
- Anderson, J.M.; McNally, A.K. Biocompatibility of implants: Lymphocyte/macrophage interactions. Semin. Immunopathol. 2011, 33, 221–233. [Google Scholar] [PubMed]
- Babior, B.M. Phagocytes and oxidative stress. Am. J. Med. 2000, 109, 33–44. [Google Scholar] [PubMed]
- Ischiropoulos, H.; Zhu, L.; Beckman, J.S. Peroxynitrite formation from macrophage-derived nitric oxide. Arch. Biochem. Biophys. 1992, 298, 446–451. [Google Scholar] [PubMed]
- Christian, A.J.; Lin, H.; Alferiev, I.S.; Connolly, J.M.; Ferrari, G.; Hazen, S.L.; Ischiropoulos, H.; Levy, R.J. The susceptibility of bioprosthetic heart valve leaflets to oxidation. Biomaterials 2014, 35, 2097–2102. [Google Scholar] [PubMed]
- Brown, B.N.; Ratner, B.D.; Goodman, S.B.; Amar, S.; Badylak, S.F. Macrophage polarization: An opportunity for improved outcomes in biomaterials and regenerative medicine. Biomaterials 2012, 33, 3792–3802. [Google Scholar] [PubMed]
- Brown, B.N.; Londono, R.; Tottey, S.; Zhang, L.; Kukla, K.A.; Wolf, M.T.; Daly, K.A.; Reing, J.E.; Badylak, S.F. Macrophage phenotype as a predictor of constructive remodeling following the implantation of biologically derived surgical mesh materials. Acta Biomater. 2012, 8, 978–987. [Google Scholar] [PubMed]
- Marco, M.; Fortin, C.; Fulop, T. Membrane-type matrix metalloproteinases: Key mediators of leukocyte function. J. Leukoc. Biol. 2013, 94, 237–246. [Google Scholar] [PubMed]
- Parks, W.C.; Wilson, C.L.; López-Boado, Y.S. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat. Rev. Immunol. 2004, 4, 617–629. [Google Scholar] [PubMed]
- Nissinen, L.; Kähäri, V.-M. Matrix metalloproteinases in inflammation. Biochim. Biophys. Acta 2014, 1840, 2571–2580. [Google Scholar] [PubMed]
- Benbow, U.; Brinckerhoff, C.E. The AP-1 site and MMP gene regulation: What is all the fuss about? Matrix Biol. 1997, 15, 519–526. [Google Scholar]
- Reunanen, N.; Li, S.-P.; Ahonen, M.; Foschi, M.; Han, J.; Kähäri, V.-M. Activation of p38 alpha MAPK enhances collagenase-1 (matrix metalloproteinase (MMP)-1) and stromelysin-1 (MMP-3) expression by mRNA stabilization. J. Biol. Chem. 2002, 277, 32360–32368. [Google Scholar] [PubMed]
- Khokha, R.; Murthy, A.; Weiss, A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 649–665. [Google Scholar] [PubMed]
- Mohan, M.J.; Seaton, T.; Mitchell, J.; Howe, A.; Blackburn, K.; Burkhart, W.; Moyer, M.; Patel, I.; Waitt, G.M.; Becherer, J.D.; et al. The tumor necrosis factor-alpha converting enzyme (TACE): A unique metalloproteinase with highly defined substrate selectivity. Biochemistry 2002, 41, 9462–9469. [Google Scholar] [PubMed]
- English, W.R.; Puente, X.S.; Freije, J.M.; Knauper, V.; Amour, A.; Merryweather, A.; Lopez-Otin, C.; Murphy, G. Membrane type 4 matrix metalloproteinase (MMP17) has tumor necrosis factor-alpha convertase activity but does not activate pro-MMP2. J. Biol. Chem. 2000, 275, 14046–14055. [Google Scholar] [PubMed]
- Schönbeck, U.; Mach, F.; Libby, P. Generation of biologically active IL-1 beta by matrix metalloproteinases: A novel caspase-1-independent pathway of IL-1 beta processing. J. Immunol. 1998, 161, 3340–3346. [Google Scholar] [PubMed]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [PubMed]
- Maeda, S.; Dean, D.D.; Gomez, R.; Schwartz, Z.; Boyan, B.D. The first stage of transforming growth factor beta1 activation is release of the large latent complex from the extracellular matrix of growth plate chondrocytes by matrix vesicle stromelysin-1 (MMP-3). Calcif. Tissue Int. 2002, 70, 54–65. [Google Scholar] [PubMed]
- Karsdal, M.A.; Larsen, L.; Engsig, M.T.; Lou, H.; Ferreras, M.; Lochter, A.; Delaissé, J.-M.; Foged, N.T. Matrix metalloproteinase-dependent activation of latent transforming growth factor-beta controls the conversion of osteoblasts into osteocytes by blocking osteoblast apoptosis. J. Biol. Chem. 2002, 277, 44061–44067. [Google Scholar] [PubMed]
- McQuibban, G.A.; Gong, J.H.; Tam, E.M.; McCulloch, C.A.; Clark-Lewis, I.; Overall, C.M. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science 2000, 289, 1202–1206. [Google Scholar] [PubMed]
- Franz, S.; Rammelt, S.; Scharnweber, D.; Simon, J.C. Immune responses to implants—A review of the implications for the design of immunomodulatory biomaterials. Biomaterials 2011, 32, 6692–6709. [Google Scholar] [PubMed]
- Göpferich, A. Mechanisms of polymer degradation and erosion. Biomaterials 1996, 17, 103–114. [Google Scholar] [PubMed]
- Brash, J.L.; Lyman, D.J. Adsorption of plasma proteins in solution to uncharged, hydrophobic polymer surfaces. J. Biomed. Mater. Res. 1969, 3, 175–189. [Google Scholar] [PubMed]
- Shastri, V.P. Non-degradable biocompatible polymers in medicine: Past, present and future. Curr. Pharm. Biotechnol. 2003, 4, 331–337. [Google Scholar] [PubMed]
- Morais, J.M.; Papadimitrakopoulos, F.; Burgess, D.J. Biomaterials/tissue interactions: Possible solutions to overcome foreign body response. AAPS J. 2010, 12, 188–196. [Google Scholar] [PubMed]
- Schulte, V.A.; Díez, M.; Möller, M.; Lensen, M.C. Surface topography induces fibroblast adhesion on intrinsically nonadhesive poly(ethylene glycol) substrates. Biomacromolecules 2009, 10, 2795–2801. [Google Scholar] [PubMed]
- Andersson, A.-S.; Bäckhed, F.; von Euler, A.; Richter-Dahlfors, A.; Sutherland, D.; Kasemo, B. Nanoscale features influence epithelial cell morphology and cytokine production. Biomaterials 2003, 24, 3427–3436. [Google Scholar] [PubMed]
- Dalby, M.J.; Riehle, M.O.; Johnstone, H.; Affrossman, S.; Curtis, A.S.G. In vitro reaction of endothelial cells to polymer demixed nanotopography. Biomaterials 2002, 23, 2945–2954. [Google Scholar] [PubMed]
- Kalwerisky, K.; Mihora, L.; Czyz, C.N.; Foster, J.A.; Holck, D.E.E. Rate of vascularization and exposure of silicone-capped porous polyethylene spherical implants: An animal model. Ophthal. Plast. Reconstr. Surg. 2013, 29, 350–356. [Google Scholar] [PubMed]
- Sussman, E.M.; Halpin, M.C.; Muster, J.; Moon, R.T.; Ratner, B.D. Porous implants modulate healing and induce shifts in local macrophage polarization in the foreign body reaction. Ann. Biomed. Eng. 2014, 42, 1508–1516. [Google Scholar] [PubMed]
- Brauker, J.H.; Carr-Brendel, V.E.; Martinson, L.A.; Crudele, J.; Johnston, W.D.; Johnson, R.C. Neovascularization of synthetic membranes directed by membrane microarchitecture. J. Biomed. Mater. Res. 1995, 29, 1517–1524. [Google Scholar] [PubMed]
- Thomson, K.S.; Korte, F.S.; Giachelli, C.M.; Ratner, B.D.; Regnier, M.; Scatena, M. Prevascularized microtemplated fibrin scaffolds for cardiac tissue engineering applications. Tissue Eng. Part A 2013, 19, 967–977. [Google Scholar]
- Madden, L.R.; Mortisen, D.J.; Sussman, E.M.; Dupras, S.K.; Fugate, J.A.; Cuy, J.L.; Hauch, K.D.; Laflamme, M.A.; Murry, C.E.; Ratner, B.D. Proangiogenic scaffolds as functional templates for cardiac tissue engineering. Proc. Natl. Acad. Sci. USA 2010, 107, 15211–15216. [Google Scholar] [PubMed]
- Zhang, L.; Cao, Z.; Bai, T.; Carr, L.; Ella-Menye, J.-R.; Irvin, C.; Ratner, B.D.; Jiang, S. Zwitterionic hydrogels implanted in mice resist the foreign-body reaction. Nat. Biotechnol. 2013, 31, 553–556. [Google Scholar] [PubMed]
- Carr, L.; Cheng, G.; Xue, H.; Jiang, S. Engineering the polymer backbone to strengthen nonfouling sulfobetaine hydrogels. Langmuir 2010, 26, 14793–14798. [Google Scholar] [PubMed]
- Carr, L.R.; Xue, H.; Jiang, S. Functionalizable and nonfouling zwitterionic carboxybetaine hydrogels with a carboxybetaine dimethacrylate crosslinker. Biomaterials 2011, 32, 961–968. [Google Scholar] [PubMed]
- Carr, L.R.; Zhou, Y.; Krause, J.E.; Xue, H.; Jiang, S. Uniform zwitterionic polymer hydrogels with a nonfouling and functionalizable crosslinker using photopolymerization. Biomaterials 2011, 32, 6893–6899. [Google Scholar] [PubMed]
- Williams, D.F. On the mechanisms of biocompatibility. Biomaterials 2008, 29, 2941–2953. [Google Scholar] [PubMed]
- Slee, J.B.; Pugh, R.; Lowe-Krentz, L.J. Beyond anticoagulation : Roles for heparin in the vasculature. In Heparin: Properties, Uses and Side Effects; Piyathilake, D.E., Liang, R., Eds.; Nova Science Publishers: Hauppauge, NY, USA, 2012; pp. 59–81. [Google Scholar]
- Anastase-Ravion, S.; Blondin, C.; Cholley, B.; Haeffner-Cavaillon, N.; Castellot, J.J.; Letourneur, D. Heparin inhibits lipopolysaccharide (LPS) binding to leukocytes and LPS-induced cytokine production. J. Biomed. Mater. Res. A 2003, 66, 376–384. [Google Scholar] [PubMed]
- Larm, O.; Larsson, R.; Olsson, P. A new non-thrombogenic surface prepared by selective covalent binding of heparin via a modified reducing terminal residue. Biomater. Med. Devices. Artif. Organs 1983, 11, 161–173. [Google Scholar] [PubMed]
- Bannan, S.; Danby, A.; Cowan, D.; Ashraf, S.; Martin, P.G. Low heparinization with heparin-bonded bypass circuits: Is it a safe strategy? Ann. Thorac. Surg. 1997, 63, 663–668. [Google Scholar] [CrossRef]
- Fukutomi, M.; Kobayashi, S.; Niwaya, K.; Hamada, Y.; Kitamura, S. Changes in platelet, granulocyte, and complement activation during cardiopulmonary bypass using heparin-coated equipment. Artif. Organs 1996, 20, 767–776. [Google Scholar] [PubMed]
- Kagisaki, K.; Masai, T.; Kadoba, K.; Sawa, Y.; Nomura, F.; Fukushima, N.; Ichikawa, H.; Ohata, T.; Suzuki, K.; Taketani, S.; et al. Biocompatibility of heparin-coated circuits in pediatric cardiopulmonary bypass. Artif. Organs 1997, 21, 836–840. [Google Scholar] [PubMed]
- Svenmarker, S.; Sandström, E.; Karlsson, T.; Jansson, E.; Häggmark, S.; Lindholm, R.; Appelblad, M.; Aberg, T. Clinical effects of the heparin coated surface in cardiopulmonary bypass. Eur. J. Cardiothorac. Surg. 1997, 11, 957–964. [Google Scholar] [PubMed]
- Bélanger, M.C.; Marois, Y.; Roy, R.; Mehri, Y.; Wagner, E.; Zhang, Z.; King, M.W.; Yang, M.; Hahn, C.; Guidoin, R. Selection of a polyurethane membrane for the manufacture of ventricles for a totally implantable artificial heart: Blood compatibility and biocompatibility studies. Artif. Organs 2000, 24, 879–888. [Google Scholar] [PubMed]
- Belboul, A.; Al-Khaja, N. Does heparin coating improve biocompatibility? A study on complement, blood cells and postoperative morbidity during cardiac surgery. Perfusion 1997, 12, 385–391. [Google Scholar] [PubMed]
- Li, G.; Yang, P.; Liao, Y.; Huang, N. Tailoring of the titanium surface by immobilization of heparin/fibronectin complexes for improving blood compatibility and endothelialization: An in vitro study. Biomacromolecules 2011, 12, 1155–1168. [Google Scholar] [PubMed]
- Li, G.; Yang, P.; Qin, W.; Maitz, M.F.; Zhou, S.; Huang, N. The effect of coimmobilizing heparin and fibronectin on titanium on hemocompatibility and endothelialization. Biomaterials 2011, 32, 4691–4703. [Google Scholar] [PubMed]
- Beamish, J.A.; Geyer, L.C.; Haq-Siddiqi, N.A.; Kottke-Marchant, K.; Marchant, R.E. The effects of heparin releasing hydrogels on vascular smooth muscle cell phenotype. Biomaterials 2009, 30, 6286–6294. [Google Scholar] [PubMed]
- Van Bilsen, P.H.J.; Popa, E.R.; Brouwer, L.A.; Vincent, J.; Taylor, C.E.; de Leij, L.F.M.H.; Hendriks, M.; van Luyn, M.J.A. Ongoing foreign body reaction to subcutaneous implanted (heparin) modified Dacron in rats. J. Biomed. Mater. Res. A 2004, 68, 423–427. [Google Scholar] [PubMed]
- Heidemann, W.; Ruffieux, K.; Fischer, J.H.; Jeschkeit-Schubbert, S.; Jung, H.; Krueger, G.; Wintermantel, E.; Gerlach, K.L. The effect of an admixture of sodium hydrogen phosphate or heparin-coating to poly(d,l)lactide—Results of an animal study. Biomed. Tech. (Berl.) 2003, 48, 262–268. [Google Scholar]
- De Scheerder, I.; Wang, K.; Wilczek, K.; Meuleman, D.; van Amsterdam, R.; Vogel, G.; Piessens, J.; van de Werf, F. Experimental study of thrombogenicity and foreign body reaction induced by heparin-coated coronary stents. Circulation 1997, 95, 1549–1553. [Google Scholar] [PubMed]
- Frost, M.C.; Reynolds, M.M.; Meyerhoff, M.E. Polymers incorporating nitric oxide releasing/generating substances for improved biocompatibility of blood-contacting medical devices. Biomaterials 2005, 26, 1685–1693. [Google Scholar] [PubMed]
- Mani, G.; Feldman, M.D.; Patel, D.; Agrawal, C.M. Coronary stents: A materials perspective. Biomaterials 2007, 28, 1689–1710. [Google Scholar] [PubMed]
- Kakade, S.; Mani, G. A comparative study of the effects of vitamin C, sirolimus, and paclitaxel on the growth of endothelial and smooth muscle cells for cardiovascular medical device applications. Drug Des. Devel. Ther. 2013, 7, 529–544. [Google Scholar] [PubMed]
- Ravetch, J.V.; Lanier, L.L. Immune inhibitory receptors. Science 2000, 290, 84–89. [Google Scholar] [PubMed]
- Daëron, M.; Jaeger, S.; Du Pasquier, L.; Vivier, E. Immunoreceptor tyrosine-based inhibition motifs: A quest in the past and future. Immunol. Rev. 2008, 224, 11–43. [Google Scholar] [PubMed]
- Huang, Z.-Y.; Hunter, S.; Kim, M.-K.; Indik, Z.K.; Schreiber, A.D. The effect of phosphatases SHP-1 and SHIP-1 on signaling by the ITIM- and ITAM-containing Fcgamma receptors FcgammaRIIB and FcgammaRIIA. J. Leukoc. Biol. 2003, 73, 823–829. [Google Scholar] [PubMed]
- Matozaki, T.; Murata, Y.; Okazawa, H.; Ohnishi, H. Functions and molecular mechanisms of the CD47-SIRPalpha signalling pathway. Trends Cell Biol. 2009, 19, 72–80. [Google Scholar] [PubMed]
- Barclay, A.N.; van den Berg, T.K. The interaction between signal regulatory protein alpha (SIRPα) and CD47: Structure, function, and therapeutic target. Annu. Rev. Immunol. 2013, 32, 25–50. [Google Scholar] [PubMed]
- Karra, L.; Levi-Schaffer, F. Down-regulation of mast cell responses through ITIM containing inhibitory receptors. Adv. Exp. Med. Biol. 2011, 716, 143–159. [Google Scholar] [PubMed]
- Van Beek, E.M.; Cochrane, F.; Barclay, A.N.; van den Berg, T.K. Signal regulatory proteins in the immune system. J. Immunol. 2005, 175, 7781–7787. [Google Scholar] [PubMed]
- Subramanian, S.; Parthasarathy, R.; Sen, S.; Boder, E.T.; Discher, D.E. Species- and cell type-specific interactions between CD47 and human SIRPalpha. Blood 2006, 107, 2548–2556. [Google Scholar] [PubMed]
- Subramanian, S.; Tsai, R.; Discher, D.E. The “metabolon,” CD47, and the “phagocytic synapse”: Molecular co-localization and species divergence. Transfus. Clin. Biol. 2006, 13, 31–38. [Google Scholar] [PubMed]
- Takizawa, H.; Manz, M.G. Macrophage tolerance: CD47-SIRP-alpha-mediated signals matter. Nat. Immunol. 2007, 8, 1287–1289. [Google Scholar] [PubMed]
- Tsai, R.K.; Discher, D.E. Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J. Cell Biol. 2008, 180, 989–1003. [Google Scholar] [PubMed]
- Lindberg, F.P.; Gresham, H.D.; Schwarz, E.; Brown, E.J. Molecular cloning of integrin-associated protein: An immunoglobulin family member with multiple membrane-spanning domains implicated in alpha v beta 3-dependent ligand binding. J. Cell Biol. 1993, 123, 485–496. [Google Scholar] [PubMed]
- Finley, M.J.; Rauova, L.; Alferiev, I.S.; Weisel, J.W.; Levy, R.J.; Stachelek, S.J. Diminished adhesion and activation of platelets and neutrophils with CD47 functionalized blood contacting surfaces. Biomaterials 2012, 33, 5803–5811. [Google Scholar] [PubMed]
- Barclay, A.N. Signal regulatory protein alpha (SIRP)/CD47 interaction and function. Curr. Opin. Immunol. 2009, 21, 47–52. [Google Scholar] [PubMed]
- Brown, E.J.; Frazier, W.A. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 2001, 11, 130–135. [Google Scholar]
- Oldenborg, P.A.; Zheleznyak, A.; Fang, Y.F.; Lagenaur, C.F.; Gresham, H.D.; Lindberg, F.P. Role of CD47 as a marker of self on red blood cells. Science 2000, 288, 2051–2054. [Google Scholar] [PubMed]
- Melotti, A.; Daga, A.; Marubbi, D.; Zunino, A.; Mutti, L.; Corte, G. In vitro and in vivo characterization of highly purified human mesothelioma derived cells. BMC Cancer 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [PubMed]
- Jaiswal, S.; Jamieson, C.H.M.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [PubMed]
- Chao, M.P.; Weissman, I.L.; Majeti, R. The CD47-SIRPα pathway in cancer immune evasion and potential therapeutic implications. Curr. Opin. Immunol. 2012, 24, 225–232. [Google Scholar] [PubMed]
- Cameron, C.M.; Barrett, J.W.; Mann, M.; Lucas, A.; McFadden, G. Myxoma virus M128L is expressed as a cell surface CD47-like virulence factor that contributes to the downregulation of macrophage activation in vivo. Virology 2005, 337, 55–67. [Google Scholar] [PubMed]
- Stachelek, S.J.; Finley, M.J.; Alferiev, I.S.; Wang, F.; Tsai, R.K.; Eckells, E.C.; Tomczyk, N.; Connolly, J.M.; Discher, D.E.; Eckmann, D.M.; et al. The effect of CD47 modified polymer surfaces on inflammatory cell attachment and activation. Biomaterials 2011, 32, 4317–4326. [Google Scholar] [PubMed]
- Finley, M.J.; Clark, K.A.; Alferiev, I.S.; Levy, R.J.; Stachelek, S.J. Intracellular signaling mechanisms associated with CD47 modified surfaces. Biomaterials 2013, 34, 8640–8649. [Google Scholar] [PubMed]
- Stachelek, S.J.; Alferiev, I.; Connolly, J.M.; Sacks, M.; Hebbel, R.P.; Bianco, R.; Levy, R.J. Cholesterol-modified polyurethane valve cusps demonstrate blood outgrowth endothelial cell adhesion post-seeding in vitro and in vivo. Ann. Thorac. Surg. 2006, 81, 47–55. [Google Scholar] [PubMed]
- Rodriguez, P.L.; Harada, T.; Christian, D.A.; Pantano, D.A.; Tsai, R.K.; Discher, D.E. Minimal “Self” peptides that inhibit phagocytic clearance and enhance delivery of nanoparticles. Science 2013, 339, 971–975. [Google Scholar] [PubMed]
- Lee, C.-K.; Koo, K.-T.; Park, Y.-J.; Lee, J.-Y.; Rhee, S.-H.; Ku, Y.; Rhyu, I.-C.; Chung, C.-P. Biomimetic surface modification using synthetic oligopeptides for enhanced guided bone regeneration in beagles. J. Periodontol. 2012, 83, 101–110. [Google Scholar] [PubMed]
- Lee, J.-Y.; Choi, Y.-S.; Lee, S.-J.; Chung, C.-P.; Park, Y.-J. Bioactive peptide-modified biomaterials for bone regeneration. Curr. Pharm. Des. 2011, 17, 2663–2676. [Google Scholar] [PubMed]
- Shin, H.; Jo, S.; Mikos, A.G. Biomimetic materials for tissue engineering. Biomaterials 2003, 24, 4353–4364. [Google Scholar] [PubMed]
- Sreejalekshmi, K.G.; Nair, P.D. Biomimeticity in tissue engineering scaffolds through synthetic peptide modifications-altering chemistry for enhanced biological response. J. Biomed. Mater. Res. A 2011, 96, 477–491. [Google Scholar] [PubMed]
- Hatherley, D.; Lea, S.M.; Johnson, S.; Barclay, A.N. Polymorphisms in the human inhibitory signal-regulatory protein α do not affect binding to its ligand CD47. J. Biol. Chem. 2014, 289, 10024–10028. [Google Scholar]
- Abramson, J.; Pecht, I. Regulation of the mast cell response to the type 1 Fc epsilon receptor. Immunol. Rev. 2007, 217, 231–254. [Google Scholar] [PubMed]
- Pinter, E.; Barreuther, M.; Lu, T.; Imhof, B.A.; Madri, J.A. Platelet-endothelial cell adhesion molecule-1 (PECAM-1/CD31) tyrosine phosphorylation state changes during vasculogenesis in the murine conceptus. Am. J. Pathol. 1997, 150, 1523–1530. [Google Scholar] [PubMed]
- Jackson, D.E. The unfolding tale of PECAM-1. FEBS Lett. 2003, 540, 7–14. [Google Scholar] [PubMed]
- Sun, Q.H.; DeLisser, H.M.; Zukowski, M.M.; Paddock, C.; Albelda, S.M.; Newman, P.J. Individually distinct Ig homology domains in PECAM-1 regulate homophilic binding and modulate receptor affinity. J. Biol. Chem. 1996, 271, 11090–11098. [Google Scholar] [PubMed]
- Newton, J.P.; Buckley, C.D.; Jones, E.Y.; Simmons, D.L. Residues on both faces of the first immunoglobulin fold contribute to homophilic binding sites of PECAM-1/CD31. J. Biol. Chem. 1997, 272, 20555–20563. [Google Scholar] [PubMed]
- Piali, L.; Hammel, P.; Uherek, C.; Bachmann, F.; Gisler, R.H.; Dunon, D.; Imhof, B.A. CD31/PECAM-1 is a ligand for alpha v beta 3 integrin involved in adhesion of leukocytes to endothelium. J. Cell Biol. 1995, 130, 451–460. [Google Scholar] [PubMed]
- Deaglio, S.; Morra, M.; Mallone, R.; Ausiello, C.M.; Prager, E.; Garbarino, G.; Dianzani, U.; Stockinger, H.; Malavasi, F. Human CD38 (ADP-ribosyl cyclase) is a counter-receptor of CD31, an Ig superfamily member. J. Immunol. 1998, 160, 395–402. [Google Scholar] [PubMed]
- Prager, E.; Sunder-Plassmann, R.; Hansmann, C.; Koch, C.; Holter, W.; Knapp, W.; Stockinger, H. Interaction of CD31 with a heterophilic counterreceptor involved in downregulation of human T cell responses. J. Exp. Med. 1996, 184, 41–50. [Google Scholar] [PubMed]
- Hua, C.T.; Gamble, J.R.; Vadas, M.A.; Jackson, D.E. Recruitment and activation of SHP-1 protein-tyrosine phosphatase by human platelet endothelial cell adhesion molecule-1 (PECAM-1). Identification of immunoreceptor tyrosine-based inhibitory motif-like binding motifs and substrates. J. Biol. Chem. 1998, 273, 28332–28340. [Google Scholar] [PubMed]
- Henshall, T.L.; Jones, K.L.; Wilkinson, R.; Jackson, D.E. Src homology 2 domain-containing protein-tyrosine phosphatases, SHP-1 and SHP-2, are required for platelet endothelial cell adhesion molecule-1/CD31-mediated inhibitory signaling. J. Immunol. 2001, 166, 3098–3106. [Google Scholar] [PubMed]
- Newman, D.K.; Hamilton, C.; Newman, P.J. Inhibition of antigen-receptor signaling by Platelet Endothelial Cell Adhesion Molecule-1 (CD31) requires functional ITIMs, SHP-2, and p56(lck). Blood 2001, 97, 2351–2357. [Google Scholar] [PubMed]
- Brown, S.; Heinisch, I.; Ross, E.; Shaw, K.; Buckley, C.D.; Savill, J. Apoptosis disables CD31-mediated cell detachment from phagocytes promoting binding and engulfment. Nature 2002, 418, 200–203. [Google Scholar] [PubMed]
- Chimini, G. Apoptosis: Repulsive encounters. Nature 2002, 418, 139–141. [Google Scholar] [PubMed]
- Ilan, N.; Mohsenin, A.; Cheung, L.; Madri, J.A. PECAM-1 shedding during apoptosis generates a membrane-anchored truncated molecule with unique signaling characteristics. FASEB J. 2001, 15, 362–372. [Google Scholar] [PubMed]
- Ma, L.; Mauro, C.; Cornish, G.H.; Chai, J.-G.; Coe, D.; Fu, H.; Patton, D.; Okkenhaug, K.; Franzoso, G.; Dyson, J.; et al. Ig gene-like molecule CD31 plays a nonredundant role in the regulation of T-cell immunity and tolerance. Proc. Natl. Acad. Sci. USA 2010, 107, 19461–19466. [Google Scholar]
- Papademetriou, I.; Tsinas, Z.; Hsu, J.; Muro, S. Combination-targeting to multiple endothelial cell adhesion molecules modulates binding, endocytosis, and in vivo biodistribution of drug nanocarriers and their therapeutic cargoes. J. Control. Release 2014, 188, 87–98. [Google Scholar] [PubMed]
- Chacko, A.-M.; Nayak, M.; Greineder, C.F.; Delisser, H.M.; Muzykantov, V.R. Collaborative enhancement of antibody binding to distinct PECAM-1 epitopes modulates endothelial targeting. PLoS One 2012, 7, e34958. [Google Scholar] [PubMed]
- Deddens, L.H.; van Tilborg, G.A.F.; van der Toorn, A.; de Vries, H.E.; Dijkhuizen, R.M. PECAM-1-targeted micron-sized particles of iron oxide as MRI contrast agent for detection of vascular remodeling after cerebral ischemia. Contrast Media Mol. Imaging 2013, 8, 393–401. [Google Scholar] [PubMed]
- Dan, M.; Cochran, D.B.; Yokel, R.A.; Dziubla, T.D. Binding, transcytosis and biodistribution of anti-PECAM-1 iron oxide nanoparticles for brain-targeted delivery. PLoS One 2013, 8, e81051. [Google Scholar] [PubMed]
- Minas, K.; Liversidge, J. Europe PMC funders group is the CD200/CD200 receptor interaction more than just a myeloid cell inhibitory signal? Crit. Rev. Immunol. 2008, 26, 213–230. [Google Scholar]
- Barclay, A.N.; Clark, M.J.; McCaughan, G.W. Neuronal/lymphoid membrane glycoprotein MRC OX-2 is a member of the immunoglobulin superfamily with a light-chain-like structure. Biochem. Soc. Symp. 1986, 51, 149–157. [Google Scholar] [PubMed]
- Broderick, C.; Hoek, R.M.; Forrester, J.V.; Liversidge, J.; Sedgwick, J.D.; Dick, A.D. Constitutive retinal CD200 expression regulates resident microglia and activation state of inflammatory cells during experimental autoimmune uveoretinitis. Am. J. Pathol. 2002, 161, 1669–1677. [Google Scholar] [PubMed]
- Walker, D.G.; Lue, L.-F. Understanding the neurobiology of CD200 and the CD200 receptor: A therapeutic target for controlling inflammation in human brains? Future Neurol. 2013, 8, 321–332. [Google Scholar]
- Preston, S.; Wright, G.J.; Starr, K.; Barclay, A.N.; Brown, M.H. The leukocyte/neuron cell surface antigen OX2 binds to a ligand on macrophages. Eur. J. Immunol. 1997, 27, 1911–1918. [Google Scholar] [PubMed]
- Wright, G.J.; Cherwinski, H.; Foster-Cuevas, M.; Brooke, G.; Puklavec, M.J.; Bigler, M.; Song, Y.; Jenmalm, M.; Gorman, D.; McClanahan, T.; et al. Characterization of the CD200 receptor family in mice and humans and their interactions with CD200. J. Immunol. 2003, 171, 3034–3046. [Google Scholar] [PubMed]
- Rijkers, E.S.K.; de Ruiter, T.; Baridi, A.; Veninga, H.; Hoek, R.M.; Meyaard, L. The inhibitory CD200R is differentially expressed on human and mouse T and B lymphocytes. Mol. Immunol. 2008, 45, 1126–1135. [Google Scholar] [PubMed]
- Wright, G.J.; Puklavec, M.J.; Willis, A.C.; Hoek, R.M.; Sedgwick, J.D.; Brown, M.H.; Barclay, A.N. Lymphoid/neuronal cell surface OX2 glycoprotein recognizes a novel receptor on macrophages implicated in the control of their function. Immunity 2000, 13, 233–242. [Google Scholar] [PubMed]
- Hoek, R.M.; Ruuls, S.R.; Murphy, C.A.; Wright, G.J.; Goddard, R.; Zurawski, S.M.; Blom, B.; Homola, M.E.; Streit, W.J.; Brown, M.H.; et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science 2000, 290, 1768–1771. [Google Scholar] [PubMed]
- Gorczynski, R.M.; Chen, Z.; Clark, D.A.; Kai, Y.; Lee, L.; Nachman, J.; Wong, S.; Marsden, P. Structural and functional heterogeneity in the CD200R family of immunoregulatory molecules and their expression at the feto-maternal interface. Am. J. Reprod. Immunol. 2004, 52, 147–163. [Google Scholar] [PubMed]
- Jenmalm, M.C.; Cherwinski, H.; Bowman, E.P.; Phillips, J.H.; Sedgwick, J.D. Regulation of myeloid cell function through the CD200 receptor. J. Immunol. 2006, 176, 191–199. [Google Scholar] [PubMed]
- Smith, M.J.; Hardy, W.R.; Murphy, J.M.; Jones, N.; Pawson, T. Screening for PTB domain binding partners and ligand specificity using proteome-derived NPXY peptide arrays. Mol. Cell. Biol. 2006, 26, 8461–8474. [Google Scholar] [PubMed]
- Zhang, S.; Cherwinski, H.; Sedgwick, J.D.; Phillips, J.H. Molecular mechanisms of CD200 inhibition of mast cell activation. J. Immunol. 2004, 173, 6786–6793. [Google Scholar] [PubMed]
- Viertlboeck, B.C.; Hanczaruk, M.A.; Schmitt, F.C.A.; Schmitt, R.; Göbel, T.W. Characterization of the chicken CD200 receptor family. Mol. Immunol. 2008, 45, 2097–2105. [Google Scholar] [PubMed]
- Mihrshahi, R.; Barclay, A.N.; Brown, M.H. Essential roles for Dok2 and RasGAP in CD200 receptor-mediated regulation of human myeloid cells. J. Immunol. 2009, 183, 4879–4886. [Google Scholar] [PubMed]
- Mihrshahi, R.; Brown, M.H. Downstream of tyrosine kinase 1 and 2 play opposing roles in CD200 receptor signaling. J. Immunol. 2010, 185, 7216–7222. [Google Scholar] [PubMed]
- McWhirter, J.R.; Kretz-Rommel, A.; Saven, A.; Maruyama, T.; Potter, K.N.; Mockridge, C.I.; Ravey, E.P.; Qin, F.; Bowdish, K.S. Antibodies selected from combinatorial libraries block a tumor antigen that plays a key role in immunomodulation. Proc. Natl. Acad. Sci. USA 2006, 103, 1041–1046. [Google Scholar] [PubMed]
- Moreaux, J.; Hose, D.; Reme, T.; Jourdan, E.; Hundemer, M.; Legouffe, E.; Moine, P.; Bourin, P.; Moos, M.; Corre, J.; et al. CD200 is a new prognostic factor in multiple myeloma. Blood 2006, 108, 4194–4197. [Google Scholar] [PubMed]
- Kawasaki, B.T.; Mistree, T.; Hurt, E.M.; Kalathur, M.; Farrar, W.L. Co-expression of the toleragenic glycoprotein, CD200, with markers for cancer stem cells. Biochem. Biophys. Res. Commun. 2007, 364, 778–782. [Google Scholar] [PubMed]
- Kretz-Rommel, A.; Qin, F.; Dakappagari, N.; Ravey, E.P.; McWhirter, J.; Oltean, D.; Frederickson, S.; Maruyama, T.; Wild, M.A.; Nolan, M.-J.; et al. CD200 expression on tumor cells suppresses antitumor immunity: New approaches to cancer immunotherapy. J. Immunol. 2007, 178, 5595–5605. [Google Scholar] [PubMed]
- Tonks, A.; Hills, R.; White, P.; Rosie, B.; Mills, K.I.; Burnett, A.K.; Darley, R.L. CD200 as a prognostic factor in acute myeloid leukaemia. Leukemia 2007, 21, 566–568. [Google Scholar] [PubMed]
- Foster-Cuevas, M.; Wright, G.J.; Puklavec, M.J.; Brown, M.H.; Barclay, A.N. Human herpesvirus 8 K14 protein mimics CD200 in down-regulating macrophage activation through CD200 receptor. J. Virol. 2004, 78, 7667–7676. [Google Scholar] [PubMed]
- Shiratori, I.; Yamaguchi, M.; Suzukawa, M.; Yamamoto, K.; Lanier, L.L.; Saito, T.; Arase, H. Down-regulation of basophil function by human CD200 and human herpesvirus-8 CD200. J. Immunol. 2005, 175, 4441–4449. [Google Scholar] [PubMed]
- Voigt, S.; Sandford, G.R.; Hayward, G.S.; Burns, W.H. The English strain of rat cytomegalovirus (CMV) contains a novel captured CD200 (vOX2) gene and a spliced CC chemokine upstream from the major immediate-early region: Further evidence for a separate evolutionary lineage from that of rat CMV Maastricht. J. Gen. Virol. 2005, 86, 263–274. [Google Scholar] [PubMed]
- Langlais, C.L.; Jones, J.M.; Estep, R.D.; Wong, S.W. Rhesus rhadinovirus R15 encodes a functional homologue of human CD200. J. Virol. 2006, 80, 3098–3103. [Google Scholar] [PubMed]
- Zhang, L.; Stanford, M.; Liu, J.; Barrett, C.; Jiang, L.; Barclay, A.N.; McFadden, G. Inhibition of macrophage activation by the myxoma virus M141 protein (vCD200). J. Virol. 2009, 83, 9602–9607. [Google Scholar] [PubMed]
- Vaine, C.A.; Soberman, R.J. The CD200-CD200R1 inhibitory signaling pathway: Immune regulation and host-pathogen interactions. Adv. Immunol. 2014, 121, 191–211. [Google Scholar] [PubMed]
- Rygiel, T.P.; Luijk, B.; Meyaard, L. Use of an anti-CD200 antibody for prolonging the survival of allografts: A patent evaluation of WO2012106634A1. Expert Opin. Ther. Pat. 2013, 23, 389–392. [Google Scholar] [PubMed]
- Simelyte, E.; Criado, G.; Essex, D.; Uger, R.A.; Feldmann, M.; Williams, R.O. CD200-Fc, a novel antiarthritic biologic agent that targets proinflammatory cytokine expression in the joints of mice with collagen-induced arthritis. Arthritis Rheum. 2008, 58, 1038–1043. [Google Scholar] [PubMed]
- Copland, D.A.; Calder, C.J.; Raveney, B.J.E.; Nicholson, L.B.; Phillips, J.; Cherwinski, H.; Jenmalm, M.; Sedgwick, J.D.; Dick, A.D. Monoclonal antibody-mediated CD200 receptor signaling suppresses macrophage activation and tissue damage in experimental autoimmune uveoretinitis. Am. J. Pathol. 2007, 171, 580–588. [Google Scholar] [PubMed]
- Rygiel, T.P.; Meyaard, L. CD200R signaling in tumor tolerance and inflammation: A tricky balance. Curr. Opin. Immunol. 2012, 24, 233–238. [Google Scholar] [PubMed]
- Cox, F.F.; Carney, D.; Miller, A.-M.; Lynch, M.A. CD200 fusion protein decreases microglial activation in the hippocampus of aged rats. Brain. Behav. Immun. 2012, 26, 789–796. [Google Scholar] [PubMed]
- Kim, Y.K.; Que, R.; Wang, S.-W.; Liu, W.F. Modification of biomaterials with a self-protein inhibits the macrophage response. Adv. Healthc. Mater. 2014, 3, 989–994. [Google Scholar] [PubMed]
- Mora-Solano, C.; Collier, J.H. Engaging adaptive immunity with biomaterials. J. Mater. Chem. B 2014, 2, 2409–2421. [Google Scholar]
- Liu, H.; Shalev, I.; Manuel, J.; He, W.; Leung, E.; Crookshank, J.; Liu, M.F.; Diao, J.; Cattral, M.; Clark, D.A.; et al. The FGL2-FcgammaRIIB pathway: A novel mechanism leading to immunosuppression. Eur. J. Immunol. 2008, 38, 3114–3126. [Google Scholar] [PubMed]
- Shankar, S.P.; Petrie, T.A.; García, A.J.; Babensee, J.E. Dendritic cell responses to self-assembled monolayers of defined chemistries. J. Biomed. Mater. Res. A 2010, 92, 1487–1499. [Google Scholar] [PubMed]
- Acharya, A.P.; Dolgova, N.V.; Clare-Salzler, M.J.; Keselowsky, B.G. Adhesive substrate-modulation of adaptive immune responses. Biomaterials 2008, 29, 4736–4750. [Google Scholar] [PubMed]
- Lewis, J.S.; Roche, C.; Zhang, Y.; Brusko, T.M.; Wasserfall, C.H.; Atkinson, M.; Clare-Salzler, M.J.; Keselowsky, B.G. Combinatorial delivery of immunosuppressive factors to dendritic cells using dual-sized microspheres. J. Mater. Chem. B 2014, 2, 2562–2574. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Slee, J.B.; Christian, A.J.; Levy, R.J.; Stachelek, S.J. Addressing the Inflammatory Response to Clinically Relevant Polymers by Manipulating the Host Response Using ITIM Domain-Containing Receptors. Polymers 2014, 6, 2526-2551. https://doi.org/10.3390/polym6102526
Slee JB, Christian AJ, Levy RJ, Stachelek SJ. Addressing the Inflammatory Response to Clinically Relevant Polymers by Manipulating the Host Response Using ITIM Domain-Containing Receptors. Polymers. 2014; 6(10):2526-2551. https://doi.org/10.3390/polym6102526
Chicago/Turabian StyleSlee, Joshua B., Abigail J. Christian, Robert J. Levy, and Stanley J. Stachelek. 2014. "Addressing the Inflammatory Response to Clinically Relevant Polymers by Manipulating the Host Response Using ITIM Domain-Containing Receptors" Polymers 6, no. 10: 2526-2551. https://doi.org/10.3390/polym6102526
APA StyleSlee, J. B., Christian, A. J., Levy, R. J., & Stachelek, S. J. (2014). Addressing the Inflammatory Response to Clinically Relevant Polymers by Manipulating the Host Response Using ITIM Domain-Containing Receptors. Polymers, 6(10), 2526-2551. https://doi.org/10.3390/polym6102526