3. Results and Discussion
We prepared PMPC
64 via RAFT using CPD as a water-soluble CTA. The
Mn(NMR) value, calculated from
1H NMR peak area intensities of pendant methylene protons and terminal phenyl protons (
Table 1), was 1.89 × 10
4. The
Mn(GPC) and
Mw/
Mn values were 8.62 × 10
3 and 1.19, estimated from GPC. A notable observation was the marked deviation of
Mn(GPC) from
Mn(NMR). To verify the true molecular weight of PMPC
64, SLS measurements were performed. The
Mw value of 2.04 × 10
4 determined by SLS was in fair agreement with
Mn(NMR), using
Mw/
Mn = 1.19. It should be mentioned that
Mn(GPC) values estimated by GPC are only apparent values, probably because sodium poly(styrenesulfonate) was used as a standard for molecular weight calibration, compared to PMPC
64, which has a bulky phosphorylcholine side chain.
Table 1.
Characteristics of poly(2-(methacryloyloxy)ethyl phosphorylcholine)-b-poly(N,N-diethyl acrylamide) (PMPCm-PDEAn).
Table 1.
Characteristics of poly(2-(methacryloyloxy)ethyl phosphorylcholine)-b-poly(N,N-diethyl acrylamide) (PMPCm-PDEAn).
| Sample code | Mn(NMR) a × 10−4 | Mn(GPC) b × 10−3 | Mw(SLS) c × 10−4 | Mw/Mn b | A2 × 104 (mol·mL·g−2) c | Rg (nm) d | dn/dCp mL/g |
|---|
| PMPC64 | 1.89 | 8.62 | 2.04 | 1.19 | 7.71 | 9.6 | 0.139 |
| PMPC64-PDEA22 | 2.26 | 8.10 | 2.95 | 1.26 | 7.34 | 13.7 | 0.134 |
| PMPC64-PDEA35 | 2.43 | 8.35 | 3.06 | 1.26 | 4.42 | 16.2 | 0.139 |
| PMPC64-PDEA46 | 2.57 | 8.30 | 3.46 | 1.22 | 4.09 | 18.3 | 0.138 |
| PMPC64-PDEA66 | 2.82 | 8.30 | 3.89 | 1.21 | 7.81 | 22.6 | 0.146 |
To prepare the thermo-responsive diblock copolymer, DEA was polymerized via RAFT radical polymerization in methanol using the PMPC
64 macro-chain transfer agent.
Figure 2 shows the pseudo-first-order kinetics plot for the polymerization of DEA in the presence of PMPC
64 at 70 °C under Ar in methanol containing a sealed tube of D
2O to lock the NMR frequency. Monomer consumption was monitored by
1H NMR spectroscopy as a function of polymerization time. There was an induction period of
ca. 30 min, which may have been due to a slow rate of formation of the radical fragment [
14]. A monomer conversion of 83% was reached within 5 h. The downward curvature was observed, which indicates a decrease in the concentration of propagating radicals.
Figure 2.
Time–conversion (○) and the first-order kinetic plots (△) for polymerization of DEA in the presence of PMPC64 (Mn(NMR) = 1.89 × 104, Mw/Mn = 1.19) in methanol at 70 °C. [M]0 and [M] are the concentrations of the monomer at polymerization time = 0 and the corresponding time, respectively.
Figure 2.
Time–conversion (○) and the first-order kinetic plots (△) for polymerization of DEA in the presence of PMPC64 (Mn(NMR) = 1.89 × 104, Mw/Mn = 1.19) in methanol at 70 °C. [M]0 and [M] are the concentrations of the monomer at polymerization time = 0 and the corresponding time, respectively.
The
Mn(GPC) values estimated from GPC were only apparent values because sodium poly(styrenesulfonate) was used as a standard for molecular weight calibration. To verify the true molecular weight of PMPC
m-PDEA
n, SLS measurements were performed (
Figure 3). The
Mn(NMR) for PMPC
m-PDEA
n was calculated from
1H NMR in D
2O at 25 °C (
Figure 4a). The
Mw(SLS) values of PMPC
m-PDEA
n, determined by SLS, were in fair agreement with
Mn(NMR), using
Mw/
Mn. Apparent
Mw,
Rg, and
A2 for the diblock copolymers at 25 °C, determined by SLS measurement, are listed in
Table 1. The d
n/d
Cp values at 633 nm for the polymers at 25 °C are listed in
Table 1. The same PMPC
64 macro-chain transfer agent of
Mn(NMR) = 1.89 × 10
4 was used to prepare a series of PMPC
64-PDEA
n with different PDEA block lengths. The DP values for PMPC and PDEA blocks were calculated based on
1H NMR.
Table 1 lists the molecular parameters of the polymers.
Figure 3.
Typical example of a Zimm plot for PMPC64-PDEA46 in 0.1 M NaCl at 25 °C. The polymer concentration was varied from 5 to 10 g/L. The Rayleigh ratio (Rθ) is determined by subtracting the solvent scattering from the total scattering for the solutions.
Figure 3.
Typical example of a Zimm plot for PMPC64-PDEA46 in 0.1 M NaCl at 25 °C. The polymer concentration was varied from 5 to 10 g/L. The Rayleigh ratio (Rθ) is determined by subtracting the solvent scattering from the total scattering for the solutions.
To study heat-induced association of the diblock copolymers,
1H NMR spectra for the block copolymers were measured at different temperatures in D
2O containing 0.1 M NaCl.
Figure 4 compares typical
1H NMR spectra for PMPC
64-PDEA
66 measured at 25 and 60 °C. At 25 °C the diblock copolymer chains are fully solvated and molecularly dissolved in water—
i.e., a “unimer” state—and all signals expected for each block were observed. The resonance bands in the 0.8–1.4 ppm region were attributed to the sum of the α-methyl protons in the main chain and the methyl protons in the pendant
N,
N-diethylamino group. The methylene protons in the main chain of the PMPC and PDEA blocks were observed at 1.6 and 2.0 ppm, respectively. The resonance peak at 2.6 ppm was attributed to the methine proton in the main chain of the PDEA block. The resonance bands at 3.1–3.5 ppm were assigned to the methyl protons of the pendant trimethyl ammonium group of the PMPC block and the methylene protons in the pendant
N,
N-diethylamino group of the PDEA block, respectively. The methylene protons in the pendant phosphorylcholine group of the PMPC block were observed at 3.6–4.5 ppm. The composition of the block copolymer was determined from the area intensity ratio of the resonance bands due to the methine proton in the main chain of the PDEA block at 2.6 ppm and the methylene protons of the pendant phosphorylcholine of the PMPC block at 3.6 ppm at 25 °C, because these peaks were relatively isolated from the other peaks. The intensity of resonance peaks corresponding to the PDEA block decreased at an elevated temperature of 60 °C, which implies poor solvation and reduced mobility of the PDEA block. Considering its chemical structure, the block copolymer should form a core-shell-type polymer micelle with dehydrated PDEA blocks forming a core and hydrophilic PMPC blocks forming a shell.
Figure 4.
Comparison of 1H NMR spectra for PMPC64-PDEA66 at 25 °C (a) and 60 °C (b) in D2O containing 0.1 M NaCl at Cp = 5.0 g/L. Asterisks represent peaks due to solvent.
Figure 4.
Comparison of 1H NMR spectra for PMPC64-PDEA66 at 25 °C (a) and 60 °C (b) in D2O containing 0.1 M NaCl at Cp = 5.0 g/L. Asterisks represent peaks due to solvent.
Figure 5 shows the peak intensity ratio for the methine proton at 2.6 ppm in the main chain of the PDEA block and the methylene protons of the pendant phosphorylcholine group of the PMPC block at 3.6 ppm as a function of temperature. The peak intensity ratio is normalized with the ratio at 25 °C. When the temperature is increased from 24 °C, the normalized intensity ratios for PMPC
64-PDEA
35 and PMPC
64-PDEA
22 were almost constant, independent of the temperature. The normalized peak intensity ratios for PMPC
64-PDEA
66 and PMPC
64-PDEA
46 were practically constant below 37 and 47 °C, respectively. The intensity ratios for PMPC
64-PDEA
66 and PMPC
64-PDEA
46 decreased with increasing temperature, reaching minimum values of 0.4 and 0.6, respectively. These findings indicate that the motion of the methine protons in the PDEA blocks for PMPC
64-PDEA
66 and PMPC
64-PDEA
46 was restricted above a certain temperature. These results suggest that the onset of micellization occurs at lower temperatures for polymers with longer DEA block lengths.
Figure 5.
Peak intensity ratio of I2.6/I3.6 normalized to the ratio at 25 °C for PMPC64-PDEA66 (○), PMPC64-PDEA46 (△), PMPC64-PDEA35 (◇), and PMPC64-PDEA22 (□) in D2O containing 0.1 M NaCl at Cp = 5.0 g/L as a function of temperature.
Figure 5.
Peak intensity ratio of I2.6/I3.6 normalized to the ratio at 25 °C for PMPC64-PDEA66 (○), PMPC64-PDEA46 (△), PMPC64-PDEA35 (◇), and PMPC64-PDEA22 (□) in D2O containing 0.1 M NaCl at Cp = 5.0 g/L as a function of temperature.
In 0.1 M NaCl aqueous solution, the diblock copolymer undergoes a transition from a molecularly dissolved unimer state at low temperatures to a micellar state above the critical association temperature (CAT).
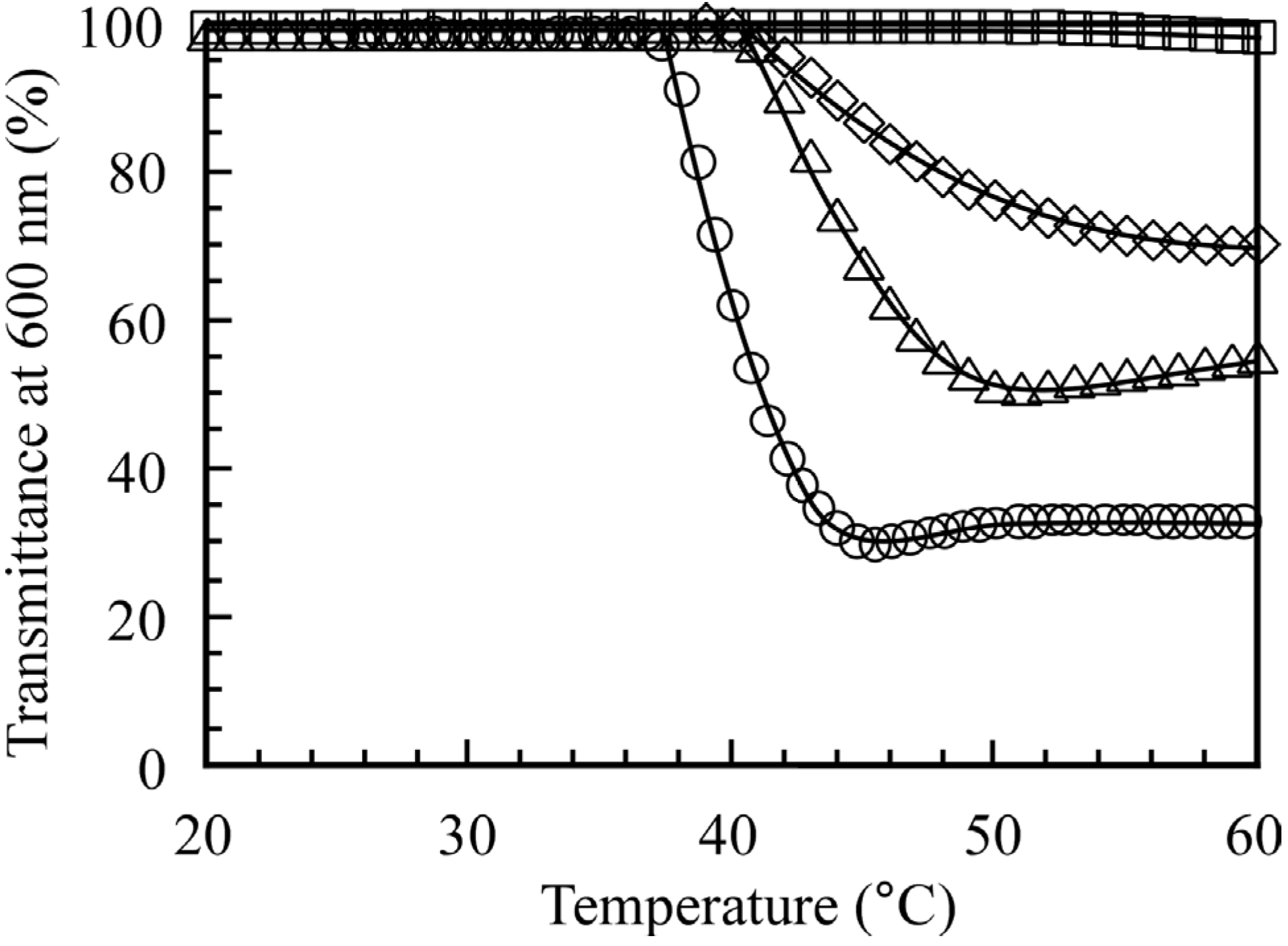
Figure 6 shows values of
T% monitored at 600 nm for an aqueous solution of the diblock copolymers at
Cp = 5.0 g/L. The
T% value for PMPC
64-PDEA
66 is 100% below 38 °C and the solution shows no Tyndall scattering.
T% decreases with increasing temperature, reaching 30% above 50 °C. Tyndall scattering at 50 °C, which is characteristic of micellar solutions, was visually confirmed. The solution, which was turbid above the CAT, became clear again when the solution was cooled below the CAT. The CAT values for the diblock copolymers were estimated from a break in the
T%
versus temperature plot. The CAT values for PMPC
64-PDEA
66, PMPC
64-PDEA
46, PMPC
64-PDEA
35, and PMPC
64-PDEA
22 were estimated at 38, 43, 44, and 54 °C, respectively. The CAT values for PMPC
m-PDEA
n increased as the thermo-responsive PDEA block length decreased. These observations are consistent with the aforementioned
1H NMR data (
Figure 5).
Figure 6.
Percent transmittance (T%) at 600 nm for 0.1 M NaCl aqueous solutions of PMPC64-PDEA66 (○), PMPC64-PDEA46 (△), PMPC64-PDEA35 (◇), and PMPC64-PDEA22 (□) at Cp = 5.0 g/L as a function of temperature.
Figure 6.
Percent transmittance (T%) at 600 nm for 0.1 M NaCl aqueous solutions of PMPC64-PDEA66 (○), PMPC64-PDEA46 (△), PMPC64-PDEA35 (◇), and PMPC64-PDEA22 (□) at Cp = 5.0 g/L as a function of temperature.
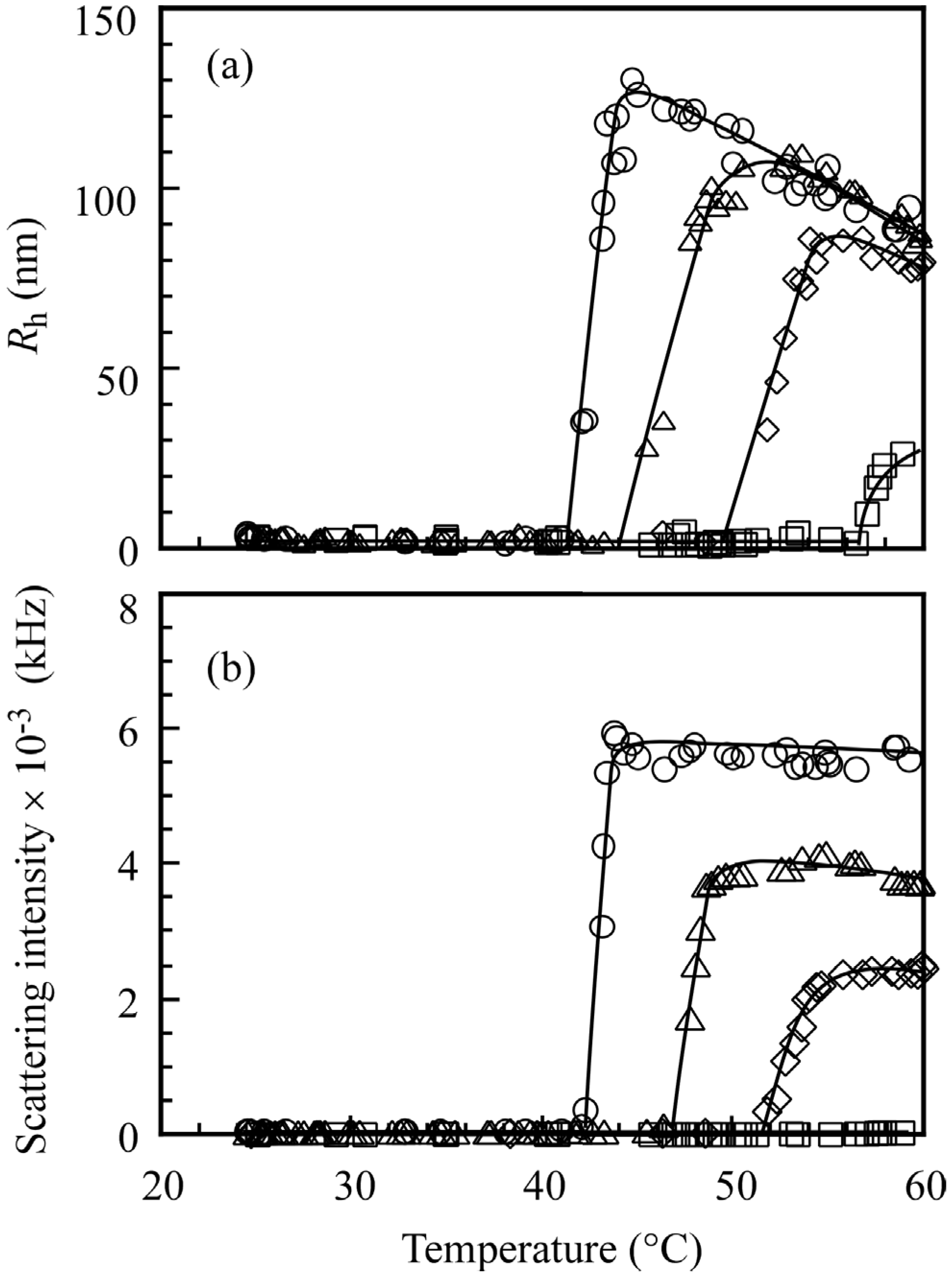
Heat-induced association behavior for the diblock copolymers was confirmed by DLS measurements. The hydrodynamic radius (
Rh) for the diblock copolymer was measured in 0.1 M NaCl aqueous solution.
Figure 7 shows DLS relaxation time distributions for PMPC
64-PDEA
66 at
Cp = 5.0 g/L at 25, 42, and 60 °C. The distributions were unimodal at 25 and 60 °C with different relaxation times. A faster relaxation mode at 25 °C was attributed to a unimer state with
Rh = 3.2 nm, whereas the slower relaxation time at 60 °C was attributed to polymer aggregates with
Rh = 95 nm. At 42 °C, the relaxation time distribution was found to be bimodal. The fast and slow modes were attributed to unimers and polymer aggregates.
Figure 7.
Typical examples of DLS relaxation time distributions for PMPC64-PDEA66 at Cp = 5.0 g/L in 0.1 M NaCl at 25 °C (a), 42 °C (b), and 60 °C (c).
Figure 7.
Typical examples of DLS relaxation time distributions for PMPC64-PDEA66 at Cp = 5.0 g/L in 0.1 M NaCl at 25 °C (a), 42 °C (b), and 60 °C (c).
The
Rh values for the diblock copolymers, determined by DLS measurement, are plotted as a function of temperature in
Figure 8a. The
Rh values for the diblock copolymers below a certain temperature were in the order of
ca. 5 nm, suggesting that all of the polymers existed in a unimer state. Upon an increase in temperature, the
Rh values for PMPC
64-PDEA
66, PMPC
64-PDEA
46, PMPC
64-PDEA
35, and PMPC
64-PDEA
22 began to increase at 42, 44, 50, and 57 °C. These observations indicate the formation of polymer micelles above certain temperatures. As temperature was further increased, the
Rh values for PMPC
64-PDEA
66, PMPC
64-PDEA
46, and PMPC
64-PDEA
35 started to decrease. These observations suggest that aggregates of the PDEA blocks became more compact due to further dehydration of the PDEA blocks as the temperature was increased beyond the CAT, or the aggregation number (
Nagg) of the multipolymer aggregate decreased with increasing temperature. The scattering intensity is proportional to
Mw, because the intensity is linearly related to
Rθ/
Cp (
i.e.,
Rθ/
Cp ∝
Mw). Therefore, the observation that the scattering intensities are nearly constant above CAT (
Figure 8b) suggests that
Nagg is practically constant. The absolute scattering intensity is not important to obtain DLS data, however for SLS measurements the absolute scattering intensity is important to determine correct
Rθ. The accurate SLS data for the polymer micelle solutions at high temperature cannot be obtained, because there is multiple scattering from the turbid solution at 60 °C (
Figure 6).
Figure 8.
(a) Hydrodynamic radius (Rh) and (b) scattering intensity for PMPC64-PDEA66 (○), PMPC64-PDEA46 (△), PMPC64-PDEA35 (◇), and PMPC64-PDEA22 (□) at Cp = 5.0 g/L in 0.1 M NaCl as a function of temperature.
Figure 8.
(a) Hydrodynamic radius (Rh) and (b) scattering intensity for PMPC64-PDEA66 (○), PMPC64-PDEA46 (△), PMPC64-PDEA35 (◇), and PMPC64-PDEA22 (□) at Cp = 5.0 g/L in 0.1 M NaCl as a function of temperature.
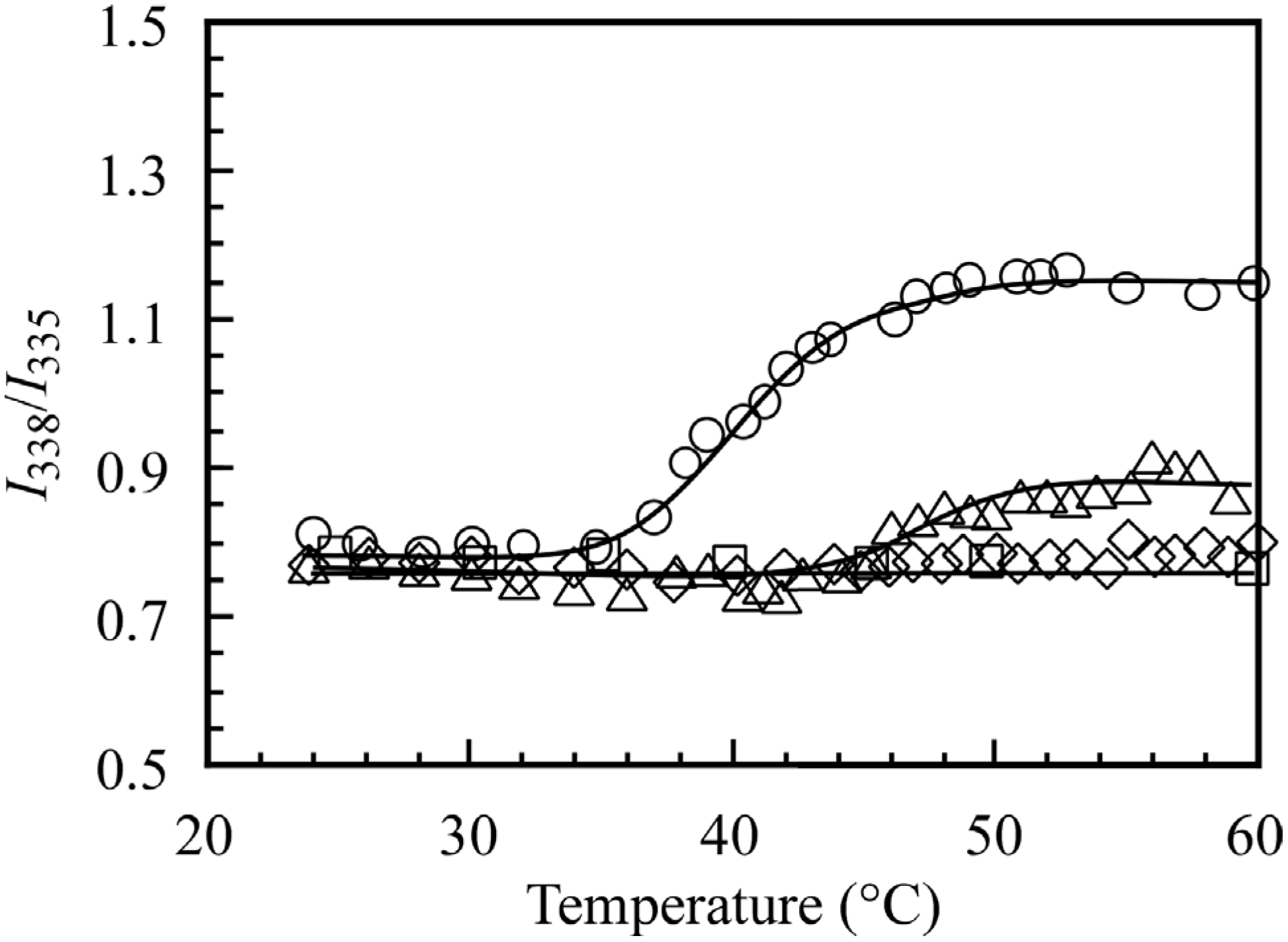
Excitation spectra of pyrene in 0.1 M NaCl aqueous solution in the presence of the block copolymers at varying temperatures were obtained. The excitation spectrum in the case of PMPC
64-PDEA
66 showed peaks associated with the (0-0) band of pyrene at 335 nm below the CAT, and the peak shifted to 338 nm at 60 °C. It is known that the (0-0) band in pyrene excitation spectra in water shifts to longer wavelengths when pyrene is solubilized in hydrophobic domains [
15]. Thus, we estimated the ratio of the intensity at 338 nm relative to that at 335 nm (
I338/
I335), and this is plotted in
Figure 9 as a function of temperature. It is known that the dithiobenzoate group at the polymer chain end prepared via RAFT is responsible for pyrene fluorescence quenching [
16]. We measured only the relative intensity ratio of the excitation emission spectra. Therefore, the quenching effect can be ignored to some degree. The
I338/
I335 values for PMPC
64-PDEA
35 and PMPC
64-PDEA
22 were almost constant independent of the temperature. This observation indicates that the hydrophobic domain formed from the PDEA blocks could not incorporate the pyrene probes, presumably because of the low hydrophobicity of the core. As temperature was increased, the
I338/
I335 value for PMPC
64-PDEA
66 and PMPC
64-PDEA
46 began to increase at 34 and 44 °C. When the temperature was increased from 25 to 60 °C and subsequently decreased to 25 °C, the thermo-responsive emission spectral changes were found to be completely reversible without hysteresis.
The CAT values for the block copolymers were measured using various methods, including 1H NMR, T%, Rh, scattering intensity, and pyrene fluorescence. These CAT values did not always coincide, depending on the measurement method. For example, the CATs for PMPC64-PDEA66 estimated from 1H NMR, Rh, and fluorescence methods were 37, 42, and 36 °C, respectively, presumably due to the difference in the sensitivity of the measurement methods.
Figure 9.
I338/I335 in pyrene excitation spectra in the presence of PMPC64-PDEA66 (○), PMPC64-PDEA46 (△), PMPC64-PDEA35 (◇), and PMPC64-PDEA22 (□) at Cp = 5.0 g/L as a function of temperature in 0.1 M NaCl aqueous solution.
Figure 9.
I338/I335 in pyrene excitation spectra in the presence of PMPC64-PDEA66 (○), PMPC64-PDEA46 (△), PMPC64-PDEA35 (◇), and PMPC64-PDEA22 (□) at Cp = 5.0 g/L as a function of temperature in 0.1 M NaCl aqueous solution.
The capture and release of small hydrophobic guest drugs by PMPC
m-PDEA
n in 0.1 M NaCl aqueous solution may be easily controlled by changing the temperature. We studied the thermo-responsive release behavior of PMPC
64-PDEA
66, applying a dialysis method, with ADR as a guest drug [
17]. Above the CAT, PMPC
64-PDEA
66 formed polymer micelles with a hydrophobic core, sufficient to keep ADR captured in the interior. Below the CAT, however, the polymer micelles dissociated, and ADR was released from the core into the aqueous bulk phase.
Figure 10 represents the time course of the cumulative permeation of ADR through the dialysis membrane at 25 and 50 °C. For comparison, data without PMPC
64-PDEA
66 at 25 and 50 °C are presented along with control data. At 25 °C, the rate of permeation of ADR through the membrane was almost same in the presence and absence of PMPC
64-PDEA
66; however, the permeation rate of ADR without PMPC
64-PDEA
66 at 50 °C was much faster than that at 25 °C. At 50 °C, the permeation rate of ADR from the polymer micelle was slow compared to the control experiment without the polymer. From these observations, it was concluded that the PMPC
64-PDEA
66 polymer micelle can retain captured ADR in its core at 50 °C. It is noteworthy that the release rate from the PMPC
64-PDEA
66 polymer micelle at 50 °C was slightly higher than that at 25 °C. This observation suggests that the polymer micelle cannot completely incorporate ADR into its core at 50 °C, because the diffusion coefficient of ADR at 50 °C is much higher than at 25 °C. To create a temperature-responsive micelle containing such a molecule without leaking the hydrophobic medication at temperatures higher than the CAT, it is necessary to improve the molecular design of the diblock copolymer—for example, by extending the hydrophobic block length.
Figure 10.
Cumulative ADR release to environment at 25 °C (
a) and 50 °C (
b). In the blank release (
![Polymers 06 00846 i002]()
), ADR (0.06 mM) solution without polymer was released into 0.1 M NaCl aqueous solution. ADR solution with PMPC
64-PDEA
66 (○) was released into 0.1 M NaCl aqueous solution.
Figure 10.
Cumulative ADR release to environment at 25 °C (
a) and 50 °C (
b). In the blank release (
![Polymers 06 00846 i002]()
), ADR (0.06 mM) solution without polymer was released into 0.1 M NaCl aqueous solution. ADR solution with PMPC
64-PDEA
66 (○) was released into 0.1 M NaCl aqueous solution.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








 ), ADR (0.06 mM) solution without polymer was released into 0.1 M NaCl aqueous solution. ADR solution with PMPC64-PDEA66 (○) was released into 0.1 M NaCl aqueous solution.
), ADR (0.06 mM) solution without polymer was released into 0.1 M NaCl aqueous solution. ADR solution with PMPC64-PDEA66 (○) was released into 0.1 M NaCl aqueous solution.
