
Water-Dispersible Silica-Polyelectrolyte Nanocomposites Prepared via Acid-Triggered Polycondensation of Silicic Acid and Directed by Polycations

Abstract
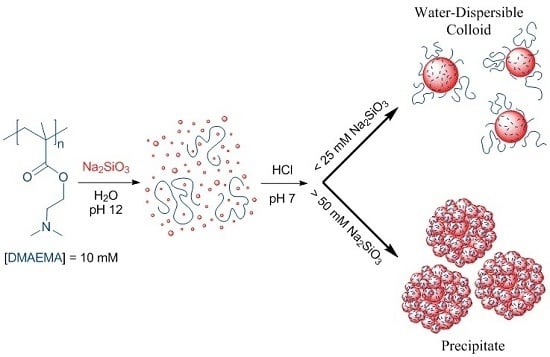
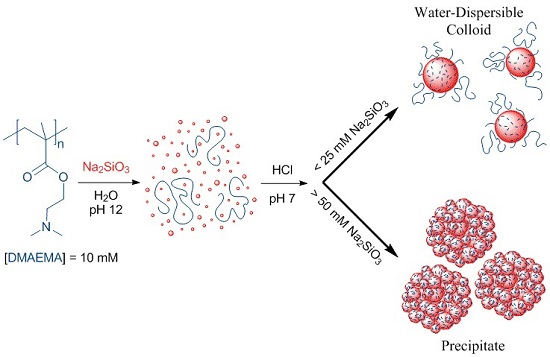
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Polymer Synthesis and Preparation of Composites
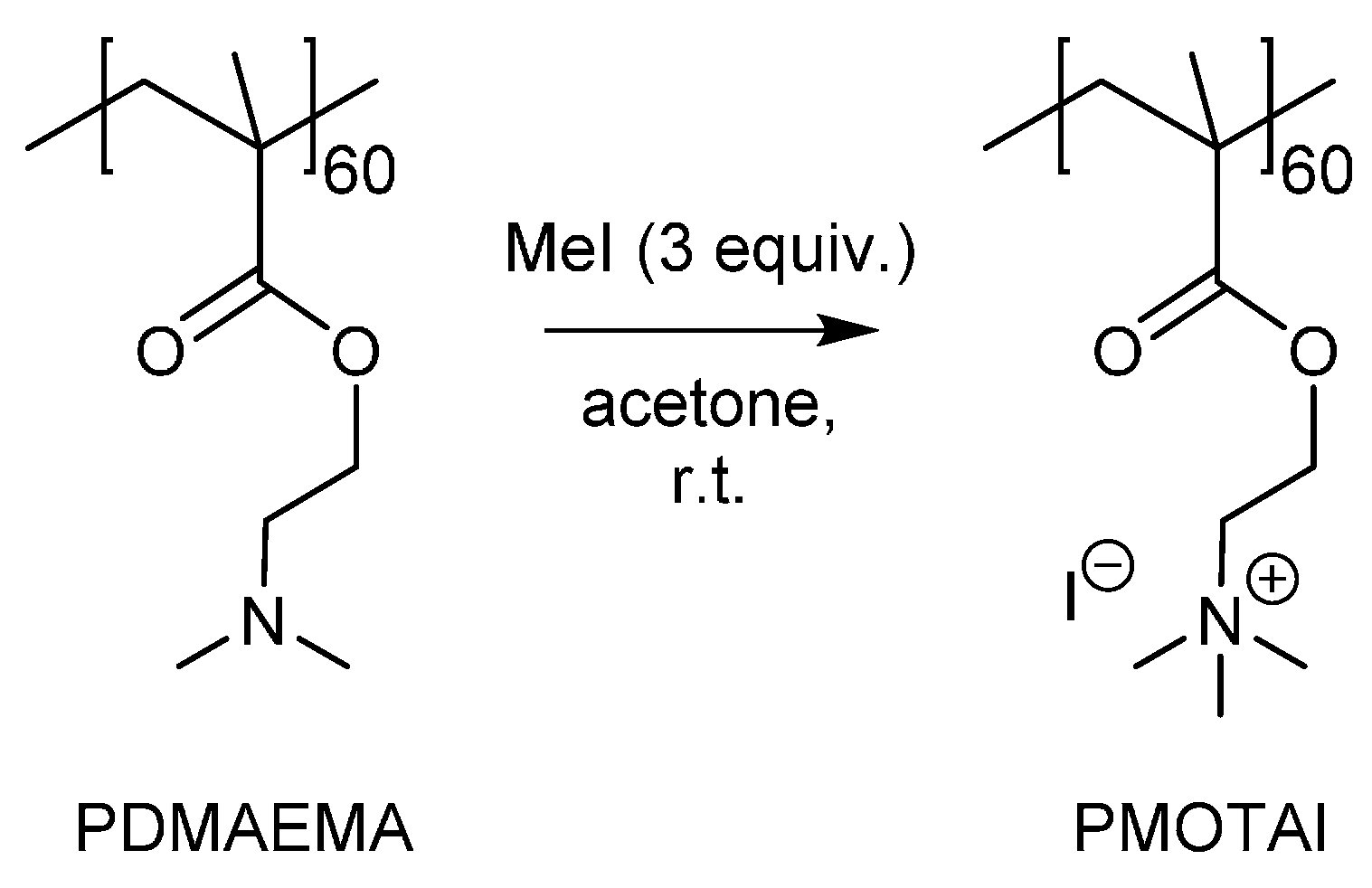
2.2.1. Synthesis of Polymers
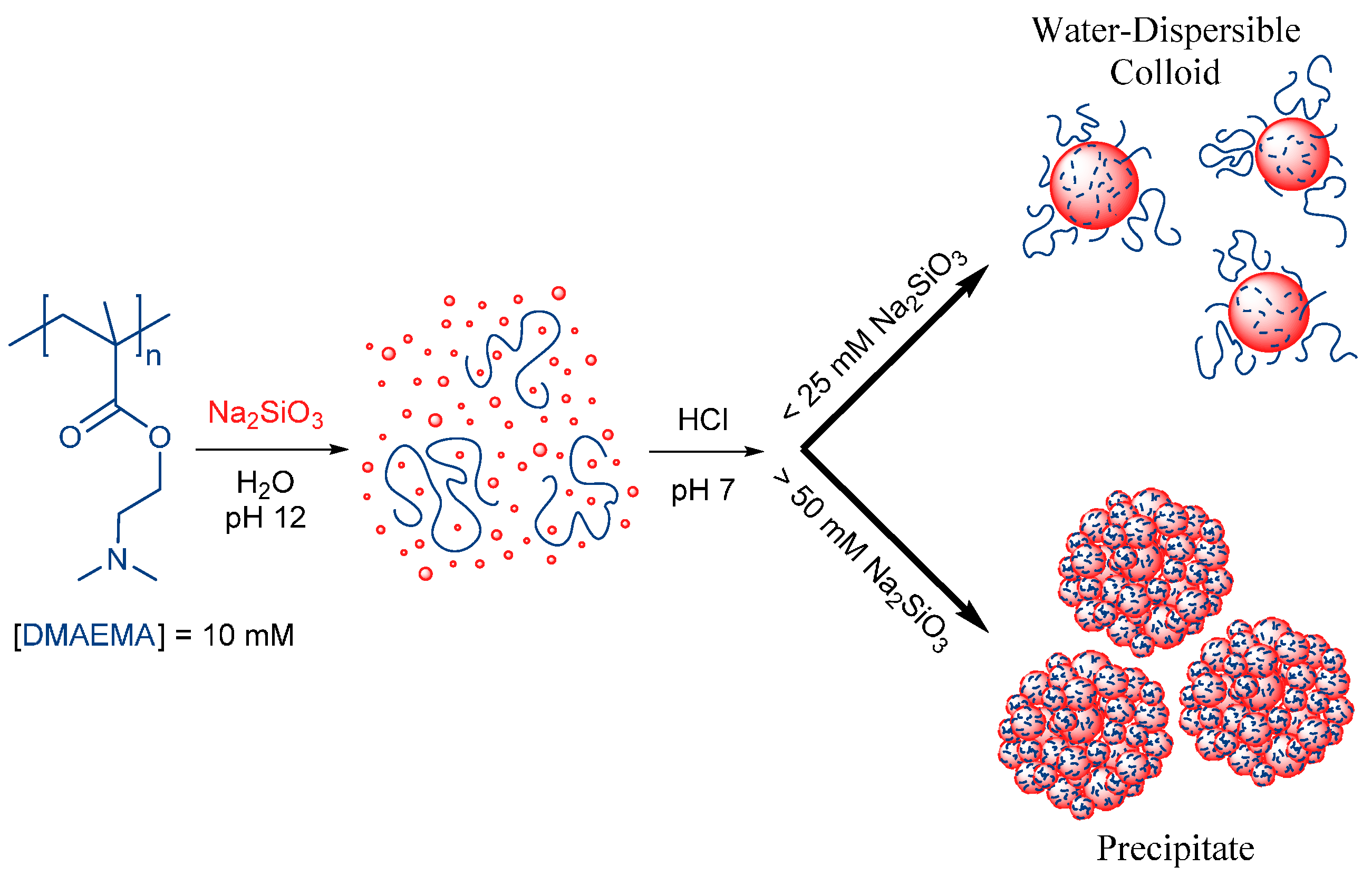
2.2.2. Preparation of Silica-Polyelectrolyte (S-PE) Precipitate Nanocomposites
2.2.3. Preparation of Silica-Polyelectrolyte (S-PE) Water-Dispersible Nanocomposites
2.3. Instrumentation
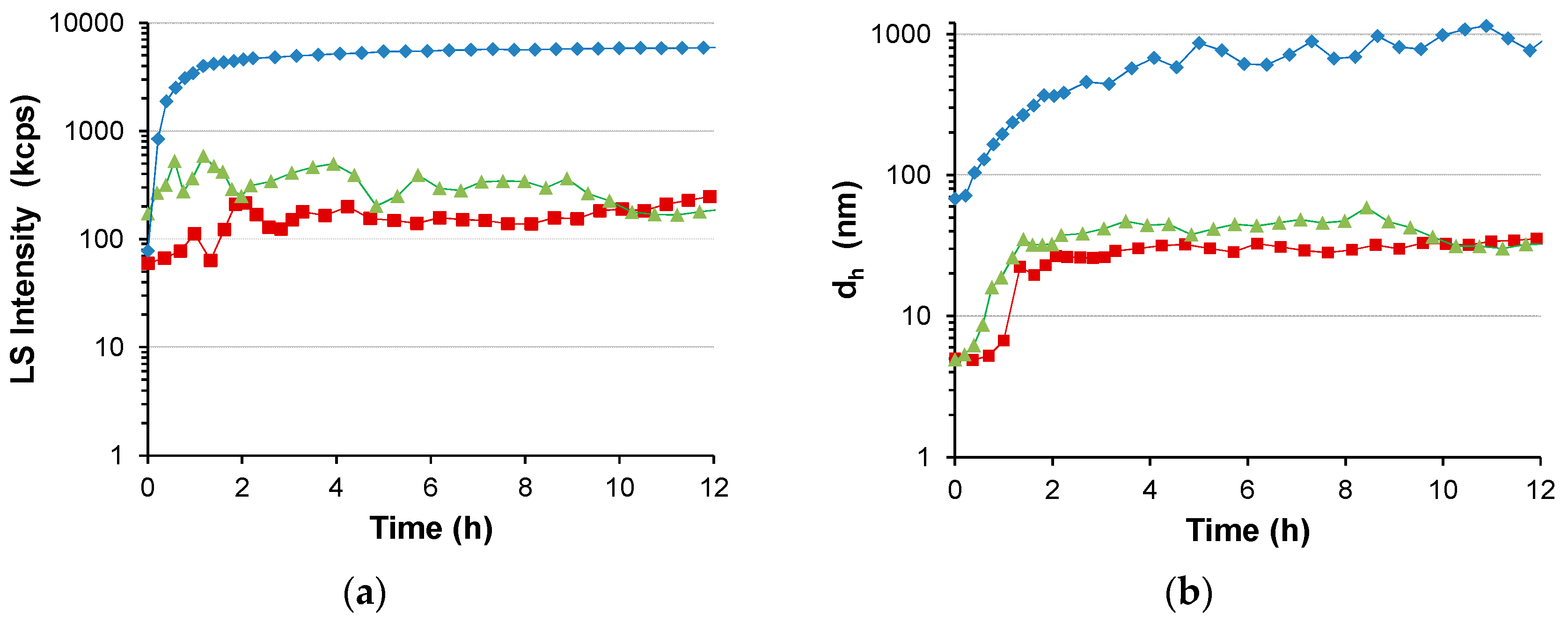
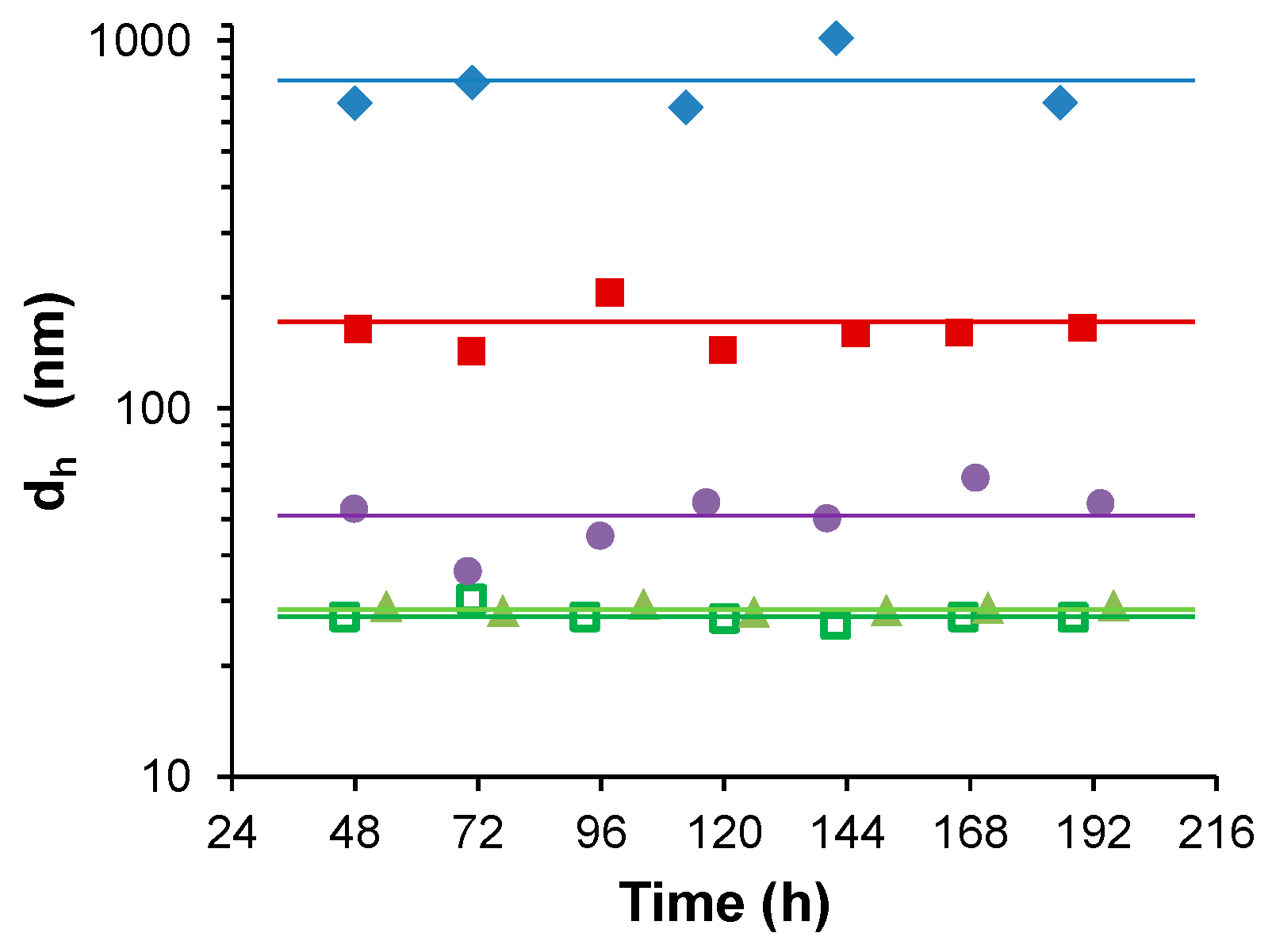
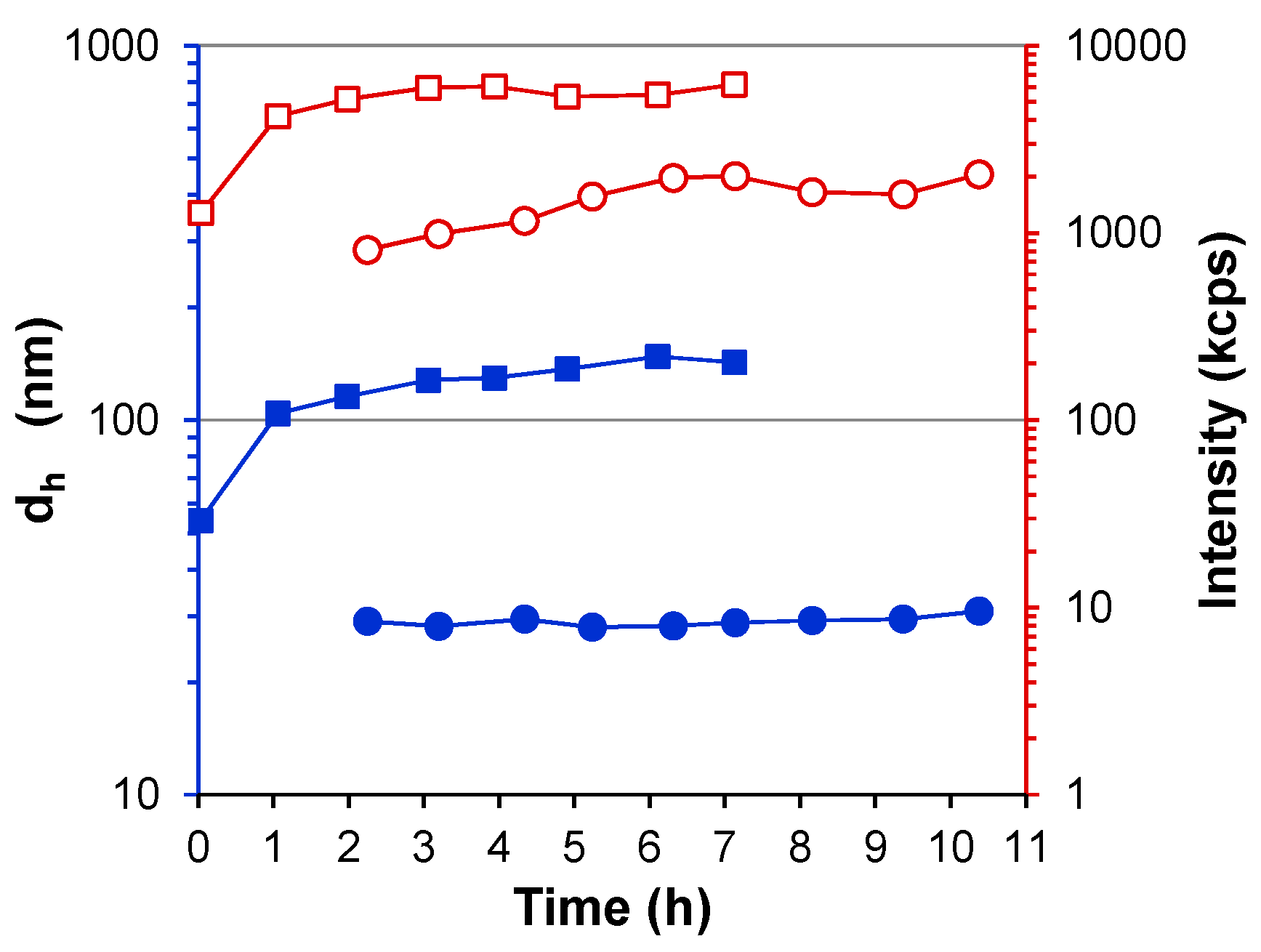
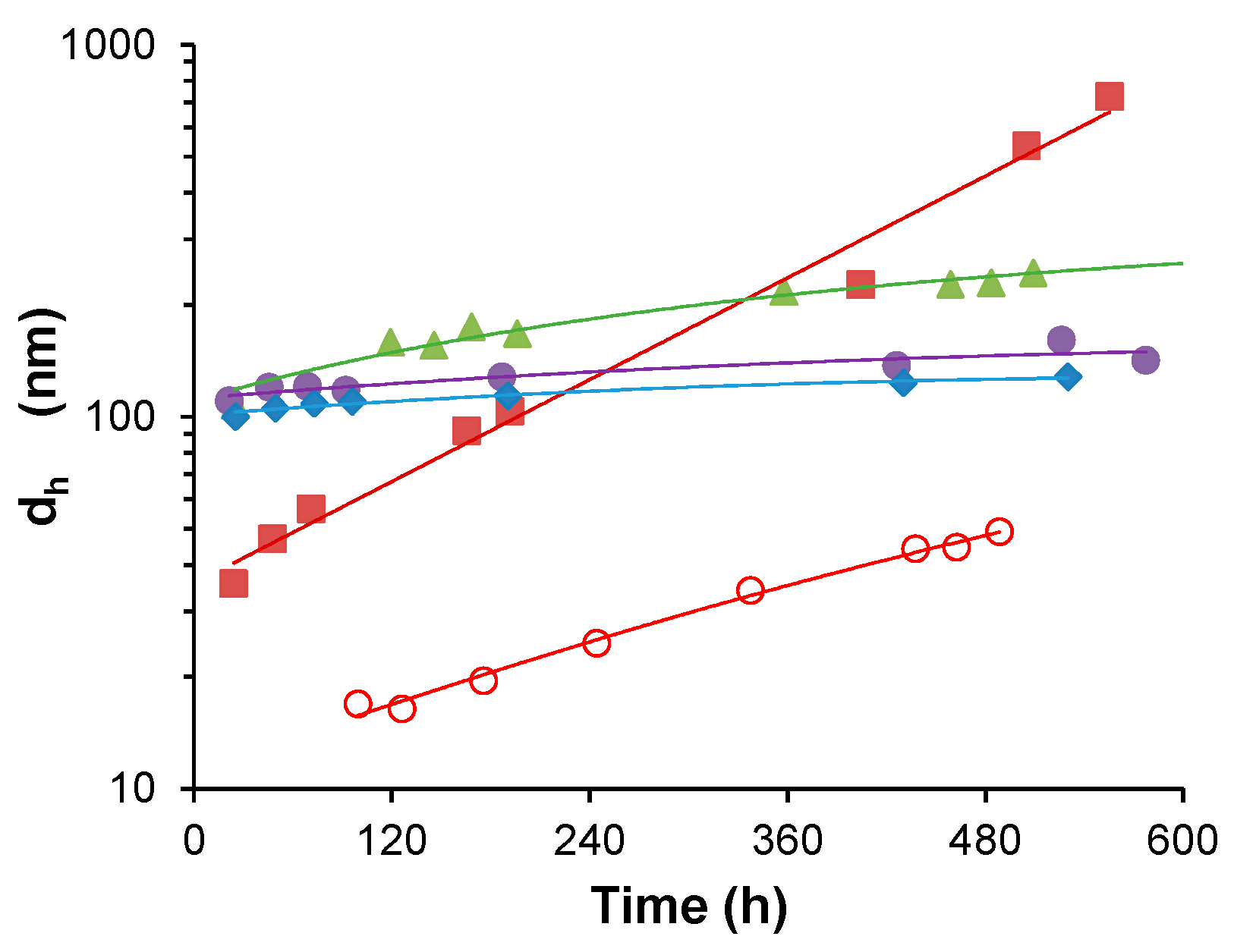
3. Results
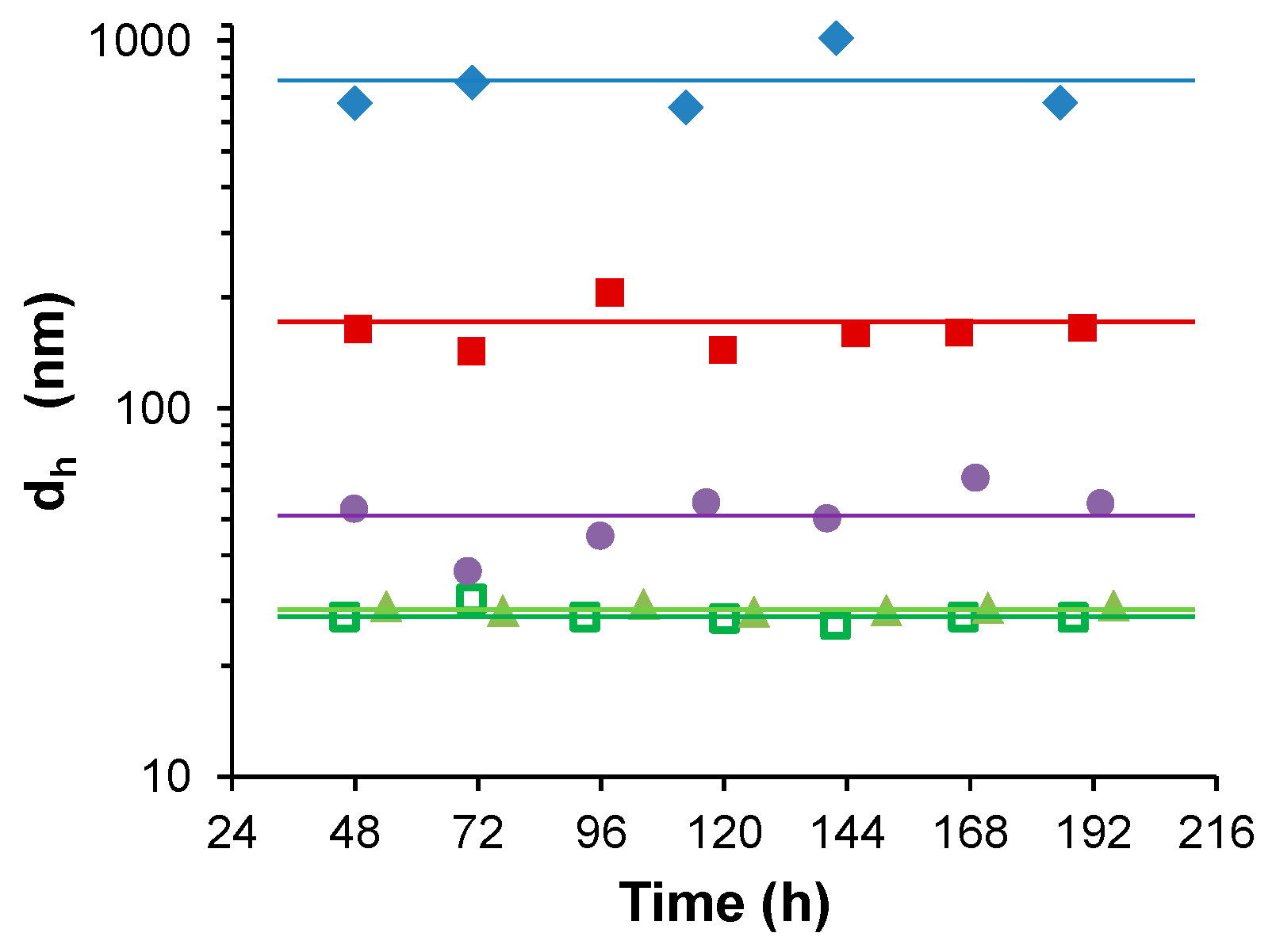
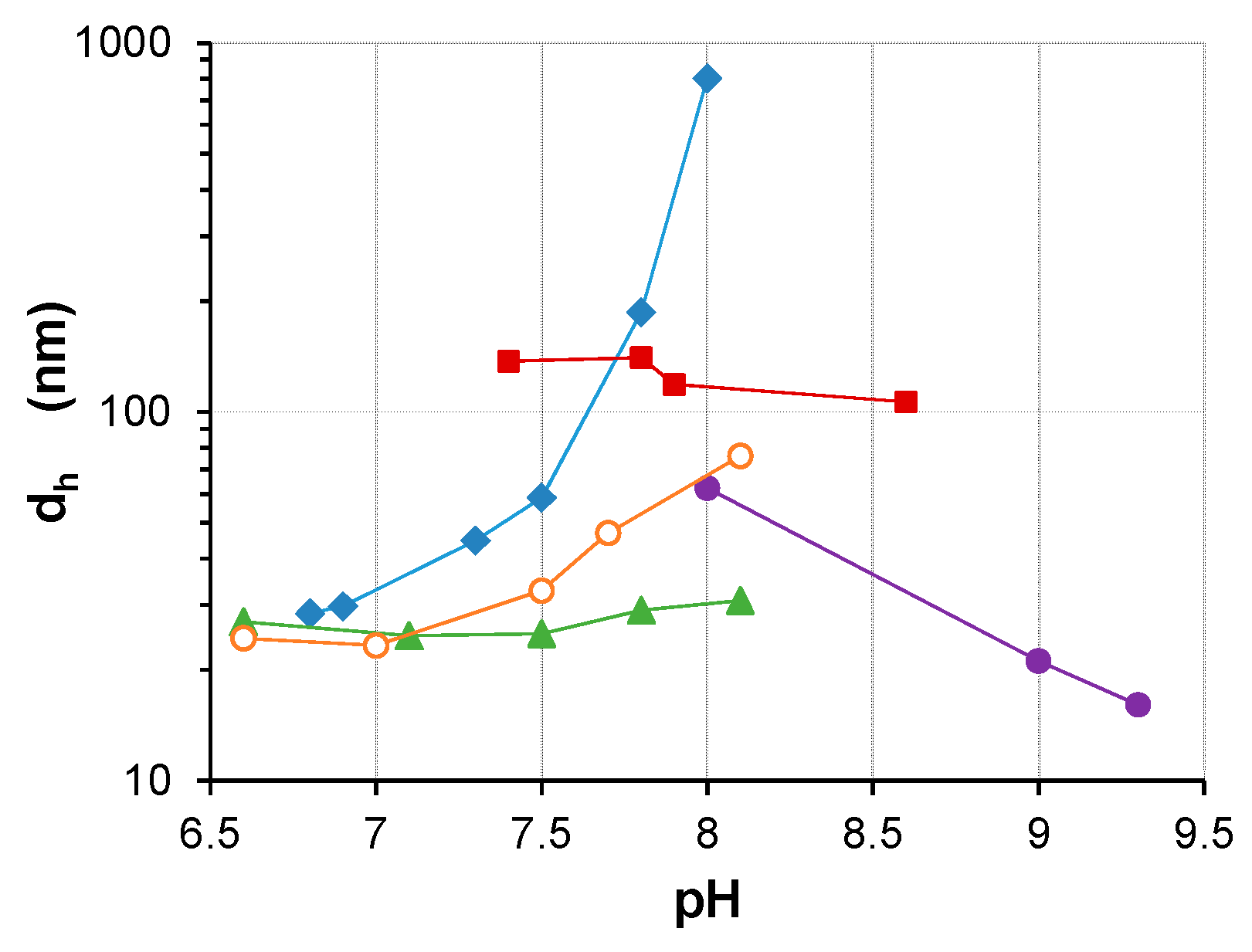
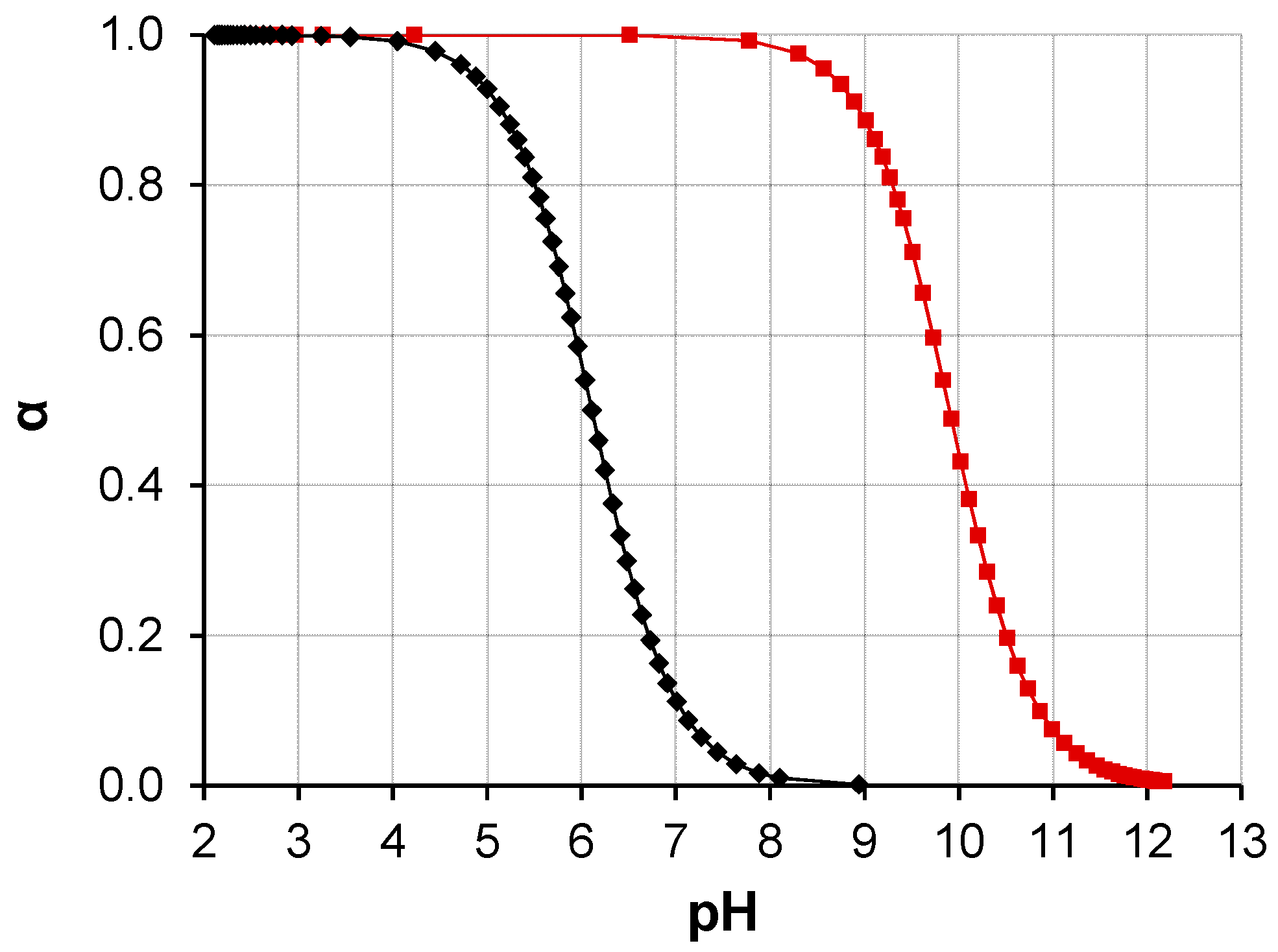
3.1. Water-Stable S-PE Nanocomposites
3.2. Precipitate S-PE Nanocomposites
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AIBN | 2,2′-azobisisobutylnitrile (or 2,2′-azobis(2-methylpropionitrile)) |
| ATRP | atom-transfer radical-polymerization |
| CPA | 4-cyanopentanoic acid dithiobenzoate |
| CTA | chain transfer agent |
| DLS | dynamic light scattering |
| EBiB | ethyl α-bromoisobutyrate |
| IR | infrared spectroscopy |
| LCPAs | long-chain polyamines |
| NMR | nuclear magnetic resonance |
| OEGMA | oligo(ethylene glycol) methyl ether methacrylate |
| PDMAEMA | poly(2-(dimethylamino)ethyl methacrylate) |
| PE | polyelectrolyte |
| PMOTAI | poly(methacryloxyethyl trimethylammonium iodide) |
| PSA | poly(silicic acid) |
| RAFT | reversible addition fragmentation chain-transfer polymerization |
| SA | silicic acid (≡orthosilicic acid, monosilicic acid) |
| SEC | size exclusion chromatography |
| SEM | scanning electron microscopy |
| SM | supplementary materials |
| S-PE | silica-polyelectrolyte (nanocomposite) |
| SS | sodium silicate (sodium water glass) |
| TBAB | tetra-n-butylammonium bromide |
| TEOS | tetraethyl orthosilicate |
| TGA | thermogravimetric analysis |
| THF | tetrahydrofuran |
| TMOS | tetramethyl orthosilicate |
References
- Amo, Y.D.; Brzezinski, M.A. The chemical form of dissolved Si taken up by marine diatoms. J. Phycol. 1999, 35, 1162–1170. [Google Scholar] [CrossRef]
- Cha, J.N.; Stucky, G.D.; Morse, D.E.; Deming, T.J. Biomimetic synthesis of ordered silica structures mediated by block copolypeptides. Nature 2000, 403, 289–292. [Google Scholar] [PubMed]
- Matsunaga, S.; Sakai, R.; Jimbo, M.; Kamiya, H. Long-chain polyamines (LCPAs) from marine sponge: Possible implication in spicule formation. ChemBioChem 2007, 8, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Kröger, N.; Deutzmann, R.; Sumper, M. Polycationic peptides from diatom biosilica that direct silica nanosphere formation. Science 1999, 286, 1129–1132. [Google Scholar] [PubMed]
- Sumper, M.; Brunner, E. Silica biomineralisation in diatoms: The model organism thalassiosira pseudonana. ChemBioChem 2008, 9, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Cha, J.; Stucky, G.D.; Morse, D.E. Silicatein α: Cathepsin L-like protein in sponge biosilica. Proc. Natl. Acad. Sci. USA 1998, 95, 6234–6238. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Lopez, P.J.; Rosengarten, G. Diatoms: Self assembled silica nanostructures, and templates for bio/chemical sensors and biomimetic membranes. Analyst 2011, 136, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Pyun, J.; Matyjaszewski, K. Synthesis of nanocomposite organic/inorganic hybrid materials using controlled/“living” radical polymerization. Chem. Mater. 2001, 13, 3436–3448. [Google Scholar] [CrossRef]
- Niskanen, J.; Karesoja, M.; Rossi, T.; Tenhu, H. Temperature and pH responsive hybrid nanoclay grafted with PDMAEMA. Polym. Chem. 2011, 2, 2027–2036. [Google Scholar] [CrossRef]
- Karesoja, M.; McKee, J.; Karjalainen, E.; Hietala, S.; Bergman, L.; Linden, M.; Tenhu, H. Mesoporous silica particles grafted with poly(ethyleneoxide-block-N-vinylcaprolactam). J. Polym. Sci. Part Polym. Chem. 2013, 51, 5012–5020. [Google Scholar] [CrossRef]
- Li, C.; Benicewicz, B.C. Synthesis of well-defined polymer brushes grafted onto silica nanoparticles via surface reversible addition-fragmentation chain transfer polymerization. Macromolecules 2005, 38, 5929–5936. [Google Scholar] [CrossRef]
- Zhang, Y.; Ye, Z. Covalent surface grafting of branched polyethylenes on silica nanoparticles by surface-initiated ethylene “living” polymerization with immobilized Pd-diimine catalysts. Macromolecules 2008, 41, 6331–6338. [Google Scholar] [CrossRef]
- Wan, Y.; Wang, H.; Zhao, Q.; Klingstedt, M.; Terasaki, O.; Zhao, D. Ordered mesoporous Pd/silica-carbon as a highly active heterogeneous catalyst for coupling reaction of chlorobenzene in aqueous media. J. Am. Chem. Soc. 2009, 131, 4541–4550. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Deng, W.; Guo, E.; Chung, P.-W.; Chen, S.; Trewyn, B.G.; Brown, R.C.; Lin, V.S.-Y. Mesoporous silica nanoparticle-stabilized and manganese-modified rhodium nanoparticles as catalysts for highly selective synthesis of ethanol and acetaldehyde from syngas. ChemCatChem 2012, 4, 674–680. [Google Scholar] [CrossRef]
- Qu, Z.; Hu, F.; Chen, K.; Duan, Z.; Gu, H.; Xu, H. A facile route to the synthesis of spherical poly(acrylic acid) brushes via RAFT polymerization for high-capacity protein immobilization. J. Colloid Interface Sci. 2013, 398, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Xue, M.; Xia, T.; Zhao, Y.-L.; Tamanoi, F.; Stoddart, J.F.; Zink, J.I.; Nel, A.E. Autonomous in vitro anticancer drug release from mesoporous silica nanoparticles by pH-sensitive nanovalves. J. Am. Chem. Soc. 2010, 132, 12690–12697. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Zakaria, M.B.; Chiang, Y.-D.; Wu, K.C.-W.; Yamauchi, Y. Thermally stable polymer composites with improved transparency by using colloidal mesoporous silica nanoparticles as inorganic fillers. Phys. Chem. Chem. Phys. 2012, 14, 7427. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Obi, S.; Kamata, Y.; Kashiwakura, T.; Kasuya, M.; Ogawa, T.; Kohri, M.; Nakahira, T. Preparation of organic/inorganic hybrid and hollow particles by catalytic deposition of silica onto core/shell heterocoagulates modified with poly[2-(N,N-dimethylamino)ethyl methacrylate]. J. Colloid Interface Sci. 2012, 368, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Kashiwakura, T.; Inada, T.; Kunisada, Y.; Kasuya, M.; Kohri, M.; Nakahira, T. Preparation of organic/inorganic composites by deposition of silica onto shell layers of polystyrene (core)/poly[2-(N,N-dimethylamino)ethyl methacrylate] (shell) particles. J. Colloid Interface Sci. 2010, 347, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.N.; Shimizu, K.; Zhou, Y.; Christiansen, S.C.; Chmelka, B.F.; Stucky, G.D.; Morse, D.E. Silicatein filaments and subunits from a marine sponge direct the polymerization of silica and silicones in vitro. Proc. Natl. Acad. Sci. USA 1999, 96, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.C. Silicification: The processes by which organisms capture and mineralize silica. Rev. Mineral. Geochem. 2003, 54, 291–327. [Google Scholar] [CrossRef]
- Knoblich, B.; Gerber, T. Aggregation in SiO2 sols from sodium silicate solutions. J. Non-Cryst. Solids 2001, 283, 109–113. [Google Scholar] [CrossRef]
- Knoblich, B.; Gerber, T. The arrangement of fractal clusters dependent on the pH value in silica gels from sodium silicate solutions. J. Non-Cryst. Solids 2001, 296, 81–87. [Google Scholar] [CrossRef]
- Preari, M.; Spinde, K.; Lazic, J.; Brunner, E.; Demadis, K.D. Bioinspired insights into silicic acid stabilization mechanisms: The dominant role of polyethylene glycol-induced hydrogen bonding. J. Am. Chem. Soc. 2014, 136, 4236–4244. [Google Scholar] [CrossRef] [PubMed]
- Danilovtseva, E.; Aseyev, V.; Karesoja, M.; Annenkov, V. Sorption of silicic acid from non-saturated aqueous solution by a complex of zinc ions with poly(vinylamine). Eur. Polym. J. 2009, 45, 1391–1396. [Google Scholar] [CrossRef]
- Si, M.; Feng, D.; Qiu, L.; Jia, D.; Elzatahry, A.A.; Zheng, G.; Zhao, D. Free-standing highly ordered mesoporous carbon-silica composite thin films. J. Mater. Chem. A 2013, 1, 13490. [Google Scholar] [CrossRef]
- Rao, K.S.; El-Hami, K.; Kodaki, T.; Matsushige, K.; Makino, K. A novel method for synthesis of silica nanoparticles. J. Colloid Interface Sci. 2005, 289, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Y.; Cao, C. Facile one-pot synthesis of mesoporous hierarchically structured silica/carbon nanomaterials. J. Mater. Chem. 2012, 22, 13918. [Google Scholar] [CrossRef]
- Kawashima, D.; Aihara, T.; Kobayashi, Y.; Kyotani, T.; Tomita, A. Preparation of mesoporous carbon from organic polymer/silica nanocomposite. Chem. Mater. 2000, 12, 3397–3401. [Google Scholar] [CrossRef]
- Park, S.K.; Kim, K.D.; Kim, H.T. Preparation of silica nanoparticles: Determination of the optimal synthesis conditions for small and uniform particles. Colloids Surf. Physicochem. Eng. Asp. 2002, 197, 7–17. [Google Scholar] [CrossRef]
- Pálmai, M.; Nagy, L.N.; Mihály, J.; Varga, Z.; Tárkányi, G.; Mizsei, R.; Szigyártó, I.C.; Kiss, T.; Kremmer, T.; Bóta, A. Preparation, purification, and characterization of aminopropyl-functionalized silica sol. J. Colloid Interface Sci. 2013, 390, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Avnir, D.; Coradin, T.; Lev, O.; Livage, J. Recent bio-applications of sol–gel materials. J. Mater. Chem. 2006, 16, 1013–1030. [Google Scholar] [CrossRef]
- Ferrer, M.L.; del Monte, F.; Levy, D.A. Novel and simple alcohol-free sol-gel route for encapsulation of labile proteins. Chem. Mater. 2002, 14, 3619–3621. [Google Scholar] [CrossRef]
- Nelson, D.M.; Tréguer, P.; Brzezinski, M.A.; Leynaert, A.; Quéguiner, B. Production and dissolution of biogenic silica in the ocean: Revised global estimates, comparison with regional data and relationship to biogenic sedimentation. Glob. Biogeochem. Cycles 1995, 9, 359–372. [Google Scholar] [CrossRef]
- Treguer, P.; Nelson, D.M.; Van Bennekom, A.J.; DeMaster, D.J.; Leynaert, A.; Queguiner, B. The silica balance in the world ocean: A reestimate. Science 1995, 268, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, M.; Volcani, B.E.; Gassmann, W.; Schroeder, J.I. A gene family of silicon transporters. Nature 1997, 385, 688–689. [Google Scholar] [CrossRef] [PubMed]
- Thamatrakoln, K.; Alverson, A.J.; Hildebrand, M. Comparative sequence analysis of diatom silicon transporters: Toward a mechanistic model of silicon transport. J. Phycol. 2006, 42, 822–834. [Google Scholar] [CrossRef]
- Choi, O.; Kim, B.-C.; An, J.-H.; Min, K.; Kim, Y.H.; Um, Y.; Oh, M.-K.; Sang, B.-I. A biosensor based on the self-entrapment of glucose oxidase within biomimetic silica nanoparticles induced by a fusion enzyme. Enzyme Microb. Technol. 2011, 49, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Brott, L.L.; Naik, R.R.; Pikas, D.J.; Kirkpatrick, S.M.; Tomlin, D.W.; Whitlock, P.W.; Clarson, S.J.; Stone, M.O. Ultrafast holographic nanopatterning of biocatalytically formed silica. Nature 2001, 413, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Luckarift, H.R.; Spain, J.C.; Naik, R.R.; Stone, M.O. Enzyme immobilization in a biomimetic silica support. Nat. Biotechnol. 2004, 22, 211–213. [Google Scholar] [CrossRef] [PubMed]
- Nam, D.H.; Lee, J.-O.; Sang, B.-I.; Won, K.; Kim, Y.H. Silaffin peptides as a novel signal enhancer for gravimetric biosensors. Appl. Biochem. Biotechnol. 2013, 170, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Brunner, E.; Lutz, K.; Sumper, M. Biomimetic synthesis of silica nanospheres depends on the aggregation and phase separation of polyamines in aqueous solution. Phys. Chem. Chem. Phys. 2004, 6, 854–857. [Google Scholar] [CrossRef]
- Annenkov, V.V.; Danilovtseva, E.N.; Pal’shin, V.A.; Aseyev, V.O.; Petrov, A.K.; Kozlov, A.S.; Patwardhan, S.V.; Perry, C.C. Poly(vinyl amine)-silica composite nanoparticles: Models of the silicic acid cytoplasmic pool and as a silica precursor for composite materials formation. Biomacromolecules 2011, 12, 1772–1780. [Google Scholar] [CrossRef] [PubMed]
- Chiefari, J.; Chong, Y.K.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T.P.T.; Mayadunne, R.T.A.; Meijs, G.F.; Moad, C.L.; Moad, G.; et al. Living free-radical polymerization by reversible addition-fragmentation chain transfer: The RAFT process. Macromolecules 1998, 31, 5559–5562. [Google Scholar] [CrossRef]
- Sahnoun, M.; Charreyre, M.-T.; Veron, L.; Delair, T.; D’Agosto, F. Synthetic and characterization aspects of dimethylaminoethyl methacrylate reversible addition fragmentation chain transfer (RAFT) polymerization. J. Polym. Sci. Polym. Chem. 2005, 43, 3551–3565. [Google Scholar] [CrossRef]
- Wang, J.-S.; Matyjaszewski, K. Controlled/“living” radical polymerization. atom transfer radical polymerization in the presence of transition-metal complexes. J. Am. Chem. Soc. 1995, 117, 5614–5615. [Google Scholar] [CrossRef]
- Deželić, G.; Kratohvil, J.P. Determination of size of small particles by light scattering. Experiments on ludox colloidal silica. Kolloid-Zeitschrift 1960, 173, 38–48. [Google Scholar] [CrossRef]
- You, Y.-Z.; Manickam, D.S.; Zhou, Q.-H.; Oupický, D. Reducible poly(2-dimethylaminoethyl methacrylate): Synthesis, cytotoxicity, and gene delivery activity. J. Controll. Release 2007, 122, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Aveston, J. 821. Hydrolysis of sodium silicate: Ultracentrifugation in chloride solutions. J. Chem. Soc. Resumed 1965, 4444–4448. [Google Scholar] [CrossRef]
- Iler, R.K. The Chemistry of Silica; John Wiley & Sons: New York, NY, USA, 1979. [Google Scholar]
- Adeogun, M.J.; Hay, J.N. Structure control in sol–gel silica synthesis using ionene polymers. 2: Evidence from spectroscopic analysis. J. Sol.-Gel. Sci. Technol. 2001, 20, 119–128. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymers | Ratio (M:I:CTA) | DMAEMA (mmol) | DMAEMA (g) | OEGMA (mmol) | OEGMA (g) | AIBN (mmol) | AIBN (mg) | CPA (mmol) | CPA (mg) | PDMAEMA (mmol) | PDMAEMA (mg) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PDMAEMA60 | 50:0.1:1.0 | 63.61 | 10.00 | - | - | 0.13 | 21.7 | 1.28 | 358.0 | - | - |
| PDMAEMA60-b-POEGMA38 | 60:0.1:1.0 | - | - | 4.73 | 2.00 | 0.01 | 1.5 | - | - | 0.08 | 745.0 |
| PDMAEMA60-b-POEGMA100 | 120:0.1:1.0 | - | - | 9.46 | 4.00 | 0.01 | 1.5 | - | - | 0.08 | 742.0 |
| Ratio SS:PDMAEMA | pH | Silica Mass Residue (%) | |
|---|---|---|---|
| 1 | 5:1 | 10.0 | 49.5 |
| 2 | 5:1 | 8.8 | 46.4 |
| 3 | 5:1 | 8.5 | 55.5 |
| 4 | 5:1 | 7.8 | 61.3 |
| 5 | 5:1 | 7.4 | 65.7 |
| 6 | 5:1 | 6.8 | 62.3 |
| 7 | 2.5:1 | 7.7 | 51.1 |
| 8 | 2:1 | 9.3 | 46.0 |
| 9 * | 1:1 | 7.8 | 65.7 |
| 10 | 1:1 | 9.6 | 57.1 |
| Polymer | Polymer Conc. (g/L) | SS Conc. (mM) | Ratio (SS:amine units) | pH | Result |
|---|---|---|---|---|---|
| PDMAEMA60 | 1.57 | 10 | 1:1 | 6.8–8.0 | colloid |
| 9.6 | precipitate | ||||
| 20 | 2:1 | 6.8–7.8 | colloid | ||
| 9.3 | precipitate | ||||
| 25 | 2.5:1 | 6.1–7.5 | colloid | ||
| 7.7 | precipitate | ||||
| 50 | 5:1 | 6.1–10.0 | precipitate | ||
| PDMAEMA300 | 1.57 | 10 | 1:1 | 6.6–8.1 | colloid |
| PDMAEMA60-b-POEGMA38 | 2.15 | 10 | 1:1 | 6.6–8.1 | colloid |
| 50 | 5:1 | 6.6–8.5 | precipitate | ||
| PMOTAI60 | 2.99 | 10 | 1:1 | 6.0–8.6 | colloid |
| 20 | 2:1 | 6.7 | colloid | ||
| 25 | 2.5:1 | 6.5–6.9 | precipitate | ||
| PMOTAI60-b-POEGMA38 | 4.79 | 10 | 1:1 | 7.5 | precipitate |
| 8.0–9.3 | colloid |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Overton, P.; Danilovtseva, E.; Karjalainen, E.; Karesoja, M.; Annenkov, V.; Tenhu, H.; Aseyev, V. Water-Dispersible Silica-Polyelectrolyte Nanocomposites Prepared via Acid-Triggered Polycondensation of Silicic Acid and Directed by Polycations. Polymers 2016, 8, 96. https://doi.org/10.3390/polym8030096
Overton P, Danilovtseva E, Karjalainen E, Karesoja M, Annenkov V, Tenhu H, Aseyev V. Water-Dispersible Silica-Polyelectrolyte Nanocomposites Prepared via Acid-Triggered Polycondensation of Silicic Acid and Directed by Polycations. Polymers. 2016; 8(3):96. https://doi.org/10.3390/polym8030096
Chicago/Turabian StyleOverton, Philip, Elena Danilovtseva, Erno Karjalainen, Mikko Karesoja, Vadim Annenkov, Heikki Tenhu, and Vladimir Aseyev. 2016. "Water-Dispersible Silica-Polyelectrolyte Nanocomposites Prepared via Acid-Triggered Polycondensation of Silicic Acid and Directed by Polycations" Polymers 8, no. 3: 96. https://doi.org/10.3390/polym8030096
APA StyleOverton, P., Danilovtseva, E., Karjalainen, E., Karesoja, M., Annenkov, V., Tenhu, H., & Aseyev, V. (2016). Water-Dispersible Silica-Polyelectrolyte Nanocomposites Prepared via Acid-Triggered Polycondensation of Silicic Acid and Directed by Polycations. Polymers, 8(3), 96. https://doi.org/10.3390/polym8030096