
A Review of Multiscale Computational Methods in Polymeric Materials




Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contents | |
| 1. Introduction | 1 |
| 2. Simulation Methods | 5 |
| 2.1. Quantum Mechanics | 5 |
| 2.2. Atomistic Techniques | 6 |
| 2.2.1. Monte Carlo | 7 |
| 2.2.2. Molecular Dynamics | 8 |
| 2.3. Mesoscale Techniques | 9 |
| 2.3.1. Brownian Dynamics | 10 |
| 2.3.2. Dissipative Particle Dynamics | 11 |
| 2.3.3. Lattice Boltzmann | 12 |
| 2.4. Macroscale Techniques | 14 |
| 2.4.1. Finite Element Method | 15 |
| 2.4.2. Finite Volume Method | 17 |
| 3. Multiscale Strategies | 19 |
| 3.1. Sequential Multiscale Approaches | 19 |
| 3.1.1. Systematic Coarse-Graining Methods | 22 |
| 3.1.1.1. Low Coarse-Graining Degrees | 23 |
| 3.1.1.2. Medium Coarse-Graining Degrees | 26 |
| 3.1.1.3. High Coarse-Graining Degrees | 29 |
| 3.1.2. Reverse Mapping | 30 |
| 3.2. Concurrent Multiscale Approaches | 33 |
| 3.2.1. The Concept of Handshaking | 34 |
| 3.2.2. Linking Atomistic and Continuum Models | 35 |
| 3.2.2.1. Quasicontinuum Approach | 37 |
| 3.2.2.2. Coarse-Grained Molecular Dynamics | 39 |
| 3.2.2.3. Finite-element/Atomistic Method | 39 |
| 3.2.2.4. Bridging Scale Method | 40 |
| 3.2.2.5. Applications in Polymeric Materials | 41 |
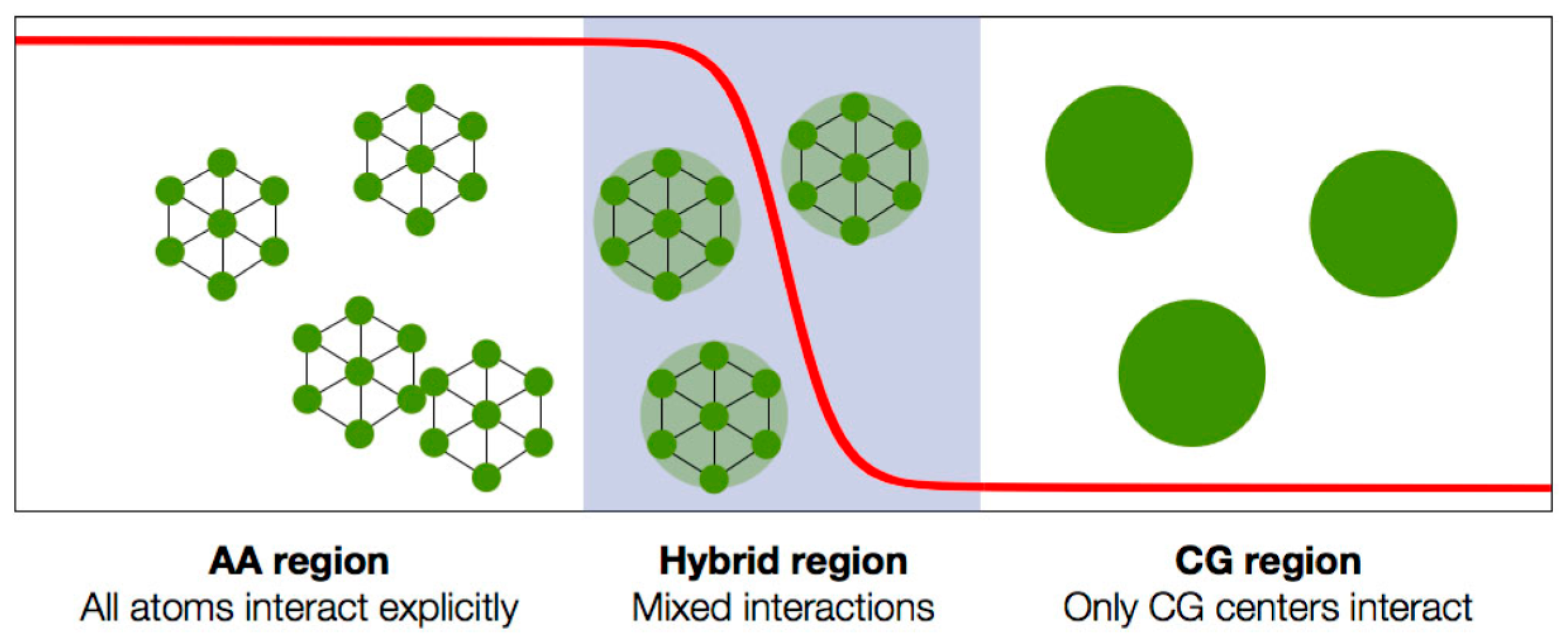
| 3.3. Adaptive Resolution Simulations | 42 |
| 3.3.1. The Adaptive Resolution Scheme | 43 |
| 3.3.2. The Hamiltonian Adaptive Resolution Scheme | 45 |
| 3.4. Extending Atomistic Simulations | 47 |
| 4. Conclusions and Outlooks | 49 |
| Appendix A. Acronyms and Nomenclature | 51 |
| References | 56 |
1. Introduction
2. Simulation Methods
2.1. Quantum Mechanics
2.2. Atomistic Techniques
2.2.1. Monte Carlo
2.2.2. Molecular Dynamics
2.3. Mesoscale Techniques
2.3.1. Brownian Dynamics
2.3.2. Dissipative Particle Dynamics
2.3.3. Lattice Boltzmann
2.4. Macroscale Techniques
2.4.1. Finite Element Method
2.4.2. Finite Volume Method
3. Multiscale Strategies
3.1. Sequential Multiscale Approaches
3.1.1. Systematic Coarse-Graining Methods
Low Coarse-Graining Degrees
Medium Coarse-Graining Degrees
High Coarse-Graining Degrees
3.1.2. Reverse Mapping
3.2. Concurrent Multiscale Approaches
3.2.1. The Concept of Handshaking
3.2.2. Linking Atomistic and Continuum Models
Quasicontinuum Approach
Coarse-Grained Molecular Dynamics
Finite-Element/Atomistic Method
Bridging Scale Method
Applications in Polymeric Materials
3.3. Adaptive Resolution Simulations
3.3.1. The Adaptive Resolution Scheme
3.3.2. The Hamiltonian Adaptive Resolution Scheme
3.4. Extending Atomistic Simulations
4. Conclusions and Outlooks
Author Contributions
Conflicts of Interest
Appendix A. Acronyms and Nomenclature
| Acronyms | |
| Acronym | Full phrase |
| AA | All-Atomistic |
| AC | Amorphous Cell method |
| AdResS | Adaptive Resolution Scheme |
| AIMD | Ab Initio Molecular Dynamics |
| AtC | Atomistic/Continuum method |
| BD | Brownian Dynamics |
| BDM | Bridging Domain Method |
| BGK-LB | Bhatnagar, Gross, And Krook LB method |
| BSM | Bridging Scale Method |
| CACM | Composite Grid Atomistic/Continuum Method |
| CADD | Coupled Atomistic and Discrete Dislocation method |
| CFD | Computational Fluid Dynamics |
| CG | Coarse-Grained |
| CGMD | Coarse-Grained Molecular Dynamics |
| CLS | Coupling of Length Scales method |
| CRW | Conditional Reversible Work |
| D2Q9 | 2-dimensional lattice with 9 allowed velocities used in LB simulations |
| D3Q19 | 3-dimensional lattice with 19 allowed velocities used in LB simulations |
| DDFT | Dynamic Density Functional Theory |
| DFT | Density Functional Theory |
| DPD | Dissipative Particle Dynamics |
| EFCG | Effective Force CG |
| EM | Energy Minimization |
| FDM | Finite Difference Method |
| FE | Finite Element |
| FEAt | Finite-Element/Atomistic method |
| FEM | Finite Element Method |
| FVM | Finite Volume Method |
| GDM | Generalized Differences Methods |
| GFEM | Galerkin Finite Element Method |
| GPU | Graphics Processing Unit |
| H-AdResS | Hamiltonian Adaptive Resolution Scheme |
| HSM | Hybrid Simulation Method |
| IBI | Iterative Boltzmann Inversion |
| IMC | Inverse Monte Carlo |
| LB | Lattice Boltzmann |
| LGCA | Lattice Gas Cellular Automata |
| LSM | Lattice Spring Model |
| MC | Monte Carlo |
| MD | Molecular Dynamics |
| Na-MMT | Sodium Montmorillonite |
| NEMS | Nano-Electro-Mechanical Systems |
| OpenFOAM | Open Source Field Operation And Manipulation |
| PA | Polyamide |
| PAC | Pseudo Amorphous Cell method |
| Pe | Peclet number |
| PE | Polyethylene |
| PNC | Polymer Nanocomposite |
| PP | Polypropylene |
| pPMF | Pair Potential of Mean Force |
| PRISM | Polymer Reference Interaction Site |
| PS | Polystyrene |
| PTT | Poly(Trimethylene Terephthalate) |
| QC | Quasicontinuum method |
| QM | Quantum Mechanics |
| QUICK | Quadratic Upstream Interpolation for Convective Kinematics |
| Re | Reynolds number |
| RVE | Representative Volume Element |
| SCFT | Self-Consistent Field Theory |
| SDPD | Smoothed Dissipative Particle Dynamics |
| SEM | Spectral Element Method |
| SPH | Smoothed Particle Hydrodynamics |
| SRF | Strain Reduction Factor model |
| SUPG | Streamline-Upwind/Petrov-Galerkin |
| TB | Tight Binding |
| TDGL | Time-Dependent Ginzburg-Landau |
| VMS | Variational Multiscale methods |
| We | Weissenberg number |
| XRD | X-Ray Diffraction |
| Nomenclature | |
| Symbol | Meaning |
| in BD method | |
| maximum repulsion between bead and bead in DPD method | |
| acceleration of th particle | |
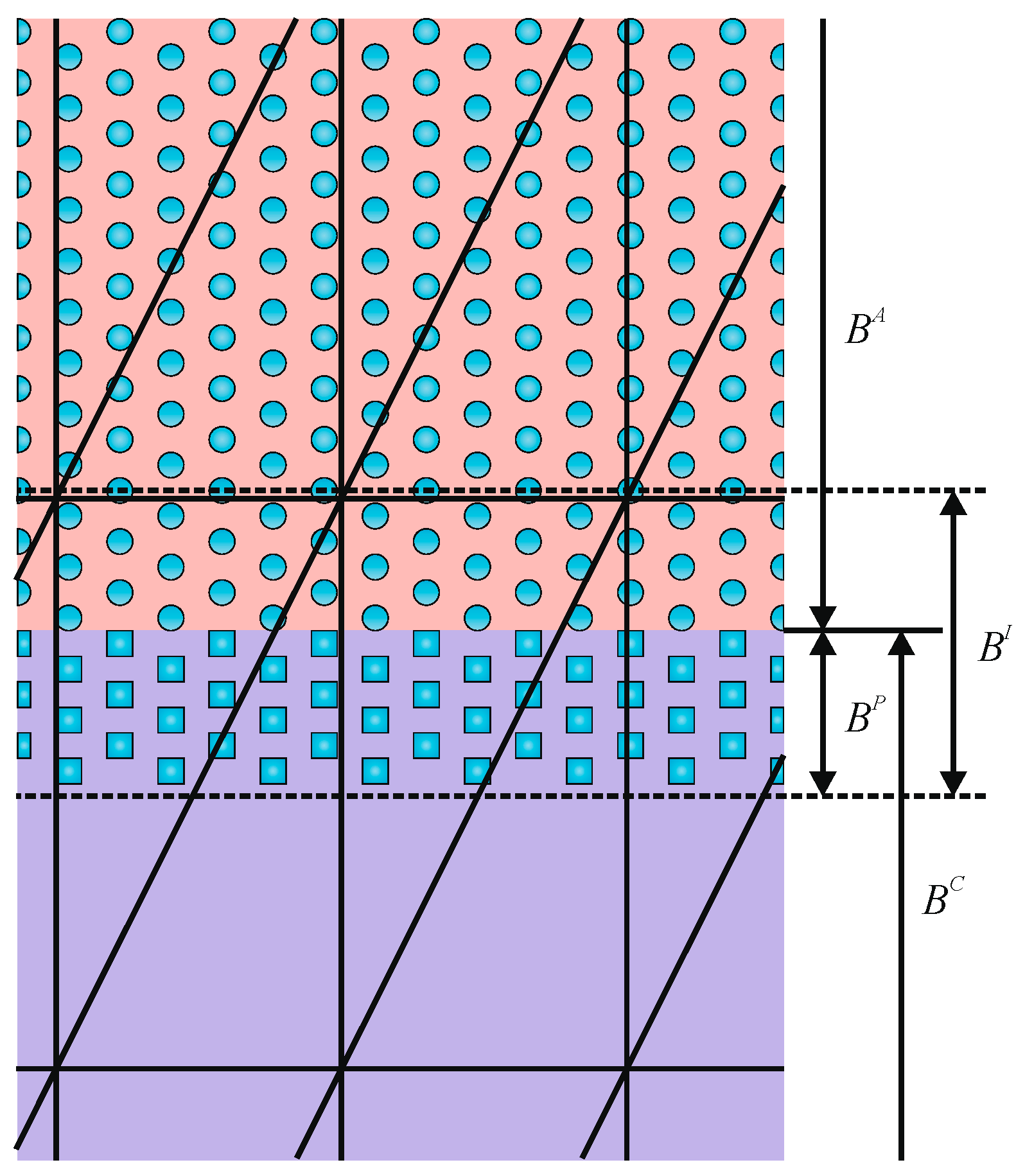
| atomistic domain in concurrent simulations | |
| continuum domain in concurrent simulations | |
| handshake region in concurrent simulations | |
| interfacial region in concurrent simulations | |
| padding region in concurrent simulations | |
| fitting parameter | |
| fitting parameter | |
| the diffusion term of | |
| center-of-mass self-diffusion coefficient | |
| element | |
| absolute unit charge of an electron | |
| Young’s modulus | |
| energy of atom, particle, or node | |
| energy of the th representative atom in QC method | |
| eigenstate of energy | |
| eigenstate energy of an electron | |
| eigenstate energy of a nucleon | |
| total energy | |
| free energy difference in H-AdResS method | |
| conservative force between bead and its neighboring bead within the force cutoff radius | |
| dissipative force between bead and its neighboring bead within the force cutoff radius | |
| random forces between bead and its neighboring bead within the force cutoff radius | |
| drift force of molecule | |
| vector of applied forces in the FE region of a concurrent simulation | |
| force acting on the th atom, particle, or node | |
| force acting between molecules and | |
| thermodynamic force | |
| Brownian random force acting on the th particle | |
| atomistic forces acting on molecule due to the interaction with molecule | |
| CG forces acting on molecule due to the interaction with molecule | |
| storage modulus | |
| loss modulus | |
| Hamiltonian of the system at system state | |
| modified Hamiltonian of the H-AdResS method | |
| change in the system Hamiltonian for going from system state to | |
| compensation term in the Hamiltonian of the H-AdResS method | |
| Hamiltonian of the FE region as a function of the nodal displacements , and time rate of nodal displacements | |
| Hamiltonian of the FE/MD handshake region as a function of the atomic positions , atomic velocities , nodal displacements , and time rate of nodal displacements | |
| Hamiltonian of the MD region as a function of the atomic positions , and atomic velocities | |
| Hamiltonian of the MD/TB handshake region as a function of the atomic positions , and atomic velocities | |
| Hamiltonian of the TB region as a function of the atomic positions , and atomic velocities | |
| total Hamiltonian | |
| Planck’s constant | |
| convection flux term in FVM formulation | |
| diffusion flux term in FVM formulation | |
| the all-atom kinetic energy of the molecules | |
| Boltzmann’s constant | |
| isothermal compressibility | |
| bond length | |
| , | molecular weight |
| mass of an atom or particle | |
| mass of an electron | |
| mass of a nucleon | |
| number of atoms, particles, or nodes | |
| number of monomers per chain | |
| number of elements | |
| number of quadrature points in the numerical integration | |
| number of representative atoms in QC method | |
| the projection matrix | |
| pressure difference along the interface in H-AdResS method | |
| probability of accepting a new configuration for going from system state to | |
| probability distribution function | |
| the target probability distribution function of AA simulations | |
| the generation/destruction of within the control volume per unit volume | |
| residual form of a partial differential equation in terms of the unknown function in FEM scheme | |
| radius of gyration | |
| center of mass coordinates of the th molecule | |
| coordinates vector of an atom, or particle, or node | |
| distance | |
| force cutoff radius | |
| spatial coordinates of an electron | |
| unit vector pointing from the center of bead to that of bead | |
| spatial coordinates of a nucleon | |
| coordinates of the Gauss point in element taken at the centroid of the triangular elements | |
| position of quadrature point of element in the reference configuration | |
| random displacement of the th particle due to the random forces during time step | |
| surface vector | |
| th subregion | |
| set of weighting functions in FEM | |
| rescaling factor for the entropy change | |
| rescaling factor for the friction change | |
| temperature | |
| time | |
| time step | |
| potential energy | |
| potential energies of the atomistic region | |
| energy functional of a systems assuming it is entirely modelled using atoms | |
| potential energies of the continuum region | |
| general form of the CG potential function in IBI method | |
| energy functional of a systems assuming it is entirely modelled using FEM | |
| potential energies of the handshake region | |
| energy of internal interactions | |
| total potential energy of the entire system | |
| bond angle potential in the blob model | |
| bond potential in the blob model | |
| potential of nonbonded interactions in the blob model | |
| potential energy of molecule in the AA representation | |
| potential energy of molecule in the CG representation | |
| vector of nodal displacements in the FE region of a concurrent simulation | |
| the unknown function in FEM which one needs to find | |
| approximation of the function under consideration in FEM | |
| displacements of atom, particle, or node | |
| rate of displacements of atom, particle, or node | |
| values of the function at node of the mesh | |
| volume of element | |
| volume element of the simulation domain in FEM | |
| surfaces surrounding the volume of element | |
| macroscopic velocity magnitude | |
| in LB method | |
| macroscopic local velocity at node at time in LB | |
| estimated velocity in the next time step using a predictor method in DPD velocity-Verlet algorithm | |
| Random velocity change of the th particle due to the random forces during time step | |
| velocity of th atom, particle, or node | |
| velocity magnitude in -direction in LB method | |
| set of prescribed velocity vectors connecting the neighboring nodes in LB method | |
| speed of sound | |
| a function of deformation gradient | |
| weighting constants used in LB method | |
| positive unit charge of a nucleon | |
| system state in a phase space at position | |
| exact solution in the projection method | |
| shear-rate | |
| coarse scale solution of a problem in the projection method | |
| fine scale solution of a problem in the projection method | |
| deformation gradient | |
| delta function | |
| chemical potential gradient in H-AdResS method | |
| neighboring cells of a specific element in FVM | |
| random number between 0 and 1 which is to determine the acceptance or rejection of a new configuration | |
| a Gaussian random number with zero mean and unit variance used in the definition of the random forces between beads and in DPD method | |
| viscosity | |
| a weighting function to link FE and atomistic models in concurrent simulations | |
| bond angle | |
| averaged initial orientation angle | |
| collision matrix used in LB method | |
| multiplication parameter in in DPD velocity-Verlet algorithm | |
| fitting parameter | |
| fitting parameter | |
| a general conserved scalar variable in FVM scheme | |
| friction coefficient between atoms or particles | |
| friction coefficient between bead and bead in DPD method | |
| friction coefficient between particles of freely-rotating chains | |
| wave function of electrons | |
| fluid density in CFD | |
| macroscopic local density at node at time in LB method | |
| molecular density profile in the th iteration step as a function of the position in the direction perpendicular to the interface, in AdResS method | |
| reference molecular density | |
| th weighting function in FEM | |
| noise amplitude between bead and bead in DPD method | |
| shape function of node evaluated at the point with coordinates | |
| characteristic collision time in LB method | |
| integral form of the weighted residuals in FEM | |
| wave function in Schrödinger’s equation | |
| wave function of the nuclei | |
| a parameter in DPD formulation which equals 1 for beads with a distance less than and equals 0 otherwise | |
| particle distribution function used in LB at node at time moving with velocity In the -direction | |
| equilibrium particle distribution function used in LB at node at time moving with velocity In the -direction | |
| spatial interpolation function in AdResS method | |
| interpolation functions in FEM for node | |
| interpolation functions in FEM for node in element | |
| simulation domain in FEM | |
| boundaries of the simulation domain in FEM | |
| dihedral angle | |
| Frequency | |
| quadrature weight signifying how many atoms a given representative atom stands for in the description of the total energy, in QC method | |
| dissipative weight function in DPD method | |
| associated Gauss quadrature weights of quadrature point of element | |
| random weight function in DPD method | |
References
- Elliott, J.A. Novel approaches to multiscale modelling in materials science. Int. Mater. Rev. 2011, 56, 207–225. [Google Scholar] [CrossRef]
- Zeng, Q.H.; Yu, A.B.; Lu, G.Q. Multiscale modeling and simulation of polymer nanocomposites. Prog. Polym. Sci. 2008, 33, 191–269. [Google Scholar] [CrossRef]
- Ayton, G.S.; Noid, W.G.; Voth, G.A. Multiscale modeling of biomolecular systems: In serial and in parallel. Curr. Opin. Struct. Biol. 2007, 17, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Ayyaswamy, P.S.; Muzykantov, V.; Eckmann, D.M.; Radhakrishnan, R. Nanocarrier hydrodynamics and binding in targeted drug delivery: Challenges in numerical modeling and experimental validation. J. Nanotechnol. Eng. Med. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Raabe, D. Challenges in computational materials science. Adv. Mater. 2002, 14, 639–650. [Google Scholar] [CrossRef]
- Kremer, K.; Müller-Plathe, F. Multiscale problems in polymer science: Simulation approaches. MRS Bull. 2001, 26, 205–210. [Google Scholar] [CrossRef]
- Murtola, T.; Bunker, A.; Vattulainen, I.; Deserno, M.; Karttunen, M. Multiscale modeling of emergent materials: Biological and soft matter. Phys. Chem. Chem. Phys. 2009, 11, 1869–1892. [Google Scholar] [CrossRef] [PubMed]
- Peter, C.; Kremer, K. Multiscale simulation of soft matter systems—From the atomistic to the coarse-grained level and back. Soft Matter 2009, 5, 4357–4366. [Google Scholar] [CrossRef]
- Sherwood, P.; Brooks, B.R.; Sansom, M.S. Multiscale methods for macromolecular simulations. Curr. Opin. Struct. Biol. 2008, 18, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Steinhauser, M.O.; Hiermaier, S. A review of computational methods in materials science: Examples from shock-wave and polymer physics. Int. J. Mol. Sci. 2009, 10, 5135–5216. [Google Scholar] [CrossRef] [PubMed]
- Fermeglia, M.; Posocco, P.; Pricl, S. Nano tools for macro problems: Multiscale molecular modeling of nanostructured polymer systems. Compos. Interfaces 2013, 20, 379–394. [Google Scholar] [CrossRef]
- Fermeglia, M.; Pricl, S. Multiscale molecular modeling in nanostructured material design and process system engineering. Comput. Chem. Eng. 2009, 33, 1701–1710. [Google Scholar] [CrossRef]
- Johnston, K.; Harmandaris, V. Hierarchical simulations of hybrid polymer-solid materials. Soft Matter 2013, 9, 6696. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.-W.; Huang, T.-K.; Wang, Y.-C.; Alamani, B.G.; Lin, J.-J. Intercalation strategies in clay/polymer hybrids. Prog. Polym. Sci. 2014, 39, 443–485. [Google Scholar] [CrossRef]
- Gao, D.; Li, R.; Lv, B.; Ma, J.; Tian, F.; Zhang, J. Flammability, thermal and physical-mechanical properties of cationic polymer/montmorillonite composite on cotton fabric. Compos. Part B 2015, 77, 329–337. [Google Scholar] [CrossRef]
- Lowe, D.J.; Chapman, A.V.; Cook, S.; Busfield, J.J.C. Micromechanical models of young’s modulus of NR/organoclay nanocomposites. J. Polym. Sci. Part B 2011, 49, 1621–1627. [Google Scholar] [CrossRef]
- Mattausch, H.; Laske, S.; Duretek, I.; Kreith, J.; Maier, G.; Holzer, C. Investigation of the influence of processing conditions on the thermal, rheological and mechanical behavior of polypropylene nanocomposites. Polym. Eng. Sci. 2013, 53, 1001–1010. [Google Scholar] [CrossRef]
- Decker, J.J.; Meyers, K.P.; Paul, D.R.; Schiraldi, D.A.; Hiltner, A.; Nazarenko, S. Polyethylene-based nanocomposites containing organoclay: A new approach to enhance gas barrier via multilayer coextrusion and interdiffusion. Polymer 2015, 61, 42–54. [Google Scholar] [CrossRef]
- Nguyen, Q.T.; Ngo, T.D.; Tran, P.; Mendis, P.; Bhattacharyya, D. Influences of clay and manufacturing on fire resistance of organoclay/thermoset nanocomposites. Compos. Part A 2015, 74, 26–37. [Google Scholar] [CrossRef]
- Gooneie, A.; Nazockdast, H.; Shahsavan, F. Effect of selective localization of carbon nanotubes in PA6 dispersed phase of PP/PA6 blends on the morphology evolution with time, part 1: Droplet deformation under simple shear flows. Polym. Eng. Sci. 2015, 55, 1504–1519. [Google Scholar] [CrossRef]
- Kotal, M.; Bhowmick, A.K. Polymer nanocomposites from modified clays: Recent advances and challenges. Prog. Polym. Sci. 2015, 51, 127–187. [Google Scholar] [CrossRef]
- Sepahvand, R.; Adeli, M.; Astinchap, B.; Kabiri, R. New nanocomposites containing metal nanoparticles, carbon nanotube and polymer. J. Nanopart. Res. 2008, 10, 1309–1318. [Google Scholar] [CrossRef]
- Pavlidou, S.; Papaspyrides, C.D. A review on polymer-layered silicate nanocomposites. Prog. Polym. Sci. 2008, 33, 1119–1198. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Winey, K.I. Polymer nanocomposites containing carbon nanotubes. Macromolecules 2006, 39, 5194–5205. [Google Scholar] [CrossRef]
- Gooneie, A.; Nazockdast, H.; Shahsavan, F. Effect of selective localization of carbon nanotubes in PA6 dispersed phase of PP/PA6 blends on the morphology evolution with time, part 2: Relaxation of deformed droplets after cessation of flow. Polym. Eng. Sci. 2016, 56, 51–60. [Google Scholar] [CrossRef]
- Sinha Ray, S.; Okamoto, M. Polymer/layered silicate nanocomposites: A review from preparation to processing. Prog. Polym. Sci. 2003, 28, 1539–1641. [Google Scholar] [CrossRef]
- Lecouvet, B.; Gutierrez, J.G.; Sclavons, M.; Bailly, C. Structure-property relationships in polyamide 12/halloysite nanotube nanocomposites. Polym. Degrad. Stab. 2011, 96, 226–235. [Google Scholar] [CrossRef]
- Akkermans, R.; Warren, P.B. Multiscale modelling of human hair. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2004, 362, 1783–1793. [Google Scholar]
- De Borst, R. Challenges in computational materials science: Multiple scales, multi-physics and evolving discontinuities. Comput. Mater. Sci. 2008, 43, 1–15. [Google Scholar] [CrossRef]
- Gates, T.S.; Odegard, G.M.; Frankland, S.J.V.; Clancy, T.C. Computational materials: Multi-scale modeling and simulation of nanostructured materials. Compos. Sci. Technol. 2005, 65, 2416–2434. [Google Scholar] [CrossRef]
- Karakasidis, T.E.; Charitidis, C.A. Multiscale modeling in nanomaterials science. Mater. Sci. Eng. C 2007, 27, 1082–1089. [Google Scholar] [CrossRef]
- Khalatur, P.G. Molecular Dynamics Simulations in Polymer Science: Methods and Main Results. In Polymer Science: A Comprehensive Reference, 10 Volume Set; Elsevier: Spain, 2012; Volume 1, pp. 417–460. [Google Scholar]
- Fredrickson, G.H. The Equilibrium Theory of Inhomogeneous Polymers, 2nd ed.; Clarendon Press: Oxford, UK, 2006. [Google Scholar]
- González, M.A. Force fields and molecular dynamics simulations. Écol. Thémat. Soc. Fr. Neutron. 2011, 12, 169–200. [Google Scholar] [CrossRef]
- Raabe, D. Computational Materials Science; Wiley-VCH Verlag: Darmstadt, Germany, 1998. [Google Scholar]
- Müller-Plathe, F. Coarse-graining in polymer simulation: From the atomistic to the mesoscopic scale and back. ChemPhysChem 2002, 3, 754–769. [Google Scholar] [CrossRef]
- Flory, P.J. Principles of Polymer Chemistry, 1st ed.; Cornell University Press: Ithaca, NY, USA, 1953. [Google Scholar]
- Ganesan, V.; Jayaraman, A. Theory and simulation studies of effective interactions, phase behavior and morphology in polymer nanocomposites. Soft Matter 2014, 10, 13–38. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Elguero, J. Review on DFT and ab initio calculations of scalar coupling constants. Int. J. Mol. Sci. 2003, 4, 64–92. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Steinbach, I. Phase-field models in materials science. Model. Simul. Mater. Sci. Eng. 2009, 17, 73001. [Google Scholar] [CrossRef]
- Wang, X.; Ouyang, J.; Zhou, W.; Liu, Z. A phase field technique for modeling and predicting flow induced crystallization morphology of semi-crystalline polymers. Polymers 2016, 8, 230. [Google Scholar] [CrossRef]
- Kim, J. Phase-field models for multi-component fluid flows. Commun. Commut. Phys. 2012, 12, 613–661. [Google Scholar] [CrossRef]
- Schrödinger, E. Quantisierung als Eigenwertproblem. Ann. Phys. 1926, 384, 361–376. [Google Scholar] [CrossRef]
- Born, M.; Oppenheimer, R. Zur quantentheorie der molekeln. Ann. Phys. 1927, 389, 457–484. [Google Scholar] [CrossRef]
- Ponosov, Y.S.; Bolotin, G.A.; Thomsen, C.; Cardona, M. Raman scattering in Os: Nonadiabatic renormalization of the optical phonon self-energies. Phys. Status Solidi B 1998, 208, 257–269. [Google Scholar] [CrossRef]
- White, J.D.; Chen, J.; Matsiev, D.; Auerbach, D.J.; Wodtke, A.M. Conversion of large-amplitude vibration to electron excitation at a metal surface. Nature 2005, 433, 503–505. [Google Scholar] [CrossRef] [PubMed]
- Pisana, S.; Lazzeri, M.; Casiraghi, C.; Novoselov, K.S.; Geim, A.K.; Ferrari, A.C.; Mauri, F. Breakdown of the adiabatic Born-Oppenheimer approximation in graphene. Nat. Mater. 2007, 6, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.G.; Molteni, C.; Marzari, N. Ab initio molecular dynamics of metal surfaces. J. Phys. 2004, 16, S2575–S2596. [Google Scholar] [CrossRef]
- Rissanou, A.N.; Power, A.J.; Harmandaris, V. Structural and dynamical properties of polyethylene/graphene nanocomposites through molecular dynamics simulations. Polymers 2015, 7, 390–417. [Google Scholar] [CrossRef]
- Car, R.; Parrinello, M. Unified approach for molecular dynamics and density-functional theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef] [PubMed]
- Hageman, J.; Meier, R.J.; Heinemann, M.; de Groot, R.A. Young modulus of crystalline polyethylene from ab initio molecular dynamics. Macromolecules 1997, 30, 5953–5957. [Google Scholar] [CrossRef]
- Serra, S.; Iarlori, S.; Tosatti, E.; Scandolo, S.; Santoro, G. Dynamical and thermal properties of polyethylene by ab initio simulation. Chem. Phys. Lett. 2000, 331, 339–345. [Google Scholar] [CrossRef]
- Saitta, A.M.; Klein, M.L. First-principles molecular dynamics study of the rupture processes of a bulklike polyethylene knot. J. Phys. Chem. B 2001, 105, 6495–6499. [Google Scholar] [CrossRef]
- Cavazzoni, C.; Colle, R.; Farchioni, R.; Grosso, G. Car-Parrinello molecular dynamics study of electronic and structural properties of neutral polyanilines. Phys. Rev. B 2002, 66, 165110. [Google Scholar] [CrossRef]
- Wallace, D.S.; Stoneham, A.M.; Hayes, W.; Fisher, A.J.; Harker, A.H. Theory of defects in conducting polymers. I. Theoretical principles and simple applications. J. Phys. 1991, 3, 3879–3903. [Google Scholar] [CrossRef]
- Wallace, D.S.; Stoneham, A.M.; Hayes, W.; Fisher, A.J.; Testa, A. Theory of defects in conducting polymers. II. Application to polyacetylene. J. Phys. 1991, 3, 3905–3920. [Google Scholar] [CrossRef]
- Röthlisberger, U.; Sprik, M.; Klein, M.L. Living polymers: Ab initio molecular dynamics study of the initiation step in the polymerization of isoprene induced by ethyl lithium. J. Chem. Soc. Faraday Trans. 1998, 94, 501–508. [Google Scholar] [CrossRef]
- Bernasconi, M.; Chiarotti, G.L.; Focher, P.; Parrinello, M.; Tosatti, E. Solid-state polymerization of acetylene under pressure: Ab Initio simulation. Phys. Rev. Lett. 1997, 78, 2008–2011. [Google Scholar] [CrossRef]
- Quarti, C.; Milani, A.; Castiglioni, C. Ab initio calculation of the IR spectrum of PTFE: Helical symmetry and defects. J. Phys. Chem. B 2013, 117, 706–718. [Google Scholar] [CrossRef] [PubMed]
- D’Amore, M.; Talarico, G.; Barone, V. Periodic and high-temperature disordered conformations of polytetrafluoroethylene chains: An ab initio modeling. J. Am. Chem. Soc. 2006, 128, 1099–1108. [Google Scholar]
- Dai, L.; Yang, S.-W.; Chen, X.-T.; Wu, P.; Tan, V. Investigation of metal diffusion into polymers by ab initio molecular dynamics. Appl. Phys. Lett. 2005, 87. [Google Scholar] [CrossRef]
- Piscitelli, F.; Posocco, P.; Toth, R.; Fermeglia, M.; Pricl, S.; Mensitieri, G.; Lavorgna, M. Sodium montmorillonite silylation: Unexpected effect of the aminosilane chain length. J. Colloid Interface Sci. 2010, 351, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Farquhar, I.E. Ergodic Theory in Statistical Mechanics; Interscience Publishers: New York, NY, USA, 1964. [Google Scholar]
- Baumgärtner, A.; Burkitt, A.N.; Ceperley, D.M.; de Raedt, H.; Heermann, D.W.; Herrmann, H.J; Landau, D.P; Levesque, D.; von der Linden, W. The Monte Carlo Method in Condensed Matter Physics; Springer: Berlin, Germany, 2012. [Google Scholar]
- Brin, M.; Stuck, G. Introduction to Dynamical Systems; Cambridge University Press: New York, NY, USA, 2002. [Google Scholar]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of state calculations by fast computing machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef]
- Potts, R.B. Some generalized order-disorder transformations. Math. Proc. Camb. Philos. Soc. 1952, 48, 106–109. [Google Scholar] [CrossRef]
- Bortz, A.B.; Kalos, M.H.; Lebowitz, J.L. A new algorithm for Monte Carlo simulation of Ising spin systems. J. Comput. Phys. 1975, 17, 10–18. [Google Scholar] [CrossRef]
- Gillespie, D.T. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 1977, 81, 2340–2361. [Google Scholar] [CrossRef]
- Makki, H.; Adema, K.N.S.; Peters, E.A.J.F.; Laven, J.; Van Der Ven, L.G.J.; Van Benthem, R.A.T.M.; de With, G. A simulation approach to study photo-degradation processes of polymeric coatings. Polym. Degrad. Stab. 2014, 105, 68–79. [Google Scholar] [CrossRef]
- Mermigkis, P.G.; Tsalikis, D.G.; Mavrantzas, V.G. Determination of the effective diffusivity of water in a poly (methyl methacrylate) membrane containing carbon nanotubes using kinetic Monte Carlo simulations. J. Chem. Phys. 2015, 143. [Google Scholar] [CrossRef] [PubMed]
- Adema, K.N.S.; Makki, H.; Peters, E.A.J.F.; Laven, J.; Van Der Ven, L.G.J.; Van Benthem, R.A.T.M.; de With, G. Kinetic Monte Carlo simulation of the photodegradation process of polyester-urethane coatings. Phys. Chem. Chem. Phys. 2015, 17, 19962–19976. [Google Scholar] [CrossRef] [PubMed]
- Kunz, L.; Kuhn, F.M.; Deutschmann, O. Kinetic Monte Carlo simulations of surface reactions on supported nanoparticles: A novel approach and computer code. J. Chem. Phys. 2015, 143. [Google Scholar] [CrossRef] [PubMed]
- Zepeda-Ruiz, L.A.; Gilmer, G.H.; Walton, C.C.; Hamza, A.V.; Chason, E. Surface morphology evolution during sputter deposition of thin films—Lattice Monte Carlo simulations. J. Cryst. Growth 2010, 312, 1183–1187. [Google Scholar] [CrossRef]
- Rysz, J. Monte Carlo simulations of phase separation in thin polymer blend films: Scaling properties of morphological measures. Polymer 2005, 46, 977–982. [Google Scholar] [CrossRef]
- Galuschko, A.; Lang, M.; Kreer, T.; Sommer, J.-U. Monte carlo simulation of thin film polymer melts. Soft Matter 2014, 12, S49–S55. [Google Scholar] [CrossRef]
- Ivanov, V.A.; Rodionova, A.S.; An, E.A.; Martemyanova, J.A.; Stukan, M.R.; Müller, M.; Paul, W.; Binder, K. Orientational ordering transitions of semiflexible polymers in thin films: A Monte Carlo simulation. Phys. Rev. E 2011, 84. [Google Scholar] [CrossRef] [PubMed]
- Müller, M. Chain conformations and correlations in thin polymer films: A Monte Carlo study. J. Chem. Phys. 2002, 116, 9930–9938. [Google Scholar] [CrossRef]
- Werner, A.; Schmid, F.; Müller, M.; Binder, K. Anomalous size-dependence of interfacial profiles between coexisting phases of polymer mixtures in thin-film geometry: A Monte Carlo simulation. J. Chem. Phys. 1997, 107, 8175–8188. [Google Scholar] [CrossRef]
- Zykova-Timan, T.; Horbach, J.; Binder, K. Monte Carlo simulations of the solid-liquid transition in hard spheres and colloid-polymer mixtures. J. Chem. Phys. 2010, 133. [Google Scholar] [CrossRef] [PubMed]
- Binder, K. Monte Carlo simulation of polymers at interfaces. Phys. A Stat. Mech. Appl. 1993, 200, 722–729. [Google Scholar] [CrossRef]
- Bitsanis, I.A.; Brinke, G.T. A lattice Monte Carlo study of long chain conformations at solid-polymer melt interfaces. J. Chem. Phys. 1993, 99, 3100–3111. [Google Scholar] [CrossRef]
- Jiang, J.; Liu, H.; Hu, Y. Lattice Monte Carlo simulation of polymer adsorption at an interface, 1: Monodisperse polymer. Macromol. Theory Simul. 1998, 7, 105–111. [Google Scholar] [CrossRef]
- Jiang, J.; Liu, H.; Hu, Y. Lattice Monte Carlo simulation of polymer adsorption at an interface, 2: Polydisperse polymer. Macromol. Theory Simul. 1998, 7, 113–117. [Google Scholar]
- John, A.; Nagel, J.; Heinrich, G. Monte Carlo simulation of polymer reactions at interfaces. Macromol. Theory Simul. 2007, 16, 430–440. [Google Scholar] [CrossRef]
- Madden, W.G. Monte Carlo studies of the melt-vacuum interface of a lattice polymer. J. Chem. Phys. 1987, 87, 1405–1422. [Google Scholar] [CrossRef]
- Müller, M. Reactions at polymer interfaces: A Monte Carlo simulation. Macromolecules 1997, 30, 6353–6357. [Google Scholar] [CrossRef]
- Müller, M.; Binder, K.; Oed, W. Structural and thermodynamic properties of interfaces between coexisting phases in polymer blends: A Monte Carlo simulation. J. Chem. Soc. Faraday Trans. 1995, 91, 2369–2379. [Google Scholar] [CrossRef]
- Müller, M.; MacDowell, L.G. Interface and surface properties of short polymers in solution: Monte Carlo simulations and self-consistent field theory. Macromolecules 2000, 33, 3902–3923. [Google Scholar] [CrossRef]
- Müller, M.; Werner, A. Interfaces between highly incompatible polymers of different stiffness: Monte Carlo simulations and self-consistent field calculations. J. Chem. Phys. 1997, 107, 10764–10776. [Google Scholar] [CrossRef]
- Reiter, J.; Zifferer, G.; Olaj, O.F. Monte Carlo studies of the interface between two polymer melts. Macromolecules 1990, 23, 224–228. [Google Scholar] [CrossRef]
- Toral, R.; Chakrabarti, A. Monte Carlo study of polymer chains end-grafted onto a spherical interface. Phys. Rev. E 1993, 47, 4240–4246. [Google Scholar] [CrossRef]
- Pandey, Y.N.; Doxastakis, M. Detailed atomistic Monte Carlo simulations of a polymer melt on a solid surface and around a nanoparticle. J. Chem. Phys. 2012, 136, 94901. [Google Scholar] [CrossRef] [PubMed]
- Shu, R.; Zha, L.; Eman, A.A.; Hu, W. Fibril crystal growth in diblock copolymer solutions studied by dynamic monte carlo simulations. J. Phys. Chem. B 2015, 119, 5926–5932. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Ma, A.; Li, J.; Jiang, X.; Ma, Y.; Toda, A.; Hu, W. Melting of polymer single crystals studied by dynamic Monte Carlo simulations. Eur. Phys. J. E 2010, 33, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Doye, J.; Frenkel, D. Kinetic Monte Carlo simulations of the growth of polymer crystals. J. Chem. Phys. 1999, 110, 2692–2702. [Google Scholar] [CrossRef]
- Zhang, J.; Muthukumar, M. Monte Carlo simulations of single crystals from polymer solutions. J. Chem. Phys. 2007, 126. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K. Monte Carlo simulation of two-dimensional growth of conductive zones in interconversion of conducting polymer films involving random morphology. J. Electroanal. Chem. 1990, 292, 63–72. [Google Scholar] [CrossRef]
- Balmer, J.A.; Mykhaylyk, O.O.; Schmid, A.; Armes, S.P.; Fairclough, J.P.A.; Ryan, A.J. Characterization of polymer-silica nanocomposite particles with core-shell morphologies using monte carlo simulations and small angle X-ray scattering. Langmuir 2011, 27, 8075–8089. [Google Scholar] [CrossRef] [PubMed]
- Ruan, C.; Liu, C.; Zheng, G. Monte Carlo Simulation for the Morphology and Kinetics of Spherulites and Shish-Kebabs in Isothermal Polymer Crystallization. Math. Probl. Eng. 2015, 2015. [Google Scholar] [CrossRef]
- Duda, Y.; Vázquez, F. Modeling of composite latex particle morphology by off-lattice Monte Carlo simulation. Langmuir 2005, 21, 1096–1102. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Liang, H.; Pan, C. Monte Carlo simulation of morphologies of self-assembled amphiphilic diblock copolymers in solution. Phys. Rev. E 2001, 63, 318041–318044. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, U.; Detcheverry, F.A.; Nealey, P.F.; De Pablo, J.J. Morphologies of linear triblock copolymers from Monte Carlo simulations. Macromolecules 2011, 44, 5490–5497. [Google Scholar] [CrossRef]
- Raj Mohan, S.; Joshi, M.P.; Singh, M.P. Charge transport in disordered organic solids: A Monte Carlo simulation study on the effects of film morphology. Org. Electron. 2008, 9, 355–368. [Google Scholar] [CrossRef]
- Narambuena, C.F.; Leiva, E.P.M.; Chávez-Páez, M.; Pérez, E. Effect of chain stiffness on the morphology of polyelectrolyte complexes. A Monte Carlo simulation study. Polymer 2010, 51, 3293–3302. [Google Scholar] [CrossRef]
- Wang, X.; Chiang, M.; Snyder, C.R. Monte-Carlo simulation for the fracture process and energy release rate of unidirectional carbon fiber-reinforced polymers at different temperatures. Compos. Part A 2004, 35, 1277–1284. [Google Scholar] [CrossRef]
- Li, J.; Ma, Y.; Hu, W. Dynamic Monte Carlo simulation of non-equilibrium Brownian diffusion of single-chain macromolecules. Mol. Simul. 2016, 42, 321–327. [Google Scholar] [CrossRef]
- Neyertz, S.; Brown, D. A trajectory-extending kinetic Monte Carlo (TEKMC) method for estimating penetrant diffusion coefficients in molecular dynamics simulations of glassy polymers. Macromolecules 2010, 43, 9210–9214. [Google Scholar] [CrossRef]
- Sikorski, A.; Adamczyk, P. Diffusion of polymer chains in porous media. A Monte Carlo study. Polymer 2010, 51, 581–586. [Google Scholar] [CrossRef]
- Tüzel, E.; Kisacikoǧlu, K.B.; Pekcan, Ö. Monitoring diffusion of reptating polymer chains by a direct energy transfer method: A Monte Carlo simulation. Macromol. Theory Simul. 2002, 11, 678–686. [Google Scholar] [CrossRef]
- Mavrantzas, V.G.; Theodorou, D.N. Atomistic simulation of polymer melt elasticity. Macromolecules 1998, 31, 6310–6332. [Google Scholar] [CrossRef]
- Baig, C.; Mavrantzas, V.G. Multiscale simulation of polymer melt viscoelasticity. Phys. Rev. B 2009, 79. [Google Scholar] [CrossRef]
- Brindle, D.; Care, C.M. Phase diagram for the lattice model of amphiphile and solvent mixtures by Monte Carlo simulation. J. Chem. Soc., Faraday Trans. 1992, 88, 2163–2166. [Google Scholar] [CrossRef]
- Ivanov, V.A.; Stukan, M.R.; Müller, M.; Paul, W.; Binder, K. Phase diagram of solutions of stiff-chain macromolecules: A Monte Carlo simulation. J. Chem. Phys. 2003, 118, 10333–10342. [Google Scholar] [CrossRef]
- Guevara-Carrion, G.; Hasse, H.; Vrabec, J. Thermodynamic properties for applications in chemical industry via classical force fields. Top. Curr. Chem. 2012, 307, 201–250. [Google Scholar] [PubMed]
- Verlet, L. Computer “experiments” on classical fluids. I. Thermodynamical properties of Lennard-Jones molecules. Phys. Rev. 1967, 159, 98–103. [Google Scholar] [CrossRef]
- Verlet, L. Computer “experiments” on classical fluids. II. Equilibrium correlation functions. Phys. Rev. 1968, 165, 201–214. [Google Scholar] [CrossRef]
- Kremer, K.; Grest, G.S. Dynamics of entangled linear polymer melts: A molecular-dynamics simulation. J. Chem. Phys. 1990, 92, 5057–5086. [Google Scholar] [CrossRef]
- Aoyagi, T.; Doi, M. Molecular dynamics simulation of entangled polymers in shear flow. Comput. Theor. Polym. Sci. 2000, 10, 317–321. [Google Scholar] [CrossRef]
- Durand, M.; Meyer, H.; Benzerara, O.; Baschnagel, J.; Vitrac, O. Molecular dynamics simulations of the chain dynamics in monodisperse oligomer melts and of the oligomer tracer diffusion in an entangled polymer matrix. J. Chem. Phys. 2010, 132. [Google Scholar] [CrossRef] [PubMed]
- Harmandaris, V.A.; Mavrantzas, V.G.; Theodorou, D.N.; Kröger, M.; Ramírez, J.; Ottinger, H.C.; Vlassopoulos, D. Crossover from the rouse to the entangled polymer melt regime: Signals from long, detailed atomistic molecular dynamics simulations, supported by rheological experiments. Macromolecules 2003, 36, 1376–1387. [Google Scholar] [CrossRef]
- Likhtman, A.E.; Sukumaran, S.K.; Ramirez, J. Linear viscoelasticity from molecular dynamics simulation of entangled polymers. Macromolecules 2007, 40, 6748–6757. [Google Scholar] [CrossRef]
- Wang, Z.; Larson, R.G. Constraint release in entangled binary blends of linear polymers: A molecular dynamics study. Macromolecules 2008, 41, 4945–4960. [Google Scholar] [CrossRef]
- Harmandaris, V.A.; Daoulas, K.; Mavrantzas, V.G. Molecular dynamics simulation of a polymer melt/solid interface: Local dynamics and chain mobility in a thin film of polyethylene melt adsorbed on graphite. Macromolecules 2005, 38, 5796–5809. [Google Scholar] [CrossRef]
- Milano, G.; Santangelo, G.; Ragone, F.; Cavallo, L.; Di Matteo, A. Gold nanoparticle/polymer interfaces: All atom structures from molecular dynamics simulations. J. Phys. Chem. C 2011, 115, 15154–15163. [Google Scholar] [CrossRef]
- Luo, T.; Lloyd, J.R. Enhancement of thermal energy transport across graphene/graphite and polymer interfaces: A molecular dynamics study. Adv. Funct. Mater. 2012, 22, 2495–2502. [Google Scholar] [CrossRef]
- Neyertz, S.; Brown, D. Molecular dynamics study of carbon dioxide sorption and plasticization at the interface of a glassy polymer membrane. Macromolecules 2013, 46, 2433–2449. [Google Scholar] [CrossRef]
- Lamas, E.J.; Balbuena, P.B. Molecular dynamics studies of a model polymer-catalyst-carbon interface. Electrochim. Acta 2006, 51, 5904–5911. [Google Scholar] [CrossRef]
- Song, Y.; Feng, W.; Liu, K.; Yang, P.; Zhang, W.; Zhang, X. Exploring the folding pattern of a polymer chain in a single crystal by combining single-molecule force spectroscopy and steered molecular dynamics simulations. Langmuir 2013, 29, 3853–3857. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T. Molecular dynamics simulations of steady-state crystal growth and homogeneous nucleation in polyethylene-like polymer. J. Chem. Phys. 2008, 129. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T. Molecular dynamics of reversible and irreversible melting in chain-folded crystals of short polyethylene-like polymer. Macromolecules 2010, 43, 9384–9393. [Google Scholar] [CrossRef]
- Hegde, G.A.; Chang, J.-F.; Chen, Y.-L.; Khare, R. Conformation and diffusion behavior of ring polymers in solution: A comparison between molecular dynamics, multiparticle collision dynamics, and lattice Boltzmann simulations. J. Chem. Phys. 2011, 135. [Google Scholar] [CrossRef] [PubMed]
- Bahlakeh, G.; Nikazar, M.; Hafezi, M.-J.; Dashtimoghadam, E.; Hasani-Sadrabadi, M.M. Molecular dynamics simulation study of proton diffusion in polymer electrolyte membranes based on sulfonated poly (ether ether ketone). Int. J. Hydrog. Energy 2012, 37, 10256–10264. [Google Scholar] [CrossRef]
- Liu, J.; Cao, D.; Zhang, L. Molecular dynamics study on nanoparticle diffusion in polymer melts: A test of the stokes-einstein law. J. Phys. Chem. C 2008, 112, 6653–6661. [Google Scholar] [CrossRef]
- Zhao, X.-T.; Yang, H.; Sheng, Y.-Z.; Li, J.-Y.; Sun, M. Molecular dynamics simulation on the effect of the distance between SWCNTs for short polymers diffusion among single wall carbon nanotubes. Comput. Mater. Sci. 2014, 95, 446–450. [Google Scholar] [CrossRef]
- Jang, S.S.; Molinero, V.; Çaǧin, T.; Goddard, W.A., III. Nanophase-Segregation and Transport in Nafion 117 from Molecular Dynamics Simulations: Effect of Monomeric Sequence. J. Phys. Chem. B 2004, 108, 3149–3157. [Google Scholar] [CrossRef]
- Błoński, S.; Brostow, W. Molecular dynamics simulations of chain relaxation and crack propagation in polymer liquid crystals. J. Chem. Phys. 1991, 95, 2890–2896. [Google Scholar]
- Brostow, W.; Cunha, A.M.; Quintanilla, J.; Simões, R. Crack formation and propagation in molecular dynamics simulations of polymer liquid crystals. Macromol. Theory Simul. 2002, 11, 308–314. [Google Scholar] [CrossRef]
- Brostow, W.; Hinze, J.A.; Simões, R. Tribological behavior of polymers simulated by molecular dynamics. J. Mater. Res. 2004, 19, 851–856. [Google Scholar] [CrossRef]
- Shagolsem, L.S.; Sommer, J.-U. Order and phase behavior of thin film of diblock copolymer-selective nanoparticle mixtures: A molecular dynamics simulation study. Macromolecules 2014, 47, 830–839. [Google Scholar] [CrossRef]
- Hartmann, L.; Gorbatschow, W.; Hauwede, J.; Kremer, F. Molecular dynamics in thin films of isotactic poly(methyl methacrylate). Eur. Phys. J. E 2002, 8, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, L.; Zhou, D.; Xu, J.; Xue, G. Effect of molecular chain architecture on dynamics of polymer thin films measured by the ac-chip calorimeter. Macromolecules 2014, 47, 3497–3501. [Google Scholar] [CrossRef]
- Gooneie, A.; Gonzalez-Gutierrez, J.; Holzer, C. Atomistic Modelling of Confined Polypropylene Chains between Ferric Oxide Substrates at Melt Temperature. Polymers 2016, 8, 361. [Google Scholar] [CrossRef]
- Egorov, E.A.; Zhizhenkov, V.V. Molecular dynamics and strengthening of liquid-crystal polymers. Phys. Solid State 2005, 47, 942–948. [Google Scholar] [CrossRef]
- Stimson, L.M.; Wilson, M.R. Molecular dynamics simulations of side chain liquid crystal polymer molecules in isotropic and liquid-crystalline melts. J. Chem. Phys. 2005, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daivis, P.J.; Matin, M.L.; Todd, B.D. Nonlinear shear and elongational rheology of model polymer melts by non-equilibrium molecular dynamics. J. Non-Newton. Fluid Mech. 2003, 111, 1–18. [Google Scholar] [CrossRef]
- Hajizadeh, E.; Todd, B.D.; Daivis, P.J. A molecular dynamics investigation of the planar elongational rheology of chemically identical dendrimer-linear polymer blends. J. Chem. Phys. 2015, 142. [Google Scholar] [CrossRef] [PubMed]
- Kairn, T.; Daivis, P.J.; Ivanov, I.; Bhattacharya, S.N. Molecular-dynamics simulation of model polymer nanocomposite rheology and comparison with experiment. J. Chem. Phys. 2005, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeng, Y.-R.; Chen, C.-C.; Shyu, S.-H. A molecular dynamics study of lubrication rheology of polymer fluids. Tribol. Lett. 2003, 15, 293–300. [Google Scholar] [CrossRef]
- Todd, B.D.; Daivis, P.J. Nonequilibrium molecular dynamics simulations of planar elongational flow with spatially and temporally periodic boundary conditions. Phys. Rev. Lett. 1998, 81, 1118–1121. [Google Scholar] [CrossRef]
- Baig, C.; Edwards, B.J.; Keffer, D.J.; Cochran, H.D.; Harmandaris, V.A. Rheological and structural studies of linear polyethylene melts under planar elongational flow using nonequilibrium molecular dynamics simulations. J. Chem. Phys. 2006, 124, 84902. [Google Scholar] [CrossRef] [PubMed]
- Chenoweth, K.; Cheung, S.; van Duin, A.; Goddard, W.A., III; Kober, E.M. Simulations on the thermal decomposition of a poly(dimethylsiloxane) polymer using the ReaxFF reactive force field. J. Am. Chem. Soc. 2005, 127, 7192–7202. [Google Scholar] [CrossRef] [PubMed]
- Chenoweth, K.; van Duin, A.; Goddard, W.A., III. ReaxFF reactive force field for molecular dynamics simulations of hydrocarbon oxidation. J. Phys. Chem. A 2008, 112, 1040–1053. [Google Scholar] [CrossRef] [PubMed]
- Van Duin, A.; Dasgupta, S.; Lorant, F.; Goddard, W.A., III. ReaxFF: A reactive force field for hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409. [Google Scholar] [CrossRef]
- Odegard, G.M.; Jensen, B.D.; Gowtham, S.; Wu, J.; He, J.; Zhang, Z. Predicting mechanical response of crosslinked epoxy using ReaxFF. Chem. Phys. Lett. 2014, 591, 175–178. [Google Scholar] [CrossRef]
- Tieleman, D.P.; Marrink, S.J.; Berendsen, H. A computer perspective of membranes: Molecular dynamics studies of lipid bilayer systems. Biochim. Biophys. Acta Rev. Biomembr. 1997, 1331, 235–270. [Google Scholar] [CrossRef]
- Heller, H.; Schaefer, M.; Schulten, K. Molecular dynamics simulation of a bilayer of 200 lipids in the gel and in the liquid-crystal phases. J. Phys. Chem. 1993, 97, 8343–8360. [Google Scholar] [CrossRef]
- Lipowsky, R.; Grotehans, S. Hydration vs. Protrusion Forces Between Lipid Bilayers. Europhys. Lett. 1993, 23, 599. [Google Scholar] [CrossRef]
- Lindahl, E.; Edholm, O. Mesoscopic undulations and thickness fluctuations in lipid bilayers from molecular dynamics simulations. Biophys. J. 2000, 79, 426–433. [Google Scholar] [CrossRef]
- Gao, L.; Shillcock, J.; Lipowsky, R. Improved dissipative particle dynamics simulations of lipid bilayers. J. Chem. Phys. 2007, 126. [Google Scholar] [CrossRef] [PubMed]
- Malevanets, A.; Kapral, R. Mesoscopic model for solvent dynamics. J. Chem. Phys. 1999, 110, 8605. [Google Scholar] [CrossRef]
- Alekseeva, U.; Winkler, R.G.; Sutmann, G. Hydrodynamics in adaptive resolution particle simulations: Multiparticle collision dynamics. J. Comput. Phys. 2016, 314, 14–34. [Google Scholar] [CrossRef]
- Satoh, A. Introduction to Molecular-Microsimulation for Colloidal Dispersions, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Satoh, A. Introduction to Practice of Molecular Simulation; Elsevier Inc.: Burlington, MA, USA, 2011. [Google Scholar]
- Pagonabarraga, I. Lattice boltzmann modeling of complex fluids: Colloidal suspensions and fluid mixtures. In Novel Methods in Soft Matter Simulations; Karttunen, M., Vattulainen, I., Lukkarinen, A., Eds.; Springer: Berlin, Germany, 2004; pp. 279–309. [Google Scholar]
- Sukop, M.; Throne, D.T. Lattice Boltzmann Modeling: An Introduction for Geoscientists and Engineers, 1st ed.; Springer: New York, NY, USA, 2006. [Google Scholar]
- Ermak, D.L.; McCammon, J.A. Brownian dynamics with hydrodynamic interactions. J. Chem. Phys. 1978, 69, 1352–1360. [Google Scholar] [CrossRef]
- Ando, T.; Chow, E.; Saad, Y.; Skolnick, J. Krylov subspace methods for computing hydrodynamic interactions in Brownian dynamics simulations. J. Chem. Phys. 2012, 137. [Google Scholar] [CrossRef] [PubMed]
- Cerbelaud, M.; Lestriez, B.; Guyomard, D.; Videcoq, A.; Ferrando, R. Brownian dynamics simulations of colloidal suspensions containing polymers as precursors of composite electrodes for lithium batteries. Langmuir 2012, 28, 10713–10724. [Google Scholar] [CrossRef] [PubMed]
- Patti, A.; Cuetos, A. Brownian dynamics and dynamic Monte Carlo simulations of isotropic and liquid crystal phases of anisotropic colloidal particles: A comparative study. Phys. Rev. E 2012, 86. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Xu, S.; Sun, Z.; Wang, J.T. Brownian dynamics simulation of the crystallization dynamics of charged colloidal particles. J. Colloid Interface Sci. 2010, 350, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.; Campanella, O.H.; Carignano, M.A. Brownian dynamics study of gel-forming colloidal particles. J. Phys. Chem. B 2010, 114, 13052–13058. [Google Scholar] [CrossRef] [PubMed]
- Saveyn, H.; de Baets, B.; Thas, O.; Hole, P.; Smith, J.; van der Meeren, P. Accurate particle size distribution determination by nanoparticle tracking analysis based on 2-D Brownian dynamics simulation. J. Colloid Interface Sci. 2010, 352, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, Y.-L.; Li, Y.-C.; Qian, H.-J.; Sun, C.-C. Self-assembly of two-patch particles in solution: A Brownian dynamics simulation study. Mol. Simul. 2014, 40, 449–457. [Google Scholar] [CrossRef]
- Mendes, M.J.; Schmidt, H.K.; Pasquali, M. Brownian dynamics simulations of single-wall carbon nanotube separation by type using dielectrophoresis. J. Phys. Chem. B 2008, 112, 7467–7477. [Google Scholar] [CrossRef] [PubMed]
- Van Den Noort, A.; Briels, W.J. Brownian dynamics simulations of concentration coupled shear banding. J. Non-Newton. Fluid Mech. 2008, 152, 148–155. [Google Scholar] [CrossRef]
- Jain, A.; Sunthar, P.; Dünweg, B.; Prakash, J.R. Optimization of a Brownian-dynamics algorithm for semidilute polymer solutions. Phys. Rev. E 2012, 85. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhu, Y.-L.; Liu, H.; Lu, Z.-Y. Brownian dynamics simulation study on the self-assembly of incompatible star-like block copolymers in dilute solution. Phys. Chem. Chem. Phys. 2012, 14, 4964–4970. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; de Pablo, J.J.; Graham, M.D. An immersed boundary method for Brownian dynamics simulation of polymers in complex geometries: Application to DNA flowing through a nanoslit with embedded nanopits. J. Chem. Phys. 2012, 136. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.T.; Bajaj, M.; Prakash, J.R. Brownian dynamics simulation of polymer collapse in a poor solvent: Influence of implicit hydrodynamic interactions. Soft Matter 2008, 4, 1196–1207. [Google Scholar] [CrossRef]
- Delong, S.; Usabiaga, F.B.; Delgado-Buscalioni, R.; Griffith, B.E.; Donev, A. Brownian dynamics without Green’s functions. J. Chem. Phys. 2014, 140. [Google Scholar] [CrossRef] [PubMed]
- Iliafar, S.; Vezenov, D.; Jagota, A. Brownian dynamics simulation of peeling a strongly-Adsorbed polymer molecule from a frictionless substrate. Langmuir 2013, 29, 1435–1445. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.-H.; Helms, V.; Geyer, T. Coarse-grained Brownian dynamics simulations of protein translocation through nanopores. J. Chem. Phys. 2012, 137. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, R.; Bhattacharya, A. Driven translocation of a semi-flexible chain through a nanopore: A Brownian dynamics simulation study in two dimensions. J. Chem. Phys. 2013, 138. [Google Scholar] [CrossRef] [PubMed]
- Hoogerbrugge, P.J.; Koelman, J.M.V.A. Simulating Microscopic Hydrodynamic Phenomena with Dissipative Particle Dynamics. Europhys. Lett. 1992, 19, 155. [Google Scholar] [CrossRef]
- Groot, R.D.; Warren, P.B. Dissipative particle dynamics: Bridging the gap between atomistic and mesoscopic simulation. J. Chem. Phys. 1997, 107, 4423–4435. [Google Scholar] [CrossRef]
- Gooneie, A.; Mattausch, H.; Witschnigg, A.; Schuschnigg, S.; Holzer, C. Multiscale simulation of polymer nanocomposites in processing: Challenges and outlooks. Key Eng. Mater. 2015, 651, 533–538. [Google Scholar] [CrossRef]
- Español, P.; Warren, P. Statistical mechanics of dissipative particle dynamics. Europhys. Lett. 1995, 30, 191. [Google Scholar] [CrossRef]
- Groot, R.D. Applications of Dissipative Particle Dynamics. In Novel Methods in Soft Matter Simulations; Karttunen, M., Vattulainen, I., Lukkarinen, A., Eds.; Springer: Berlin, Germany, 2004; pp. 5–38. [Google Scholar]
- Groot, R.D.; Madden, T.J. Dynamic simulation of diblock copolymer microphase separation. J. Chem. Phys. 1998, 108, 8713–8724. [Google Scholar] [CrossRef]
- Den Otter, W.K.; Clarke, J. The temperature in dissipative particle dynamics. Int. J. Mod. Phys. C 2000, 11, 1179–1193. [Google Scholar] [CrossRef]
- Pan, W.; Caswell, B.; Karniadakis, G.E. Rheology, microstructure and migration in brownian colloidal suspensions. Langmuir 2010, 26, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Yamanoi, M.; Pozo, O.; Maia, J.M. Linear and non-linear dynamics of entangled linear polymer melts by modified tunable coarse-grained level Dissipative Particle Dynamics. J. Chem. Phys. 2011, 135, 44904. [Google Scholar] [CrossRef] [PubMed]
- Gooneie, A.; Schuschnigg, S.; Holzer, C. Orientation of anisometric layered silicate particles in uncompatibilized and compatibilized polymer melts under shear flow: A dissipative particle dynamics study. Macromol. Theory Simul. 2016, 25, 85–98. [Google Scholar] [CrossRef]
- Gooneie, A.; Schuschnigg, S.; Holzer, C. Dissipative particle dynamics models of orientation of weakly-interacting anisometric silicate particles in polymer melts under shear flow: Comparison with the standard orientation models. Macromol. Theory Simul. 2016, 25, 287–302. [Google Scholar] [CrossRef]
- Fedosov, D.A.; Noguchi, H.; Gompper, G. Multiscale modeling of blood flow: From single cells to blood rheology. Biomech. Model. Mechanobiol. 2014, 13, 239–258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhang, N.; Deng, Y.; Bluestein, D. A multiple time stepping algorithm for efficient multiscale modeling of platelets flowing in blood plasma. J. Comput. Phys. 2015, 284, 668–686. [Google Scholar] [CrossRef] [PubMed]
- Gooneie, A.; Schuschnigg, S.; Holzer, C. Dissipative particle dynamics simulations of orientation of layered silicate particles embedded in polymer melts under shear flows. AIP Conf. Proc. 2016, 1779, 50010. [Google Scholar]
- Gai, J.-G.; Hu, G.-H.; Li, H.-L.; Zhu, S.-P.; Hoppe, S. Dissipative particle dynamics and flory-huggins theories for predicting the rheological behavior of ultrahigh molecular weight polyethylene blends. Ind. Eng. Chem. Res. 2010, 49, 11369–11379. [Google Scholar] [CrossRef]
- Goicochea, A.G. Adsorption and disjoining pressure isotherms of confined polymers using dissipative particle dynamics. Langmuir 2007, 23, 11656–11663. [Google Scholar] [CrossRef] [PubMed]
- Kacar, G.; Peters, E.; de With, G. Mesoscopic simulations for the molecular and network structure of a thermoset polymer. Soft Matter 2013, 9, 5785–5793. [Google Scholar] [CrossRef]
- Kacar, G.; Peters, E.; de With, G. Structure of a thermoset polymer near an alumina substrate as studied by dissipative particle dynamics. J. Phys. Chem. C 2013, 117, 19038–19047. [Google Scholar] [CrossRef]
- Kauzlari, D.; Meier, J.T.; Español, P.; Succi, S.; Greiner, A.; Korvink, J.G. Bottom-up coarse-graining of a simple graphene model: The blob picture. J. Chem. Phys. 2011, 134. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-T.; Mao, R.; Vishnyakov, A.; Neimark, A.V. Parametrization of chain molecules in dissipative particle dynamics. J. Phys. Chem. B 2016. [Google Scholar] [CrossRef] [PubMed]
- Maly, M.; Posocco, P.; Pricl, S.; Fermeglia, M. Self-assembly of nanoparticle mixtures in diblock copolymers: Multiscale molecular modeling. Ind. Eng. Chem. Res. 2008, 47, 5023–5038. [Google Scholar] [CrossRef]
- Posocco, P.; Posel, Z.; Fermeglia, M.; Lísal, M.; Pricl, S. A molecular simulation approach to the prediction of the morphology of self-assembled nanoparticles in diblock copolymers. J. Mater. Chem. 2010, 20, 10511–10520. [Google Scholar] [CrossRef]
- Esteves, A.C.C.; Lyakhova, K.; Van Der Ven, L.G.J.; Van Benthem, R.A.T.M.; de With, G. Surface segregation of low surface energy polymeric dangling chains in a cross-linked polymer network investigated by a combined experimental-simulation Approach. Macromolecules 2013, 46, 1993–2002. [Google Scholar] [CrossRef]
- Esteves, A.C.C.; Lyakhova, K.; van Riel, J.M.; Van Der Ven, L.G.J.; Van Benthem, R.A.T.M.; de With, G. Self-replenishing ability of cross-linked low surface energy polymer films investigated by a complementary experimental-simulation approach. J. Chem. Phys. 2014, 140, 124902. [Google Scholar] [CrossRef] [PubMed]
- Lyakhova, K.; Esteves, A.C.C.; van de Put, M.W.P.; van der Ven, L.G.J.; van Benthem, R.A.T.M.; de With, G. Simulation-Experimental Approach to Investigate the Role of Interfaces in Self-Replenishing Composite Coatings. Adv. Mater. Interfaces 2014, 1, 1400053. [Google Scholar] [CrossRef]
- Rahatekar, S.S.; Hamm, M.; Shaffer, M.; Elliott, J.A. Mesoscale modeling of electrical percolation in fiber-filled systems. J. Chem. Phys. 2005, 123. [Google Scholar] [CrossRef] [PubMed]
- Frisch, U.; Hasslacher, B.; Pomeau, Y. Lattice-gas automata for the Navier-Stokes equation. Phys. Rev. Lett. 1986, 56, 1505–1508. [Google Scholar] [CrossRef] [PubMed]
- Benzi, R.; Succi, S.; Vergassola, M. The lattice Boltzmann equation: Theory and applications. Phys. Rep. 1992, 222, 145–197. [Google Scholar] [CrossRef]
- Higuera, F.J.; Jiménez, J. Boltzmann approach to lattice gas simulations. Europhys. Lett. 1989, 9, 663–668. [Google Scholar] [CrossRef]
- Higuera, F.J.; Succi, S.; Benzi, R. Lattice gas dynamics with enhanced collisions. Europhys. Lett. 1989, 9, 345–349. [Google Scholar] [CrossRef]
- Bhatnagar, P.L.; Gross, E.P.; Krook, M. A model for collision processes in gases. I. Small amplitude processes in charged and neutral one-component systems. Phys. Rev. 1954, 94, 511–525. [Google Scholar] [CrossRef]
- Qian, Y.H.; D’Humières, D.; Lallemand, P. Lattice bgk models for navier-stokes equation. Europhys. Lett. 1992, 17, 479–484. [Google Scholar] [CrossRef]
- Dünweg, B.; Ladd, A. Lattice Boltzmann Simulations of Soft Matter Systems. arXiv, 2009; arXiv:0803.2826. [Google Scholar]
- Usta, O.B.; Ladd, A.; Butler, J.E. Lattice-Boltzmann simulations of the dynamics of polymer solutions in periodic and confined geometries. J. Chem. Phys. 2005, 122. [Google Scholar] [CrossRef]
- Ahlrichs, P.; Dünweg, B. Lattice-boltzmann simulation of polymer-solvent systems. Int. J. Mod. Phys. C 1998, 9, 1429–1438. [Google Scholar] [CrossRef]
- Aidun, C.K.; Clausen, J.R. Lattice-boltzmann method for complex flows. Annu. Rev. Fluid Mech. 2010, 42, 439–472. [Google Scholar] [CrossRef]
- Spaid, M.; Phelan, F.R., Jr. Lattice Boltzmann methods for modeling microscale flow in fibrous porous media. Phys. Fluids 1997, 9, 2468–2474. [Google Scholar] [CrossRef]
- Sinha, P.K.; Mukherjee, P.P.; Wang, C.-Y. Impact of GDL structure and wettability on water management in polymer electrolyte fuel cells. J. Mater. Chem. 2007, 17, 3089–3103. [Google Scholar] [CrossRef]
- Care, C.M.; Cleaver, D.J. Computer simulation of liquid crystals. Rep. Prog. Phys. 2005, 68, 2665–2700. [Google Scholar] [CrossRef]
- Denniston, C.; Orlandini, E.; Yeomans, J.M. Lattice Boltzmann simulations of liquid crystal hydrodynamics. Phys. Rev. E 2001, 63, 056702. [Google Scholar] [CrossRef] [PubMed]
- Marenduzzo, D.; Orlandini, E.; Cates, M.E.; Yeomans, J.M. Steady-state hydrodynamic instabilities of active liquid crystals: Hybrid lattice Boltzmann simulations. Phys. Rev. E Stat., Nonlinear Soft Matter Phys. 2007, 76. [Google Scholar] [CrossRef] [PubMed]
- Usta, O.B.; Perchak, D.; Clarke, A.; Yeomans, J.M.; Balazs, A.C. Shear and extensional deformation of droplets containing polymers and nanoparticles. J. Chem. Phys. 2009, 130. [Google Scholar] [CrossRef] [PubMed]
- Nabovati, A.; Llewellin, E.W.; Sousa, A. A general model for the permeability of fibrous porous media based on fluid flow simulations using the lattice Boltzmann method. Compos. Part A 2009, 40, 860–869. [Google Scholar] [CrossRef]
- Wang, M.; He, J.; Yu, J.; Pan, N. Lattice Boltzmann modeling of the effective thermal conductivity for fibrous materials. Int. J. Therm. Sci. 2007, 46, 848–855. [Google Scholar] [CrossRef]
- Bird, R.B.; Armstrong, R.C.; Hassager, O. Dynamics of Polymeric Liquids; John Wiley & Sons Inc.: Hoboken, NJ, USA, 1987. [Google Scholar]
- Rappaz, M.; Bellet, M.; Deville, M. Numerical Modeling in Materials Science and Engineering; Springer: Leutershausen, Germany, 2003. [Google Scholar]
- Ellero, M.; Español, P.; Flekkøy, E.G. Thermodynamically consistent fluid particle model for viscoelastic flows. Phys. Rev. E 2003, 68, 041504. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Quesada, A.; Ellero, M.; Español, P. Smoothed particle hydrodynamic model for viscoelastic fluids with thermal fluctuations. Phys. Rev. E 2009, 79. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.; Litvinov, S.; Qian, R.; Ellero, M.; Adams, N.A. Multiscale modeling of particle in suspension with smoothed dissipative particle dynamics. Phys. Fluids 2012, 24. [Google Scholar] [CrossRef]
- Vázquez-Quesada, A.; Ellero, M.; Español, P. Consistent scaling of thermal fluctuations in smoothed dissipative particle dynamics. J. Chem. Phys. 2009, 130. [Google Scholar] [CrossRef] [PubMed]
- Petsev, N.D.; Leal, L.G.; Shell, M.S. Multiscale simulation of ideal mixtures using smoothed dissipative particle dynamics. J. Chem. Phys. 2016, 144. [Google Scholar] [CrossRef] [PubMed]
- Pirondi, A.; Giuliese, G.; Moroni, F. Fatigue debonding three-dimensional simulation with cohesive zone. J. Adhes. 2016, 92, 553–571. [Google Scholar] [CrossRef]
- Schiel, M.; Reh, S.; Schwienhorst, M.; Welters, T.; Stammen, E.; Dilger, K. Finite element modelling of cure-dependent mechanical properties by model-free kinetic analysis using a cohesive zone approach. J. Adhes. 2016, 92, 572–585. [Google Scholar] [CrossRef]
- Heydari-Meybodi, M.; Saber-Samandari, S.; Sadighi, M. 3D multiscale modeling to predict the elastic modulus of polymer/nanoclay composites considering realistic interphase property. Compos. Interfaces 2016, 23, 641–661. [Google Scholar] [CrossRef]
- Sun, M.-K.; Shieh, J.; Chen, C.-S.; Chiang, H.; Huang, C.-W.; Chen, W.-S. Effects of an implant on temperature distribution in tissue during ultrasound diathermy. Ultrason. Sonochem. 2016, 32, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Gooneie, A.; Schuschnigg, S.; Duretek, I.; Holzer, C. Numerical simulations of the flow of wood polypropylene composites with wall slipping in a profile die: The significance of material data. Arch. Iran. Med. 2015, 1664, 50014. [Google Scholar]
- Duretek, I.; Schuschnigg, S.; Gooneie, A.; Langecker, G.R.; Holzer, C.; Gomze, L.A. Rheological properties of wood polymer composites and their role in extrusion. J. Phys. Conf. Ser. 2015, 602, 12014. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, D.; Yang, X.; Cui, X.; Zhang, X.; Wang, Q. Research on torsional friction behavior and fluid load support of PVA/HA composite hydrogel. J. Mech. Behav. Biomed. Mater. 2016, 62, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Ehrenhofer, A.; Bingel, G.; Paschew, G.; Tietze, M.; Schröder, R.; Richter, A.; Wallmersperger, T. Permeation control in hydrogel-layered patterned PET membranes with defined switchable pore geometry—Experiments and numerical simulation. Sens. Actuators B 2016, 232, 499–505. [Google Scholar] [CrossRef]
- Baaijens, F. Mixed finite element methods for viscoelastic flow analysis: A review. J. Non-Newton. Fluid Mech. 1998, 79, 361–385. [Google Scholar] [CrossRef]
- Verbeeten, W.; Peters, G.; Baaijens, F. Differential constitutive equations for polymer melts: The extended Pom-Pom model. J. Rheol. 2001, 45, 823–843. [Google Scholar] [CrossRef]
- Yue, P.; Zhou, C.; Feng, J.J.; Ollivier-Gooch, C.F.; Hu, H.H. Phase-field simulations of interfacial dynamics in viscoelastic fluids using finite elements with adaptive meshing. J. Comput. Phys. 2006, 219, 47–67. [Google Scholar] [CrossRef]
- Wilkes, E.D.; Phillips, S.D.; Basaran, O.A. Computational and experimental analysis of dynamics of drop formation. Phys. Fluids 1999, 11, 3577–3598. [Google Scholar] [CrossRef]
- Patera, A.T. A spectral element method for fluid dynamics. J. Comput. Phys. 1984, 54, 468–488. [Google Scholar] [CrossRef]
- Pozrikidis, C. Introduction to Finite and Spectral Element Methods Using MATLAB, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar]
- Van Os, R.; Phillips, T.N. The prediction of complex flows of polymer melts using spectral elements. J. Non-Newton. Fluid Mech. 2004, 122, 287–301. [Google Scholar] [CrossRef]
- Fiétier, N.; Deville, M.O. Time-dependent algorithms for the simulation of viscoelastic flows with spectral element methods. J. Comput. Phys. 2003, 186, 93–121. [Google Scholar] [CrossRef]
- Li, R.; Chen, Z.; Wu, W. Generalized Difference Methods for Differential Equations: Numerical Analysis of Finite Volume Methods; Marcel Dekker: New York, NY, USA, 2000. [Google Scholar]
- Hughes, T.; Brooks, A. Multi-dimensional upwind scheme with no crosswind diffusion. Am. Soc. Mech. Eng. 1979, 34, 19–35. [Google Scholar]
- Hughes, T.; Mallet, M.; Akira, M. A new finite element formulation for computational fluid dynamics: II. Beyond SUPG. Comput. Methods Appl. Mech. Eng. 1986, 54, 341–355. [Google Scholar] [CrossRef]
- Jiang, B.; Liao, G. The Least-Squares Meshfree Finite Element Method. In Proceedings of the Computational Mechanics: International Symposium on Computational Mechanic, Beijing, China, 30 July–1 August 2009; p. 341.
- Kumar, R.; Dennis, B.H. A Least-Squares Galerkin Split Finite Element Method for Compressible Navier-Stokes Equations. In Proceedings of the 29th Computers and Information in Engineering Conference, San Diego, CA, USA, 30 August 30–2 September 2009.
- Baliga, B.R.; Patankar, S.V. A control volume finite-element method for two-dimensional fluid flow and heat transfer. Numer. Heat Transf. 1983, 6, 245–261. [Google Scholar] [CrossRef]
- Patankar, S.V. A calculation procedure for two-dimensional elliptic situations. Numer. Heat Transf. 1981, 4, 409–425. [Google Scholar] [CrossRef]
- Prakash, C.; Patankar, S.V. A control volume-based finite-element method for solving the navier-stokes equations using equal-order velocity-pressure interpolation. Numer. Heat Transf. 1985, 8, 259–280. [Google Scholar] [CrossRef]
- Balsara, D.S. Divergence-free reconstruction of magnetic fields and WENO schemes for magnetohydrodynamics. J. Comput. Phys. 2009, 228, 5040–5056. [Google Scholar] [CrossRef]
- Powell, K.G.; Roe, P.L.; Linde, T.J.; Gombosi, T.I.; de Zeeuw, D.L. A Solution-Adaptive Upwind Scheme for Ideal Magnetohydrodynamics. J. Comput. Phys. 1999, 154, 284–309. [Google Scholar] [CrossRef]
- Tóth, G. The ▽·B = 0 Constraint in Shock-Capturing Magnetohydrodynamics Codes. J. Comput. Phys. 2000, 161, 605–652. [Google Scholar] [CrossRef]
- Liu, F.; Cai, J.; Zhu, Y.; Tsai, H.M.; Wong, A. Calculation of wing flutter by a coupled fluid-structure method. J. Aircr. 2001, 38, 334–342. [Google Scholar] [CrossRef]
- Makhijani, V.B.; Yang, H.Q.; Dionne, P.J.; Thubrikar, M.J. Three-dimensional coupled fluid-structure simulation of pericardial bioprosthetic aortic valve function. ASAIO J. 1997, 43, M387–M392. [Google Scholar] [CrossRef] [PubMed]
- Narumanchi, S.; Murthy, J.Y.; Amon, C.H. Submicron heat transport model in silicon accounting for phonon dispersion and polarization. J. Heat Transf. 2004, 126, 946–955. [Google Scholar] [CrossRef]
- Voicolescu, I.; Andrew McGill, R.; Zaghloul, M.E.; Mott, D.; Stepnowski, J.; Stepnowski, S.; Summers, H.; Nguyen, V.; Ross, S.; Walsh, K.; et al. Micropreconcentrator for enhanced trace detection of explosives and chemical agents. IEEE Sens. J. 2006, 6, 1094–1103. [Google Scholar] [CrossRef]
- Moukalled, F.; Mangani, L.; Darwish, M. The Finite Volume Method in Computational Fluid Dynamics: An Advanced Introduction with OpenFOAM® and Matlab®; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Patankar, S.V. Numerical Heat Transfer and Fluid Flow; Hemisphere Publishing: Washington, DC, USA, 1980. [Google Scholar]
- Leonard, B.P. A stable and accurate convective modelling procedure based on quadratic upstream interpolation. Comput. Methods Appl. Mech. Eng. 1979, 19, 59–98. [Google Scholar] [CrossRef]
- Weller, H.G.; Tabor, G.; Jasak, H.; Fureby, C. A tensorial approach to computational continuum mechanics using object-oriented techniques. Comput. Phys. 1998, 12, 620–631. [Google Scholar] [CrossRef]
- Jasak, H. OpenFOAM: Open source CFD in research and industry. Int. J. Nav. Archit. Ocean Eng. 2009, 1, 89–94. [Google Scholar]
- Favero, J.L.; Secchi, A.R.; Cardozo, N.; Jasak, H. Viscoelastic flow analysis using the software OpenFOAM and differential constitutive equations. J. Non-Newton. Fluid Mech. 2010, 165, 1625–1636. [Google Scholar] [CrossRef]
- Favero, J.L.; Secchi, A.R.; Cardozo, N.; Jasak, H. Viscoelastic flow simulation: Development of a methodology of analysis using the software OpenFOAM and differential constitutive equations. Comput. Aided Chem. Eng. 2009, 27, 915–920. [Google Scholar]
- Favero, J.L.; Secchi, A.R.; Cardozo, N.; Jasak, H. Viscoelastic fluid analysis in internal and in free surface flows using the software OpenFOAM. Comput. Chem. Eng. 2010, 34, 1984–1993. [Google Scholar] [CrossRef]
- Holmes, L.; Favero, J.; Osswald, T. Numerical simulation of three-dimensional viscoelastic planar contraction flow using the software OpenFOAM. Comput. Chem. Eng. 2012, 37, 64–73. [Google Scholar] [CrossRef]
- Yang, W.-J.; Yi, W.; Ren, X.-G.; Xu, L.-Y.; Xu, X.-H.; Yuan, X.-F. Toward large scale parallel computer simulation of viscoelastic fluid flow: A study of benchmark flow problems. J. Non-Newton. Fluid Mech. 2015, 222, 82–95. [Google Scholar] [CrossRef]
- Lima, N.C.; D’ávila, M.A. Numerical simulation of electrohydrodynamic flows of Newtonian and viscoelastic droplets. J. Non-Newton. Fluid Mech. 2014, 213, 1–14. [Google Scholar] [CrossRef]
- Cao, Y.; Ren, X.-G.; Guo, X.-W.; Wang, M.; Wang, Q.; Xu, X.-H.; Yang, X.-J. A new method to simulate free surface flows for Viscoelastic fluid. Adv. Mater. Sci. Eng. 2015, 2015. [Google Scholar] [CrossRef]
- Habla, F.; Marschall, H.; Hinrichsen, O.; Dietsche, L.; Jasak, H.; Favero, J.L. Numerical simulation of viscoelastic two-phase flows using openFOAM®. Chem. Eng. Sci. 2011, 66, 5487–5496. [Google Scholar] [CrossRef]
- Zhang, K.; Kuang, T.; Liu, H.; Zeng, X.; Deng, Y.; Jasak, H. Simulation and analysis of mold filling in water-assisted injection molding of viscoelastic polymers. Gaofenzi Cailiao Kexue Yu Gongcheng 2014, 30, 93–96. [Google Scholar]
- Haddadi Sisakht, B.; Jordan, C.; Schretter, P.; Lassmann, T.; Harasek, M. Designing Better Membrane Modules Using CFD. Chem. Prod. Process Model. 2016, 11, 57–66. [Google Scholar] [CrossRef]
- Wu, W.-T.; Yang, F.; Antaki, J.F.; Aubry, N.; Massoudi, M. Study of blood flow in several benchmark micro-channels using a two-fluid approach. Int. J. Eng. Sci. 2015, 95, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Carolan, D.; Chong, H.M.; Ivankovic, A.; Kinloch, A.J.; Taylor, A.C. Co-continuous polymer systems: A numerical investigation. Comput. Mater. Sci. 2015, 98, 24–33. [Google Scholar] [CrossRef]
- Shou, Z.; Buxton, G.A.; Balazs, A.C. Predicting the self-assembled morphology and mechanical properties of mixtures of diblocks and rod-like nanoparticles. Compos. Interfaces 2003, 10, 343–368. [Google Scholar] [CrossRef]
- Travasso, R.; Buxton, G.A.; Kuksenok, O.; Good, K.; Balazs, A.C. Modeling the morphology and mechanical properties of sheared ternary mixtures. J. Chem. Phys. 2005, 122, 194906. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Deng, S.; Huang, Y.; Liu, H.; Hu, Y. Simulation of morphologies and mechanical properties of A/B polymer blend film. Chin. J. Chem. Eng. 2011, 19, 549–557. [Google Scholar] [CrossRef]
- Smith, K.A.; Tyagi, S.; Balazs, A.C. Healing surface defects with nanoparticle-filled polymer coatings: Effect of particle geometry. Macromolecules 2005, 38, 10138–10147. [Google Scholar] [CrossRef]
- Tyagi, S.; Lee, J.Y.; Buxton, G.A.; Balazs, A.C. Using nanocomposite coatings to heal surface defects. Macromolecules 2004, 37, 9160–9168. [Google Scholar] [CrossRef]
- Brown, J.R.; Seo, Y.; Maula, T.; Hall, L.M. Fluids density functional theory and initializing molecular dynamics simulations of block copolymers. J. Chem. Phys. 2016, 144. [Google Scholar] [CrossRef] [PubMed]
- Buxton, G.A.; Balazs, A.C. Predicting the mechanical and electrical properties of nanocomposites formed from polymer blends and nanorods. Mol. Simul. 2004, 30, 249–257. [Google Scholar] [CrossRef]
- Buxton, G.A.; Balazs, A.C. Simulating the morphology and mechanical properties of filled diblock copolymers. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2003, 67, 31802. [Google Scholar] [CrossRef] [PubMed]
- Suter, J.L.; Groen, D.; Coveney, P.V. Chemically specifi C multiscale modeling of clay-polymer nanocomposites reveals intercalation dynamics, tactoid self-assembly and emergent materials properties. Adv. Mater. 2015, 27, 966–984. [Google Scholar] [CrossRef] [PubMed]
- Scocchi, G.; Posocco, P.; Fermeglia, M.; Pricl, S. Polymer—Clay nanocomposites: A multiscale molecular modeling approach. J. Phys. Chem. B 2007, 111, 2143–2151. [Google Scholar] [CrossRef] [PubMed]
- Scocchi, G.; Posocco, P.; Handgraaf, J.-W.; Fraaije, J.G.E.M.; Fermeglia, M.; Pricl, S. A complete multiscale modelling approach for polymer-clay nanocomposites. Chem. Eur. J. 2009, 15, 7586–7592. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.P.; Scocchi, G.; Toth, R.; Posocco, P.; Nieto, D.R.; Pricl, S.; Fermeglia, M. Multiscale Modeling of Polymer/Clay Nanocomposites. J. Multiscale Model. 2011, 3, 151–176. [Google Scholar] [CrossRef]
- Kalligiannaki, E.; Chazirakis, A.; Tsourtis, A.; Katsoulakis, M.A.; Plecháč, P.; Harmandaris, V. Parametrizing coarse grained models for molecular systems at equilibrium. Eur. Phys. J. Spec. Top. 2016, 225, 1347–1372. [Google Scholar] [CrossRef]
- Harmandaris, V.; Kalligiannaki, E.; Katsoulakis, M.; Plecháč, P. Path-space variational inference for non-equilibrium coarse-grained systems. J. Comput. Phys. 2016, 314, 355–383. [Google Scholar] [CrossRef]
- Li, Y.; Abberton, B.C.; Kröger, M.; Liu, W.K. Challenges in multiscale modeling of polymer dynamics. Polymers 2013, 5, 751–832. [Google Scholar] [CrossRef]
- Brini, E.; Algaer, E.A.; Ganguly, P.; Li, C.; Rodríguez-Ropero, F.; van der Vegt, N. Systematic coarse-graining methods for soft matter simulations-a review. Soft Matter 2013, 9, 2108–2119. [Google Scholar] [CrossRef]
- Hess, B.; Holm, C.; van der Vegt, N. Modeling multibody effects in ionic solutions with a concentration dependent dielectric permittivity. Phys. Rev. Lett. 2006, 96. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.-W.; Li, C.; van der Vegt, N.; Peter, C. Transferability of coarse grained potentials: Implicit solvent models for hydrated ions. J. Chem. Theory Comput. 2011, 7, 1916–1927. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Noid, W.G.; Liu, P.; Voth, G.A. Effective force coarse-graining. Phys. Chem. Chem. Phys. 2009, 11, 2002–2015. [Google Scholar] [CrossRef] [PubMed]
- Brini, E.; Marcon, V.; van der Vegt, N. Conditional reversible work method for molecular coarse graining applications. Phys. Chem. Chem. Phys. 2011, 13, 10468–10474. [Google Scholar] [CrossRef] [PubMed]
- Brini, E.; van der Vegt, N. Chemically transferable coarse-grained potentials from conditional reversible work calculations. J. Chem. Phys. 2012, 137. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Peter, C.; van der Vegt, N. Self-assembling dipeptides: Conformational sampling in solvent-free coarse-grained simulation. Phys. Chem. Chem. Phys. 2009, 11, 2077–2086. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Shen, J.; Peter, C.; van der Vegt, N. A chemically accurate implicit-solvent coarse-grained model for polystyrenesulfonate solutions. Macromolecules 2012, 45, 2551–2561. [Google Scholar] [CrossRef]
- Tschöp, W.; Kremer, K.; Hahn, O.; Batoulis, J.; Bürger, T. Simulation of polymer melts. I. coarse-graining procedure for polycarbonates. Acta Polym. 1998, 49, 61–74. [Google Scholar] [CrossRef]
- Lyubartsev, A.P.; Laaksonen, A. Calculation of effective interaction potentials from radial distribution functions: A reverse Monte Carlo approach. Phys. Rev. E 1995, 52, 3730–3737. [Google Scholar] [CrossRef]
- Lyubartsev, A.P.; Laaksonen, A. Osmotic and activity coefficients from effective potentials for hydrated ions. Phys. Rev. E 1997, 55, 5689–5696. [Google Scholar] [CrossRef]
- Reith, D.; Pütz, M.; Müller-Plathe, F. Deriving effective mesoscale potentials from atomistic simulations. J. Comput. Chem. 2003, 24, 1624–1636. [Google Scholar] [CrossRef] [PubMed]
- Peter, C.; Delle Site, L.; Kremer, K. Classical simulations from the atomistic to the mesoscale and back: Coarse graining an azobenzene liquid crystal. Soft Matter 2008, 4, 859–869. [Google Scholar] [CrossRef]
- Murtola, T.; Karttunen, M.; Vattulainen, I. Systematic coarse graining from structure using internal states: Application to phospholipid/cholesterol bilayer. J. Chem. Phys. 2009, 131. [Google Scholar] [CrossRef] [PubMed]
- Savelyev, A.; Papoian, G.A. Molecular renormalization group coarse-graining of electrolyte solutions: Application to aqueous NaCl and KCl. J. Phys. Chem. B 2009, 113, 7785–7793. [Google Scholar] [CrossRef] [PubMed]
- Savelyev, A.; Papoian, G.A. Molecular renormalization group coarse-graining of polymer chains: Application to double-stranded DNA. Biophys. J. 2009, 96, 4044–4052. [Google Scholar] [CrossRef] [PubMed]
- Savelyev, A.; Papoian, G.A. Chemically accurate coarse graining of double-stranded DNA. Proc. Natl. Acad. Sci. USA 2010, 107, 20340–20345. [Google Scholar] [CrossRef] [PubMed]
- Megariotis, G.; Vyrkou, A.; Leygue, A.; Theodorou, D.N. Systematic coarse graining of 4-Cyano-4′-pentylbiphenyl. Ind. Eng. Chem. Res. 2011, 50, 546–556. [Google Scholar] [CrossRef]
- Mukherjee, B.; Delle Site, L.; Kremer, K.; Peter, C. Derivation of coarse grained models for multiscale simulation of liquid crystalline phase transitions. J. Phys. Chem. B 2012, 116, 8474–8484. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.; Biermann, O.; Faller, R.; Reith, D.; Müller-Plathe, F. Coarse graining of nonbonded inter-particle potentials using automatic simplex optimization to fit structural properties. J. Chem. Phys. 2000, 113, 6264–6275. [Google Scholar] [CrossRef]
- Ganguly, P.; Mukherji, D.; Junghans, C.; van der Vegt, N. Kirkwood-buff coarse-grained force fields for aqueous solutions. J. Chem. Theory Comput. 2012, 8, 1802–1807. [Google Scholar] [CrossRef] [PubMed]
- Shell, M.S. The relative entropy is fundamental to multiscale and inverse thermodynamic problems. J. Chem. Phys. 2008, 129. [Google Scholar] [CrossRef] [PubMed]
- Chaimovich, A.; Shell, M.S. Relative entropy as a universal metric for multiscale errors. Phys. Rev. E 2010, 81. [Google Scholar] [CrossRef] [PubMed]
- Chaimovich, A.; Shell, M.S. Coarse-graining errors and numerical optimization using a relative entropy framework. J. Chem. Phys. 2011, 134. [Google Scholar] [CrossRef] [PubMed]
- Chaimovich, A.; Shell, M.S. Anomalous waterlike behavior in spherically-symmetric water models optimized with the relative entropy. Phys. Chem. Chem. Phys. 2009, 11, 1901–1915. [Google Scholar] [CrossRef] [PubMed]
- Mullinax, J.W.; Noid, W.G. Reference state for the generalized Yvon-Born-Green theory: Application for coarse-grained model of hydrophobic hydration. J. Chem. Phys. 2010, 133. [Google Scholar] [CrossRef] [PubMed]
- Ercolesi, F.; Adams, J.B. Interatomic potentials from first-principles calculations: The force-matching method. Europhys. Lett. 1994, 26, 583–588. [Google Scholar] [CrossRef]
- Rühle, V.; Junghans, C.; Lukyanov, A.; Kremer, K.; Andrienko, D. Versatile object-oriented toolkit for coarse-graining applications. J. Chem. Theory Comput. 2009, 5, 3211–3223. [Google Scholar] [CrossRef] [PubMed]
- Izvekov, S.; Voth, G.A. A multiscale coarse-graining method for biomolecular systems. J. Phys. Chem. B 2005, 109, 2469–2473. [Google Scholar] [CrossRef] [PubMed]
- Izvekov, S.; Chung, P.W.; Rice, B.M. The multiscale coarse-graining method: Assessing its accuracy and introducing density dependent coarse-grain potentials. J. Chem. Phys. 2010, 133. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Thorpe, I.F.; Izvekov, S.; Voth, G.A. Coarse-grained peptide modeling using a systematic multiscale approach. Biophys. J. 2007, 92, 4289–4303. [Google Scholar] [CrossRef] [PubMed]
- Hills, R.D., Jr.; Lu, L.; Voth, G.A. Multiscale coarse-graining of the protein energy landscape. PLoS Comput. Biol. 2010, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Izvekov, S.; Voth, G.A. Multiscale coarse graining of liquid-state systems. J. Chem. Phys. 2005, 123. [Google Scholar] [CrossRef] [PubMed]
- Noid, W.G.; Chu, J.-W.; Ayton, G.S.; Krishna, V.; Izvekov, S.; Voth, G.A.; Das, A.; Andersen, H.C. The multiscale coarse-graining method. I. A rigorous bridge between atomistic and coarse-grained models. J. Chem. Phys. 2008, 128, 244114. [Google Scholar] [CrossRef] [PubMed]
- Noid, W.G.; Chu, J.-W.; Ayton, G.S.; Voth, G.A. Multiscale coarse-graining and structural correlations: Connections to liquid-state theory. J. Phys. Chem. B 2007, 111, 4116–4127. [Google Scholar] [CrossRef] [PubMed]
- Noid, W.G.; Liu, P.; Wang, Y.; Chu, J.-W.; Ayton, G.S.; Izvekov, S.; Andersen, H.C.; Voth, G.A. The multiscale coarse-graining method. II. Numerical implementation for coarse-grained molecular models. J. Chem. Phys. 2008, 128, 244115. [Google Scholar] [CrossRef] [PubMed]
- Wu, C. Phase morphologies of binary polymer blends predicted by systematically coarse-grained models. Macromol. Theory Simul. 2016. [Google Scholar] [CrossRef]
- Potestio, R.; Peter, C.; Kremer, K. Computer simulations of soft matter: Linking the scales. Entropy 2014, 16, 4199–4245. [Google Scholar] [CrossRef]
- Rzepiela, A.J.; Louhivuori, M.; Peter, C.; Marrink, S.J. Hybrid simulations: Combining atomistic and coarse-grained force fields using virtual sites. Phys. Chem. Chem. Phys. 2011, 13, 10437–10448. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tang, S.; Abberton, B.C.; Kröger, M.; Burkhart, C.; Jiang, B.; Papakonstantopoulos, G.J.; Poldneff, M.; Liu, W.K. A predictive multiscale computational framework for viscoelastic properties of linear polymers. Polymer 2012, 53, 5935–5952. [Google Scholar] [CrossRef]
- Noid, W.G. Perspective: Coarse-grained models for biomolecular systems. J. Chem. Phys. 2013, 139. [Google Scholar] [CrossRef] [PubMed]
- Rudzinski, J.F.; Noid, W.G. Investigation of coarse-grained mappings via an iterative generalized Yvon-Born-Green method. J. Phys. Chem. B 2014, 118, 8295–8312. [Google Scholar] [CrossRef] [PubMed]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Den Otter, W.K.; Briels, W.J. The calculation of free-energy differences by constrained molecular-dynamics simulations. J. Chem. Phys. 1998, 109, 4139–4146. [Google Scholar] [CrossRef]
- Villa, A.; Peter, C.; van der Vegt, N. Transferability of nonbonded interaction potentials for coarse-grained simulations: Benzene in water. J. Chem. Theory Comput. 2010, 6, 2434–2444. [Google Scholar] [CrossRef] [PubMed]
- Hahn, O.; Site, L.D.; Kremer, K. Simulation of polymer melts: From spherical to ellipsoidal beads. Macromol. Theory Simul. 2001, 10, 288–303. [Google Scholar] [CrossRef]
- Xie, G.-L.; Zhang, Y.-H.; Huang, S.-P. Glass formation of n-butanol: Coarse-grained molecular dynamics simulations using gay-berne potential model. Chin. J. Chem. Phys. 2012, 25, 177–185. [Google Scholar] [CrossRef]
- Müller-Plathe, F. Local structure and dynamics in solvent-swollen polymers. Macromolecules 1996, 29, 4782–4791. [Google Scholar] [CrossRef]
- Milano, G.; Müller-Plathe, F. Mapping atomistic simulations to mesoscopic models: A systematic coarse-graining procedure for vinyl polymer chains. J. Phys. Chem. B 2005, 109, 18609–18619. [Google Scholar] [CrossRef] [PubMed]
- Milano, G.; Goudeau, S.; Müller-Plathe, F. Multicentered Gaussian-based potentials for coarse-grained polymer simulations: Linking atomistic and mesoscopic scales. J. Polym. Sci. Part B 2005, 43, 871–885. [Google Scholar] [CrossRef]
- Spyriouni, T.; Tzoumanekas, C.; Theodorou, D.; Müller-Plathe, F.; Milano, G. Coarse-grained and reverse-mapped united-atom simulations of long-chain atactic polystyrene melts: Structure, thermodynamic properties, chain conformation, and entanglements. Macromolecules 2007, 40, 3876–3885. [Google Scholar] [CrossRef]
- Sun, Q.; Faller, R. Systematic coarse-graining of atomistic models for simulation of polymeric systems. Comput. Chem. Eng. 2005, 29, 2380–2385. [Google Scholar] [CrossRef]
- Sun, Q.; Faller, R. Crossover from unentangled to entangled dynamics in a systematically coarse-grained polystyrene melt. Macromolecules 2006, 39, 812–820. [Google Scholar] [CrossRef]
- Qian, H.-J.; Carbone, P.; Xiaoyu, C.; Karimi-Varzaneh, H.A.; Liew, C.C.; Müller-Plathe, F. Temperature-Transferable Coarse-Grained potentials for ethylbenzene, polystyrene, and their mixtures. Macromolecules 2008, 41, 9919–9929. [Google Scholar] [CrossRef]
- Harmandaris, V.A.; Adhikari, N.P.; van der Vegt, N.; Kremer, K. Hierarchical modeling of polystyrene: From atomistic to coarse-grained simulations. Macromolecules 2006, 39, 6708–6719. [Google Scholar] [CrossRef]
- Harmandaris, V.A.; Reith, D.; van der Vegt, N.; Kremer, K. Comparison between coarse-graining models for polymer systems: Two mapping schemes for polystyrene. Macromol. Chem. Phys. 2007, 208, 2109–2120. [Google Scholar] [CrossRef]
- Fritz, D.; Harmandaris, V.A.; Kremer, K.; van der Vegt, N. Coarse-grained polymer melts based on isolated atomistic chains: Simulation of polystyrene of different tacticities. Macromolecules 2009, 42, 7579–7588. [Google Scholar] [CrossRef]
- Mulder, T.; Harmandaris, V.A.; Lyulin, A.V.; van der Vegt, N.; Vorselaars, B.; Michels, M. Equilibration and deformation of amorphous polystyrene: Scale-jumping simulational approach. Macromol. Theory Simul. 2008, 17, 290–300. [Google Scholar] [CrossRef]
- Mulder, T.; Harmandaris, V.A.; Lyulin, A.V.; van der Vegt, N.; Kremer, K.; Michels, M. Structural properties of atactic polystyrene of different thermal history obtained from a multiscale simulation. Macromolecules 2009, 42, 384–391. [Google Scholar] [CrossRef]
- Harmandaris, V.A.; Kremer, K. Predicting polymer dynamics at multiple length and time scales. Soft Matter 2009, 5, 3920–3926. [Google Scholar] [CrossRef]
- Harmandaris, V.A.; Kremer, K. Dynamics of polystyrene melts through hierarchical multiscale simulations. Macromolecules 2009, 42, 791–802. [Google Scholar] [CrossRef]
- Karimi-Varzaneh, H.A.; van der Vegt, N.; Müller-Plathe, F.; Carbone, P. How good are coarse-grained polymer models? A comparison for atactic polystyrene. ChemPhysChem 2012, 13, 3428–3439. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, G.K. An Introduction to Fluid Dynamics; Cambridge University Press: New York, NY, USA, 2000. [Google Scholar]
- Lyubimov, I.; Guenza, M.G. First-principle approach to rescale the dynamics of simulated coarse-grained macromolecular liquids. Phys. Rev. E 2011, 84. [Google Scholar] [CrossRef] [PubMed]
- Lyubimov, I.Y.; McCarty, J.; Clark, A.; Guenza, M.G. Analytical rescaling of polymer dynamics from mesoscale simulations. J. Chem. Phys. 2010, 132. [Google Scholar] [CrossRef] [PubMed]
- Lyubimov, I.Y.; Guenza, M.G. Theoretical reconstruction of realistic dynamics of highly coarse-grained cis-1,4-polybutadiene melts. J. Chem. Phys. 2013, 138. [Google Scholar] [CrossRef] [PubMed]
- Fritz, D.; Koschke, K.; Harmandaris, V.A.; van der Vegt, N.; Kremer, K. Multiscale modeling of soft matter: Scaling of dynamics. Phys. Chem. Chem. Phys. 2011, 13, 10412–10420. [Google Scholar] [CrossRef] [PubMed]
- Colmenero, J.; Arbe, A. Segmental dynamics in miscible polymer blends: Recent results and open questions. Soft Matter 2007, 3, 1474–1485. [Google Scholar] [CrossRef]
- Roland, C.M.; Ngai, K.L. Dynamical heterogeneity in a miscible polymer blend. Macromolecules 1991, 24, 2261–2265. [Google Scholar] [CrossRef]
- Harmandaris, V.A.; Kremer, K.; Floudas, G. Dynamic heterogeneity in fully miscible blends of polystyrene with oligostyrene. Phys. Rev. Lett. 2013, 110. [Google Scholar] [CrossRef] [PubMed]
- Louis, A.A. Beware of density dependent pair potentials. J. Phys. 2002, 14, 9187–9206. [Google Scholar] [CrossRef]
- Eslami, H.; Karimi-Varzaneh, H.A.; Müller-Plathe, F. Coarse-grained computer simulation of nanoconfined polyamide-6,6. Macromolecules 2011, 44, 3117–3128. [Google Scholar] [CrossRef]
- Bayramoglu, B.; Faller, R. Coarse-grained modeling of polystyrene in various environments by iterative Boltzmann inversion. Macromolecules 2012, 45, 9205–9219. [Google Scholar] [CrossRef]
- Fukunaga, H.; Takimoto, J.-I.; Doi, M. A coarse-graining procedure for flexible polymer chains with bonded and nonbonded interactions. J. Chem. Phys. 2002, 116, 8183–8190. [Google Scholar] [CrossRef]
- Carbone, P.; Varzaneh, H.; Chen, X.; Müller-Plathe, F. Transferability of coarse-grained force fields: The polymer case. J. Chem. Phys. 2008, 128. [Google Scholar] [CrossRef] [PubMed]
- Harmandaris, V.A.; Floudas, G.; Kremer, K. Temperature and pressure dependence of polystyrene dynamics through molecular dynamics simulations and experiments. Macromolecules 2011, 44, 393–402. [Google Scholar] [CrossRef]
- Strauch, T.; Yelash, L.; Paul, W. A coarse-graining procedure for polymer melts applied to 1,4-polybutadiene. Phys. Chem. Chem. Phys. 2009, 11, 1942–1948. [Google Scholar] [CrossRef] [PubMed]
- Vettorel, T.; Meyer, H. Coarse graining of short polythylene chains for studying polymer crystallization. J. Chem. Theory Comput. 2006, 2, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Yelash, L.; Müller, M.; Paul, W.; Binder, K. How well can coarse-grained models of real polymers describe their structure? The case of polybutadiene. J. Chem. Theory Comput. 2006, 2, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.-C.; Kulkarni, P.M.; Scott Shell, M.; Gary Leal, L. A test of systematic coarse-graining of molecular dynamics simulations: Thermodynamic properties. J. Chem. Phys. 2012, 137. [Google Scholar] [CrossRef] [PubMed]
- Baron, R.; Trzesniak, D.; de Vries, A.H.; Elsener, A.; Marrink, S.J.; van Gunsteren, W.F. Comparison of thermodynamic properties of coarse-grained and atomic-level simulation models. ChemPhysChem 2007, 8, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Betancourt, M.R.; Omovie, S.J. Pairwise energies for polypeptide coarse-grained models derived from atomic force fields. J. Chem. Phys. 2009, 130. [Google Scholar] [CrossRef] [PubMed]
- Mullinax, J.W.; Noid, W.G. Extended ensemble approach for deriving transferable coarse-grained potentials. J. Chem. Phys. 2009, 131. [Google Scholar] [CrossRef]
- Patrone, P.N.; Rosch, T.W.; Phelan, F.R., Jr. Bayesian calibration of coarse-grained forces: Efficiently addressing transferability. J. Chem. Phys. 2016, 144. [Google Scholar] [CrossRef] [PubMed]
- Unadkat, J.D.; Beal, S.L.; Sheiner, L.B. Bayesian calibration. Anal. Chim. Acta 1986, 181, 27–36. [Google Scholar] [CrossRef]
- Padding, J.T.; Briels, W.J. Uncrossability constraints in mesoscopic polymer melt simulations: Non-rouse behavior of C120H242. J. Chem. Phys. 2001, 115, 2846–2859. [Google Scholar] [CrossRef]
- Padding, J.T.; Briels, W.J. Time and length scales of polymer melts studied by coarse-grained molecular dynamics simulations. J. Chem. Phys. 2002, 117, 925–943. [Google Scholar] [CrossRef]
- Padding, J.T.; Briels, W.J.; Stukan, M.R.; Boek, E.S. Review of multi-scale particulate simulation of the rheology of wormlike micellar fluids. Soft Matter 2009, 5, 4367–4375. [Google Scholar] [CrossRef]
- Padding, J.T.; Briels, W.J. Coarse-grained molecular dynamics simulations of polymer melts in transient and steady shear flow. J. Chem. Phys. 2003, 118, 10276–10286. [Google Scholar] [CrossRef]
- Pérez-Aparicio, R.; Colmenero, J.; Alvarez, F.; Padding, J.T.; Briels, W.J. Chain dynamics of poly(ethylene-alt-propylene) melts by means of coarse-grained simulations based on atomistic molecular dynamics. J. Chem. Phys. 2010, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Padding, J.T.; Den Otter, W.K.; Briels, W.J. Coarse-grained simulations of moderately entangled star polyethylene melts. J. Chem. Phys. 2013, 138. [Google Scholar] [CrossRef] [PubMed]
- Maiti, A.; McGrother, S. Bead-bead interaction parameters in dissipative particle dynamics: Relation to bead-size, solubility parameter, and surface tension. J. Chem. Phys. 2004, 120, 1594–1601. [Google Scholar] [CrossRef] [PubMed]
- Kacar, G.; Peters, E.; de With, G. A generalized method for parameterization of dissipative particle dynamics for variable bead volumes. Europhys. Lett. 2013, 102. [Google Scholar] [CrossRef]
- Johnston, K.; Harmandaris, V. Hierarchical multiscale modeling of polymer-solid interfaces: Atomistic to coarse-grained description and structural and conformational properties of polystyrene-gold systems. Macromolecules 2013, 46, 5741–5750. [Google Scholar] [CrossRef]
- Pan, G.; Manke, C.W. Developments toward simulation of entangled polymer melts by dissipative particle dynamics (DPD). Int. J. Mod. Phys. B 2003, 17, 231–235. [Google Scholar] [CrossRef]
- Sirk, T.W.; Slizoberg, Y.R.; Brennan, J.K.; Lisal, M.; Andzelm, J.W. An enhanced entangled polymer model for dissipative particle dynamics. J. Chem. Phys. 2012, 136, 134903. [Google Scholar] [CrossRef] [PubMed]
- Nikunen, P.; Vattulainen, I.; Karttunen, M. Reptational dynamics in dissipative particle dynamics simulations of polymer melts. Phys. Rev. E 2007, 75, 36713. [Google Scholar] [CrossRef] [PubMed]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon Press: Oxford, UK, 1986. [Google Scholar]
- Gooneie, A.; Schuschnigg, S.; Holzer, C. Coupled orientation and stretching of chains in mesoscale models of polydisperse linear polymers in startup of steady shear flow simulations. Macromol. Theory Simul. 2016, 25, 170–186. [Google Scholar] [CrossRef]
- Murat, M.; Kremer, K. From many monomers to many polymers: Soft ellipsoid model for polymer melts and mixtures. J. Chem. Phys. 1998, 108, 4340–4348. [Google Scholar] [CrossRef]
- D’Adamo, G.; Pelissetto, A.; Pierleoni, C. Coarse-graining strategies in polymer solutions. Soft Matter 2012, 8, 5151–5167. [Google Scholar] [CrossRef]
- D’Adamo, G.; Pelissetto, A.; Pierleoni, C. Polymers as compressible soft spheres. J. Chem. Phys. 2012, 136. [Google Scholar] [CrossRef] [PubMed]
- Vettorel, T.; Besold, G.; Kremer, K. Fluctuating soft-sphere approach to coarse-graining of polymer models. Soft Matter 2010, 6, 2282–2292. [Google Scholar] [CrossRef]
- Zhang, G.; Daoulas, K.C.; Kremer, K. A New Coarse Grained Particle-To-Mesh Scheme for Modeling Soft Matter. Macromol. Chem. Phys. 2013, 214, 214–224. [Google Scholar] [CrossRef]
- Kindt, P.; Briels, W.J. A single particle model to simulate the dynamics of entangled polymer melts. J. Chem. Phys. 2007, 127. [Google Scholar] [CrossRef] [PubMed]
- Briels, W.J. Transient forces in flowing soft matter. Soft Matter 2009, 5, 4401–4411. [Google Scholar] [CrossRef]
- Zhu, Y.-L.; Liu, H.; Lu, Z.-Y. A highly coarse-grained model to simulate entangled polymer melts. J. Chem. Phys. 2012, 136. [Google Scholar] [CrossRef] [PubMed]
- Sprakel, J.; Padding, J.T.; Briels, W.J. Transient forces and non-equilibrium states in sheared polymer networks. Europhys. Lett. 2011, 93. [Google Scholar] [CrossRef]
- Sprakel, J.; Spruijt, E.; van der Gucht, J.; Padding, J.T.; Briels, W.J. Failure-mode transition in transient polymer networks with particle-based simulations. Soft Matter 2009, 5, 4748–4756. [Google Scholar] [CrossRef]
- Savin, T.; Briels, W.J.; öttinger, H.C. Thermodynamic formulation of flowing soft matter with transient forces. Rheol. Acta 2013, 52, 23–32. [Google Scholar] [CrossRef]
- Padding, J.T.; van Ruymbeke, E.; Vlassopoulos, D.; Briels, W.J. Computer simulation of the rheology of concentrated star polymer suspensions. Rheol. Acta 2010, 49, 473–484. [Google Scholar] [CrossRef]
- Padding, J.T.; Mohite, L.V.; Auhl, D.; Briels, W.J.; Bailly, C. Mesoscale modeling of the rheology of pressure sensitive adhesives through inclusion of transient forces. Soft Matter 2011, 7, 5036–5046. [Google Scholar] [CrossRef]
- Padding, J.T.; Mohite, L.V.; Auhl, D.; Schweizer, T.; Briels, W.J.; Bailly, C. Quantitative mesoscale modeling of the oscillatory and transient shear rheology and the extensional rheology of pressure sensitive adhesives. Soft Matter 2012, 8, 7967–7981. [Google Scholar] [CrossRef]
- Schweizer, K.S.; Curro, J.G. Integral equation theories of the structure, thermodynamics, and phase transitions of polymer fluids. Adv. Chem. Phys. 1997, 98, 1–142. [Google Scholar]
- McCarty, J.; Guenza, M.G. Multiscale modeling of binary polymer mixtures: Scale bridging in the athermal and thermal regime. J. Chem. Phys. 2010, 133. [Google Scholar] [CrossRef] [PubMed]
- McCarty, J.; Lyubimov, I.Y.; Guenza, M.G. Effective soft-core potentials and mesoscopic simulations of binary polymer mixtures. Macromolecules 2010, 43, 3964–3979. [Google Scholar] [CrossRef]
- McCarty, J.; Lyubimov, I.Y.; Guenza, M.G. Multiscale modeling of coarse-grained macromolecular liquids. J. Phys. Chem. B 2009, 113, 11876–11886. [Google Scholar] [CrossRef] [PubMed]
- Guenza, M.G. Theoretical models for bridging timescales in polymer dynamics. J. Phys.: Condens. Matter 2007, 20, 033101. [Google Scholar] [CrossRef]
- Clark, A.J.; McCarty, J.; Lyubimov, I.Y.; Guenza, M.G. Thermodynamic consistency in variable-level coarse graining of polymeric liquids. Phys. Rev. Lett. 2012, 109. [Google Scholar] [CrossRef] [PubMed]
- McCarty, J.; Clark, A.J.; Lyubimov, I.Y.; Guenza, M.G. Thermodynamic consistency between analytic integral equation theory and coarse-grained molecular dynamics simulations of homopolymer melts. Macromolecules 2012, 45, 8482–8493. [Google Scholar] [CrossRef]
- Santangelo, G.; Di Matteo, A.; Müller-Plathe, F.; Milano, G. From mesoscale back to atomistic models: A fast reverse-mapping procedure for vinyl polymer chains. J. Phys. Chem. B 2007, 111, 2765–2773. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.J.; Rottler, J. Using soft potentials for the simulation of block copolymer morphologies. Macromol. Theory Simul. 2014, 23, 401–409. [Google Scholar] [CrossRef]
- Sliozberg, Y.R.; Kröger, M.; Chantawansri, T.L. Fast equilibration protocol for million atom systems of highly entangled linear polyethylene chains. J. Chem. Phys. 2016, 144. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Carbone, P.; Santangelo, G.; Di Matteo, A.; Milano, G.; Müller-Plathe, F. Backmapping coarse-grained polymer models under sheared nonequilibrium conditions. Phys. Chem. Chem. Phys. 2009, 11, 1977–1988. [Google Scholar] [CrossRef] [PubMed]
- Carbone, P.; Ali Karimi-Varzaneh, H.; Müller-Plathe, F. Fine-graining without coarse-graining: An easy and fast way to equilibrate dense polymer melts. Faraday Discuss. 2009, 144, 25–42. [Google Scholar] [CrossRef]
- Tschöp, W.; Kremer, K.; Halm, O.; Batoulis, J.; Bürger, T. Simulation of polymer melts. II. From coarse-grained models back to atomistic description. Acta Polym. 1998, 49, 75–79. [Google Scholar] [CrossRef]
- Kotelyanskii, M.; Wagner, N.J.; Paulaitis, M.E. Building large amorphous polymer structures: Atomistic simulation of glassy polystyrene. Macromolecules 1996, 29, 8497–8506. [Google Scholar] [CrossRef]
- Harmandaris, V.A.; Mavrantzas, V.G.; Theodorou, D.N. Atomistic molecular dynamics simulation of stress relaxation upon cessation of steady-state uniaxial elongational flow. Macromolecules 2000, 33, 8062–8076. [Google Scholar] [CrossRef]
- Queyroy, S.; Neyertz, S.; Brown, D.; Müller-Plathe, F. Preparing relaxed systems of amorphous polymers by multiscale simulation: Application to cellulose. Macromolecules 2004, 37, 7338–7350. [Google Scholar] [CrossRef]
- Hess, B.; León, S.; van der Vegt, N.; Kremer, K. Long time atomistic polymer trajectories from coarse grained simulations: Bisphenol-A polycarbonate. Soft Matter 2006, 2, 409–414. [Google Scholar] [CrossRef]
- Karimi-Varzaneh, H.A.; Carbone, P.; Müller-Plathe, F. Fast dynamics in coarse-grained polymer models: The effect of the hydrogen bonds. J. Chem. Phys. 2008, 129. [Google Scholar] [CrossRef] [PubMed]
- Wu, C. Multiscale simulations of the structure and dynamics of stereoregular poly(methyl methacrylate)s. J. Mol. Model. 2014, 20. [Google Scholar] [CrossRef] [PubMed]
- Handgraaf, J.-W.; Serral Gracia, R.; Nath, S.K.; Chen, Z.; Chou, S.-H.; Ross, R.B.; Schultz, N.E.; Fraaije, J. A multiscale modeling protocol to generate realistic polymer surfaces. Macromolecules 2011, 44, 1053–1061. [Google Scholar] [CrossRef]
- Bleha, T.; Gajdos, J.; Karasz, F.E. Energetics of strain-induced conformational transitions in polymethylene chains. Macromolecules 1990, 23, 4076–4082. [Google Scholar] [CrossRef]
- Ghanbari, A.; Böhm, M.C.; Müller-Plathe, F. A simple reverse mapping procedure for coarse-grained polymer models with rigid side groups. Macromolecules 2011, 44, 5520–5526. [Google Scholar] [CrossRef]
- Abraham, F.F.; Broughton, J.Q.; Bernstein, N.; Kaxiras, E. Spanning the length scales in dynamic simulation. Comput. Phys. 1998, 12, 538–546. [Google Scholar] [CrossRef]
- Stillinger, F.H.; Weber, T.A. Computer simulation of local order in condensed phases of silicon. Phys. Rev. B Condens. Matter Mater. Phys. 1985, 31, 5262–5271. [Google Scholar] [CrossRef]
- Rudd, R.E.; Broughton, J.Q. Concurrent coupling of length scales in solid state systems. Phys. Status Solidi B 2000, 217, 251–291. [Google Scholar] [CrossRef]
- Nakano, A.; Bachlechner, M.E.; Kalia, R.K.; Lidorikis, E.; Vashishta, P.; Voyiadjis, G.Z.; Campbell, T.J.; Ogata, S.; Shimojo, F. Multiscale simulation of nanosystems. Comput. Sci. Eng. 2001, 3, 56–66. [Google Scholar] [CrossRef]
- Broughton, J.Q.; Abraham, F.F.; Bernstein, N.; Kaxiras, E. Concurrent coupling of length scales: Methodology and application. Phys. Rev. B Condens. Matter Mater. Phys. 1999, 60, 2391–2403. [Google Scholar] [CrossRef]
- Abraham, F.F.; Broughton, J.Q.; Bernstein, N.; Kaxiras, E. Spanning the continuum to quantum length scales in a dynamic simulation of brittle fracture. Europhys. Lett. 1998, 44, 783–787. [Google Scholar] [CrossRef]
- Curtin, W.A.; Miller, R.E. Atomistic/continuum coupling in computational materials science. Model. Simul. Mater. Sci. Eng. 2003, 11, R33–R68. [Google Scholar] [CrossRef]
- Miller, R.E.; Tadmor, E.B. A unified framework and performance benchmark of fourteen multiscale atomistic/continuum coupling methods. Model. Simul. Mater. Sci. Eng. 2009, 17, 53001. [Google Scholar] [CrossRef]
- Badia, S.; Parks, M.; Bochev, P.; Gunzburger, M.; Lehouc, Q. On atomistic-to-continuum coupling by blending. Multiscale Model. Simul. 2008, 7, 381–406. [Google Scholar] [CrossRef]
- Shenoy, V.B.; Miller, R.; Tadmor, E.B.; Rodney, D.; Phillips, R.; Ortiz, M. An adaptive finite element approach to atomic-scale mechanics—The quasicontinuum method. J. Mech. Phys. Solids 1999, 47, 611–642. [Google Scholar] [CrossRef]
- Li, X.; Ming, P. On the effect of ghost force in the quasicontinuum method: Dynamic problems in one dimension. Commun. Comput. Phys. 2014, 15, 647–676. [Google Scholar] [CrossRef]
- Ortner, C.; Zhang, L. Atomistic/continuum blending with ghost force correction. Siam J. Sci. Comput. 2016, 38, A346–A375. [Google Scholar] [CrossRef]
- Shimokawa, T.; Mortensen, J.J.; Schiøtz, J.; Jacobsen, K.W. Matching conditions in the quasicontinuum method: Removal of the error introduced at the interface between the coarse-grained and fully atomistic region. Phys. Rev. B Condens. Matter Mater. Phys. 2004, 69, 214104. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Yang, J.Z. Uniform accuracy of the quasicontinuum method. Phys. Rev. B 2006, 74. [Google Scholar] [CrossRef]
- Klein, P.A.; Zimmerman, J.A. Coupled atomistic-continuum simulations using arbitrary overlapping domains. J. Comput. Phys. 2006, 213, 86–116. [Google Scholar] [CrossRef]
- Tadmor, E.B.; Ortiz, M.; Phillips, R. Quasicontinuum analysis of defects in solids. Philos. Mag. A Phys. Condens. Matter Struct. Defects Mech. Prop. 1996, 73, 1529–1563. [Google Scholar] [CrossRef]
- Tadmor, E.B.; Phillips, R.; Ortiz, M. Mixed atomistic and continuum models of deformation in solids. Langmuir 1996, 12, 4529–4532. [Google Scholar] [CrossRef]
- Rudd, R.E.; Broughton, J.Q. Coarse-grained molecular dynamics and the atomic limit of finite elements. Phys. Rev. B 1998, 58, R5893–R5896. [Google Scholar] [CrossRef]
- Shenoy, V.B.; Miller, R.; Tadmor, E.B.; Phillips, R.; Ortiz, M. Quasicontinuum models of interfacial structure and deformation. Phys. Rev. Lett. 1998, 80, 742–745. [Google Scholar] [CrossRef]
- Rodney, D.; Martin, G. Dislocation pinning by small interstitial loops: A molecular dynamics study. Phys. Rev. Lett. 1999, 82, 3272–3275. [Google Scholar] [CrossRef]
- Rodney, D.; Phillips, R. Structure and Strength of Dislocation Junctions: An Atomic Level Analysis. Phys. Rev. Lett. 1999, 82, 1704–1707. [Google Scholar] [CrossRef]
- Tadmor, E.B.; Smith, G.S.; Bernstein, N.; Kaxiras, E. Mixed finite element and atomistic formulation for complex crystals. Phys. Rev. B 1999, 59, 235–245. [Google Scholar] [CrossRef]
- Miller, R.; Ortiz, M.; Phillips, R.; Shenoy, V.; Tadmor, E.B. Quasicontinuum models of fracture and plasticity. Eng. Fract. Mech. 1998, 61, 427–444. [Google Scholar] [CrossRef]
- Miller, R.; Tadmor, E.B.; Phillips, R.; Ortiz, M. Quasicontinuum simulation of fracture at the atomic scale. Model. Simul. Mater. Sci. Eng. 1998, 6, 607–638. [Google Scholar] [CrossRef]
- Tadmor, E.B.; Miller, R.; Phillips, R.; Ortiz, M. Nanoindentation and incipient plasticity. J. Mater. Res. 1999, 14, 2233–2250. [Google Scholar] [CrossRef]
- Binder, A.; Luskin, M.; Perez, D.; Voter, A.F. Analysis of transition state theory rates upon spatial coarse-graining. Multiscale Model. Simul. 2015, 13, 890–915. [Google Scholar] [CrossRef]
- Eidel, B.; Stukowski, A. A variational formulation of the quasicontinuum method based on energy sampling in clusters. J. Mech. Phys. Solids 2009, 57, 87–108. [Google Scholar] [CrossRef]
- Miller, R.E.; Tadmor, E.B. Hybrid continuum mechanics and atomistic methods for simulating materials deformation and failure. MRS Bull. 2007, 32, 920–926. [Google Scholar] [CrossRef]
- Iacobellis, V.; Behdinan, K. Comparison of concurrent multiscale methods in the application of fracture in nickel. J. Appl. Mech. Trans. ASME 2013, 80. [Google Scholar] [CrossRef]
- Xiao, S.P.; Belytschko, T. A bridging domain method for coupling continua with molecular dynamics. Comput. Methods Appl. Mech. Eng. 2004, 193, 1645–1669. [Google Scholar] [CrossRef]
- Datta, D.K.; Picu, C.; Shephard, M.S. Composite Grid Atomistic Continuum Method: An Adaptive Approach to Bridge Continuum with Atomistic Analysis. Int. J. Multiscale Comput. Eng. 2004, 2, 71–90. [Google Scholar] [CrossRef]
- Rudd, R.E.; Broughton, J.Q. Coarse-grained molecular dynamics: Nonlinear finite elements and finite temperature. Phys. Rev. B Condens. Matter Mater. Phys. 2005, 72. [Google Scholar] [CrossRef]
- Curtarolo, S.; Ceder, G. Dynamics of an inhomogeneously coarse grained multiscale system. Phys. Rev. Lett. 2002, 88, 2555041–2555044. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, L.M.; Tadmor, E.B.; Miller, R.E.; Phillips, R. Finite-temperature quasicontinuum: Molecular dynamics without all the atoms. Phys. Rev. Lett. 2005, 95. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Zhao, H.; Li, G.; Aluru, N.R. Finite-temperature quasicontinuum method for multiscale analysis of silicon nanostructures. Phys. Rev. B Condens. Matter Mater. Phys. 2006, 74. [Google Scholar] [CrossRef]
- Marian, J.; Venturini, G.; Hansen, B.L.; Knap, J.; Ortiz, M.; Campbell, G.H. Finite-temperature extension of the quasicontinuum method using Langevin dynamics: Entropy losses and analysis of errors. Model. Simul. Mater. Sci. Eng. 2010, 18. [Google Scholar] [CrossRef]
- Kohlhoff, S.; Gumbsch, P.; Fischmeister, H.F. Crack propagation in b.c.c. crystals studied with a combined finite-element and atomistic model. Philos. Mag. A Phys. Condens. Matter Struct. Defects Mech. Prop. 1991, 64, 851–878. [Google Scholar] [CrossRef]
- Shilkrot, L.E.; Miller, R.E.; Curtin, W.A. Coupled atomistic and discrete dislocation plasticity. Phys. Rev. Lett. 2002, 89, 255011–255014. [Google Scholar] [CrossRef] [PubMed]
- Shilkrot, L.E.; Miller, R.E.; Curtin, W.A. Multiscale plasticity modeling: Coupled atomistics and discrete dislocation mechanics. J. Mech. Phys. Solids 2004, 52, 755–787. [Google Scholar] [CrossRef]
- Luan, B.Q.; Hyun, S.; Molinari, J.F.; Bernstein, N.; Robbins, M.O. Multiscale modeling of two-dimensional contacts. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2006, 74. [Google Scholar] [CrossRef] [PubMed]
- Badia, S.; Bochev, P.; Lehoucq, R.; Parks, M.L.; Fish, J.; Nuggehally, M.A.; Gunzburger, M. A force-based blending model foratomistic-to-continuum coupling. Int. J. Multiscale Comput. Eng. 2007, 5, 387–406. [Google Scholar] [CrossRef]
- Fish, J.; Nuggehally, M.A.; Shephard, M.S.; Picu, C.R.; Badia, S.; Parks, M.L.; Gunzburger, M. Concurrent AtC coupling based on a blend of the continuum stress and the atomistic force. Comput. Methods Appl. Mech. Eng. 2007, 196, 4548–4560. [Google Scholar] [CrossRef]
- Hughes, T.; Feijóo, G.R.; Mazzei, L.; Quincy, J.-B. The variational multiscale method—A paradigm for computational mechanics. Comput. Methods Appl. Mech. Eng. 1998, 166, 3–24. [Google Scholar] [CrossRef]
- Qian, D.; Wagner, G.J.; Liu, W.K. A multiscale projection method for the analysis of carbon nanotubes. Comput. Methods Appl. Mech. Eng. 2004, 193, 1603–1632. [Google Scholar] [CrossRef]
- Wagner, G.J.; Liu, W.K. Coupling of atomistic and continuum simulations using a bridging scale decomposition. J. Comput. Phys. 2003, 190, 249–274. [Google Scholar] [CrossRef]
- Park, H.S.; Liu, W.K. An introduction and tutorial on multiple-scale analysis in solids. Comput. Methods Appl. Mech. Eng. 2004, 193, 1733–1772. [Google Scholar] [CrossRef]
- Tan, V.; Zeng, X.S.; Deng, M.; Lim, K.M.; Tay, T.E. Multiscale modeling of polymers—The Pseudo Amorphous Cell. Comput. Methods Appl. Mech. Eng. 2008, 197, 536–554. [Google Scholar] [CrossRef]
- Theodorou, D.N.; Suter, U.W. Detailed molecular structure of a vinyl polymer glass. Macromolecules 1985, 18, 1467–1478. [Google Scholar] [CrossRef]
- Theodorou, D.N.; Suter, U.W. Geometrical considerations in model systems with periodic boundaries. J. Chem. Phys. 1985, 82, 955–966. [Google Scholar] [CrossRef]
- Su, Z.C.; Tay, T.-E.; Chen, Y.; Tan, V. Multiscale modeling for amorphous materials—Mapping atomistic displacements to macroscopic deformation. Intl. J. Appl. Mech. 2012, 4. [Google Scholar] [CrossRef]
- Codina, R. Stabilization of incompressibility and convection through orthogonal sub-scales in finite element methods. Comput. Methods Appl. Mech. Eng. 2000, 190, 1579–1599. [Google Scholar] [CrossRef]
- Castillo, E.; Codina, R. Stabilized stress-velocity-pressure finite element formulations of the Navier-Stokes problem for fluids with non-linear viscosity. Comput. Methods Appl. Mech. Eng. 2014, 279, 554–578. [Google Scholar] [CrossRef]
- Castillo, E.; Codina, R. Variational multi-scale stabilized formulations for the stationary three-field incompressible viscoelastic flow problem. Comput. Methods Appl. Mech. Eng. 2014, 279, 579–605. [Google Scholar] [CrossRef]
- Koo, B.; Subramanian, N.; Chattopadhyay, A. Molecular dynamics study of brittle fracture in epoxy-based thermoset polymer. Compos. Part B 2016, 95, 433–439. [Google Scholar] [CrossRef]
- Büyüköztürk, O.; Buehler, M.J.; Lau, D.; Tuakta, C. Structural solution using molecular dynamics: Fundamentals and a case study of epoxy-silica interface. Int. J. Solids Struct. 2011, 48, 2131–2140. [Google Scholar] [CrossRef]
- Jo, W.H.; Yang, J.S. Molecular simulation approaches for multiphase polymer systems. In Molecular Simulation Fracture Gel Theory; Springer: Berlin, Germany, 2002. [Google Scholar]
- Li, C.; Chou, T.-W. Elastic moduli of multi-walled carbon nanotubes and the effect of van der Waals forces. Compos. Sci. Technol. 2003, 63, 1517–1524. [Google Scholar] [CrossRef]
- Li, C.; Chou, T.-W. Multiscale modeling of compressive behavior of carbon nanotube/polymer composites. Compos. Sci. Technol. 2006, 66, 2409–2414. [Google Scholar] [CrossRef]
- Montazeri, A.; Naghdabadi, R. Study the effect of viscoelastic matrix model on the stability of CNT/polymer composites by multiscale modeling. Polym. Compos. 2009, 30, 1545–1551. [Google Scholar] [CrossRef]
- De, S.; Fish, J.; Shephard, M.S.; Keblinski, P.; Kumar, S.K. Multiscale modeling of polymer rheology. Phys. Rev. E 2006, 74, 30801. [Google Scholar] [CrossRef] [PubMed]
- Guenza, M.G. Advancements in multi scale modeling: Adaptive resolution simulations and related issues. Eur. Phys. J. 2015, 224, 2491–2495. [Google Scholar] [CrossRef]
- Kreis, K.; Fogarty, A.C.; Kremer, K.; Potestio, R. Advantages and challenges in coupling an ideal gas to atomistic models in adaptive resolution simulations. Eur. Phys. J. 2015, 224, 2289–2304. [Google Scholar] [CrossRef]
- Zavadlav, J.; Podgornik, R.; Praprotnik, M. Adaptive resolution simulation of a DNA molecule in salt solution. J. Chem. Theory Comput. 2015, 11, 5035–5044. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Agarwal, A. Adaptive resolution simulation in equilibrium and beyond. Eur. Phys. J. 2015, 224, 2269–2287. [Google Scholar] [CrossRef]
- Youn Park, J.; Park, C.-H.; Shin Park, J.; Kong, K.-J.; Chang, H.; Im, S. Multiscale computations for carbon nanotubes based on a hybrid QM/QC (quantum mechanical and quasicontinuum) approach. J. Mech. Phys. Solids 2010, 58, 86–102. [Google Scholar] [CrossRef]
- Praprotnik, M.; Delle Site, L.; Kremer, K. A macromolecule in a solvent: Adaptive resolution molecular dynamics simulation. J. Chem. Phys. 2007, 126, 134902. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.O.; Srinivas, G.; Klein, M.L. Incorporating a hydrophobic solid into a coarse grain liquid framework: Graphite in an aqueous amphiphilic environment. J. Chem. Phys. 2005, 123, 124907. [Google Scholar] [CrossRef] [PubMed]
- Praprotnik, M.; Delle Site, L.; Kremer, K. Adaptive resolution molecular-dynamics simulation: Changing the degrees of freedom on the fly. J. Chem. Phys. 2005, 123. [Google Scholar] [CrossRef] [PubMed]
- Praprotnik, M.; Delle Site, L.; Kremer, K. Adaptive resolution scheme for efficient hybrid atomistic-mesoscale molecular dynamics simulations of dense liquids. Phys. Rev. E 2006, 73. [Google Scholar] [CrossRef] [PubMed]
- Praprotnik, M.; Matysiak, S.; Site, L.D.; Kremer, K.; Clementi, C. Adaptive resolution simulation of liquid water. J. Phys. 2007, 19. [Google Scholar] [CrossRef]
- Praprotnik, M.; Site, L.D.; Kremer, K. Multiscale simulation of soft matter: From scale bridging to adaptive resolution. Annu. Rev. Phys. Chem. 2008, 59, 545–571. [Google Scholar] [CrossRef] [PubMed]
- Praprotnik, M.; Poblete, S.; Delle Site, L.; Kremer, K. Comment on “adaptive multiscale molecular dynamics of macromolecular fluids”. Phys. Rev. Lett. 2011, 107. [Google Scholar] [CrossRef] [PubMed]
- Delle Site, L.; Leon, S.; Kremer, K. BPA-PC on a Ni(111) Surface: The Interplay between Adsorption Energy and Conformational Entropy for Different Chain-End Modifications. J. Am. Chem. Soc. 2004, 126, 2944–2955. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, S.; Poblete, S.; Junghans, C.; Ciccotti, G.; Delle Site, L.; Kremer, K. Adaptive resolution molecular dynamics simulation through coupling to an internal particle reservoir. Phys. Rev. Lett. 2012, 108. [Google Scholar] [CrossRef] [PubMed]
- Delle Site, L. Some fundamental problems for an energy-conserving adaptive-resolution molecular dynamics scheme. Phys. Rev. E 2007, 76. [Google Scholar] [CrossRef] [PubMed]
- Poma, A.B.; Delle Site, L. Classical to path-integral adaptive resolution in molecular simulation: Towards a smooth quantum-classical coupling. Phys. Rev. Lett. 2010, 104. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, S.; Junghans, C.; Kremer, K. Structure formation of toluene around C60: Implementation of the adaptive resolution scheme (AdResS) into GROMACS. J. Chem. Theory Comput. 2012, 8, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Poblete, S.; Praprotnik, M.; Kremer, K.; Delle Site, L. Coupling different levels of resolution in molecular simulations. J. Chem. Phys. 2010, 132. [Google Scholar] [CrossRef] [PubMed]
- Mukherji, D.; van der Vegt, N.; Kremer, K. Preferential solvation of triglycine in aqueous urea: An open boundary simulation approach. J. Chem. Theory Comput. 2012, 8, 3536–3541. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, B.P., Jr.; Junghans, C.; Kremer, K.; Clementi, C.; Site, L.D. Communication: On the locality of Hydrogen bond networks at hydrophobic interfaces. J. Chem. Phys. 2010, 133, 221101. [Google Scholar] [CrossRef] [PubMed]
- Poma, A.B.; Site, L.D. Adaptive resolution simulation of liquid para-hydrogen: Testing the robustness of the quantum-classical adaptive coupling. Phys. Chem. Chem. Phys. 2011, 13, 10510–10519. [Google Scholar] [CrossRef] [PubMed]
- Potestio, R.; Delle Site, L. Quantum locality and equilibrium properties in low-temperature parahydrogen: A multiscale simulation study. J. Chem. Phys. 2012, 136, 54101. [Google Scholar] [CrossRef] [PubMed]
- Mukherji, D.; van der Vegt, N.; Kremer, K.; Delle Site, L. Kirkwood-buff analysis of liquid mixtures in an open boundary simulation. J. Chem. Theory Comput. 2012, 8, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Mukherji, D.; Kremer, K. Coil-globule-coil transition of PNIPAm in aqueous methanol: Coupling all-atom simulations to semi-grand canonical coarse-grained reservoir. Macromolecules 2013, 46, 9158–9163. [Google Scholar] [CrossRef]
- Agarwal, A.; Zhu, J.; Hartmann, C.; Wang, H.; Site, L.D. Molecular dynamics in a grand ensemble: Bergmann-Lebowitz model and adaptive resolution simulation. New J. Phys. 2015, 17. [Google Scholar] [CrossRef]
- Wang, H.; Hartmann, C.; Schütte, C.; Site, L.D. Grand-canonical-like molecular-dynamics simulations by using an adaptive-resolution technique. Phys. Rev. X 2013, 3. [Google Scholar] [CrossRef]
- Nielsen, S.O.; Moore, P.B.; Ensing, B. Adaptive multiscale molecular dynamics of macromolecular fluids. Phys. Rev. Lett. 2010, 105. [Google Scholar] [CrossRef] [PubMed]
- Heyden, A.; Truhlar, D.G. Conservative algorithm for an adaptive change of resolution in mixed atomistic/ coarse-grained multiscale simulations. J. Chem. Theory Comput. 2008, 4, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Heyden, A. Solving the equations of motion for mixed atomistic and coarse-grained systems. Mol. Simul. 2009, 35, 962–973. [Google Scholar] [CrossRef]
- Potestio, R.; Fritsch, S.; Español, P.; Delgado-Buscalioni, R.; Kremer, K.; Everaers, R.; Donadio, D. Hamiltonian adaptive resolution simulation for molecular liquids. Phys. Rev. Lett. 2013, 110. [Google Scholar] [CrossRef] [PubMed]
- Potestio, R.; Español, P.; Delgado-Buscalioni, R.; Everaers, R.; Kremer, K.; Donadio, D. Monte carlo adaptive resolution simulation of multicomponent molecular liquids. Phys. Rev. Lett. 2013, 111. [Google Scholar] [CrossRef] [PubMed]
- Voter, A.F. A method for accelerating the molecular dynamics simulation of infrequent events. J. Chem. Phys. 1996, 106, 4665–4677. [Google Scholar] [CrossRef]
- Voter, A.F. Hyperdynamics: Accelerated molecular dynamics of infrequent events. Phys. Rev. Lett. 1997, 78, 3908–3911. [Google Scholar] [CrossRef]
- Voter, A.F.; Montalenti, F.; Germann, T.C. Extending the time scale in atomistic simulation of materials. Annu. Rev. Mater. Sci. 2002, 32, 321–346. [Google Scholar] [CrossRef]
- Voter, A.F. Parallel replica method for dynamics of infrequent events. Phys. Rev. B 1998, 57, R13985–R13988. [Google Scholar] [CrossRef]
- Sørensen, M.R.; Voter, A.F. Temperature-accelerated dynamics for simulation of infrequent events. J. Chem. Phys. 2000, 112, 9599–9606. [Google Scholar] [CrossRef]
- Hänggi, P.; Talkner, P.; Borkovec, M. Reaction-rate theory: Fifty years after Kramers. Rev. Mod. Phys. 1990, 62, 251–341. [Google Scholar] [CrossRef]
- Elber, R.; Karplus, M. A method for determining reaction paths in large molecules: Application to myoglobin. Chem. Phys. Lett. 1987, 139, 375–380. [Google Scholar] [CrossRef]
- Elber, R.; Karplus, M. Multiple conformational states of proteins: A molecular dynamics analysis of myoglobin. Science 1987, 235, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Elber, R.; Ghosh, A.; Cárdenas, A. Long time dynamics of complex systems. Acc. Chem. Res. 2002, 35, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Dellago, C.; Bolhuis, P.G.; Csajka, F.S.; Chandler, D. Transition path sampling and the calculation of rate constants. J. Chem. Phys. 1998, 108, 1964–1977. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L., III; MacKerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Christen, M.; van Gunsteren, W.F. On searching in, sampling of, and dynamically moving through conformational space of biomolecular systems: A review. J. Comput. Chem. 2008, 29, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Escobedo, F.A.; Borrero, E.E.; Araque, J.C. Transition path sampling and forward flux sampling. Applications to biological systems. J. Phys. Condens. Matter 2009, 21. [Google Scholar] [CrossRef] [PubMed]
- Klenin, K.; Strodel, B.; Wales, D.J.; Wenzel, W. Modelling proteins: Conformational sampling and reconstruction of folding kinetics. Biochim. Biophys. Acta Proteins Proteomics 2011, 1814, 977–1000. [Google Scholar] [CrossRef] [PubMed]
- E, W.; Ren, W.; Vanden-Eijnden, E. Finite temperature string method for the study of rare events. J. Phys. Chem. B 2005, 109, 6688–6693. [Google Scholar] [CrossRef] [PubMed]
- Vanden-Eijnden, E.; Ren, W.; Vanden-Eijnden, E. Transition pathways in complex systems: Reaction coordinates, isocommittor surfaces, and transition tubes. Chem. Phys. Lett. 2005, 413, 242–247. [Google Scholar]
- Weinan, E.; Vanden-Eijnden, E. Transition-path theory and path-finding algorithms for the study of rare events. Annu. Rev. Phys. Chem. 2010, 61, 391–420. [Google Scholar]
- Ren, W.; Vanden-Eijnden, E.; Maragakis, P.; Vanden-Eijnden, E. Transition pathways in complex systems: Application of the finite-temperature string method to the alanine dipeptide. J. Chem. Phys. 2005, 123, 134109. [Google Scholar] [CrossRef] [PubMed]
- Passerone, D.; Parrinello, M. Action-derived molecular dynamics in the study of rare events. Phys. Rev. Lett. 2001, 87, 108302. [Google Scholar] [CrossRef] [PubMed]
- Branduardi, D.; Gervasio, F.L.; Parrinello, M. From A to B in free energy space. J. Chem. Phys. 2007, 126. [Google Scholar] [CrossRef] [PubMed]
- Dellago, C.; Bolhuis, P.G. Transition path sampling and other advanced simulation techniques for rare events. In Advanced Computer Simulation Approaches for Soft Matter Sciences III; Springer: Berlin, Germany, 2009. [Google Scholar]
- Dellago, C.; Bolhuis, P.G.; Geissler, P.L. Transition path sampling. Annu. Rev. Phys. Chem. 2002, 53, 291–318. [Google Scholar]
- Schwetlick, H.; Zimmer, J. Calculation of long time classical trajectories: Algorithmic treatment and applications for molecular systems. J. Chem. Phys. 2009, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wales, D.J. Discrete path sampling. Mol. Phys. 2002, 100, 3285–3305. [Google Scholar] [CrossRef]
- Zaloj, V.; Elber, R. Parallel computations of molecular dynamics trajectories using the stochastic path approach. Comput. Phys. Commun. 2000, 128, 118–127. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed]
- Kamerlin, S.; Vicatos, S.; Dryga, A.; Warshel, A. Coarse-grained (multiscale) simulations in studies of biophysical and chemical systems. Annu. Rev. Phys. Chem. 2011, 62, 41–64. [Google Scholar] [CrossRef] [PubMed]
- Neri, M.; Anselmi, C.; Cascella, M.; Maritan, A.; Carloni, P. Coarse-grained model of proteins incorporating atomistic detail of the active site. Phys. Rev. Lett. 2005, 95. [Google Scholar] [CrossRef] [PubMed]
- Van Gunsteren, W.F.; Bakowies, D.; Baron, R.; Chandrasekhar, I.; Christen, M.; Daura, X.; Gee, P.; Geerke, D.P.; Glättli, A.; Hünenberger, P.H.; et al. Biomolecular modeling: Goals, problems, perspectives. Angew. Chem. Int. Ed. 2006, 45, 4064–4092. [Google Scholar] [CrossRef] [PubMed]
- Tuckerman, M.E.; Martyna, G.J.; Berne, B.J. Molecular dynamics algorithm for condensed systems with multiple time scales. J. Chem. Phys. 1990, 93, 1287–1291. [Google Scholar] [CrossRef]



















© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gooneie, A.; Schuschnigg, S.; Holzer, C. A Review of Multiscale Computational Methods in Polymeric Materials. Polymers 2017, 9, 16. https://doi.org/10.3390/polym9010016
Gooneie A, Schuschnigg S, Holzer C. A Review of Multiscale Computational Methods in Polymeric Materials. Polymers. 2017; 9(1):16. https://doi.org/10.3390/polym9010016
Chicago/Turabian StyleGooneie, Ali, Stephan Schuschnigg, and Clemens Holzer. 2017. "A Review of Multiscale Computational Methods in Polymeric Materials" Polymers 9, no. 1: 16. https://doi.org/10.3390/polym9010016
APA StyleGooneie, A., Schuschnigg, S., & Holzer, C. (2017). A Review of Multiscale Computational Methods in Polymeric Materials. Polymers, 9(1), 16. https://doi.org/10.3390/polym9010016