Novel Random PBS-Based Copolymers Containing Aliphatic Side Chains for Sustainable Flexible Food Packaging



 ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Polymer Synthesis
2.3. Film Preparation
2.4. Molecular and Thermal Characterization
2.5. Wide-Angle X-ray Analysis
2.6. Stress–Strain Measurements
2.7. Gas Transport Measurements
3. Results and Discussion
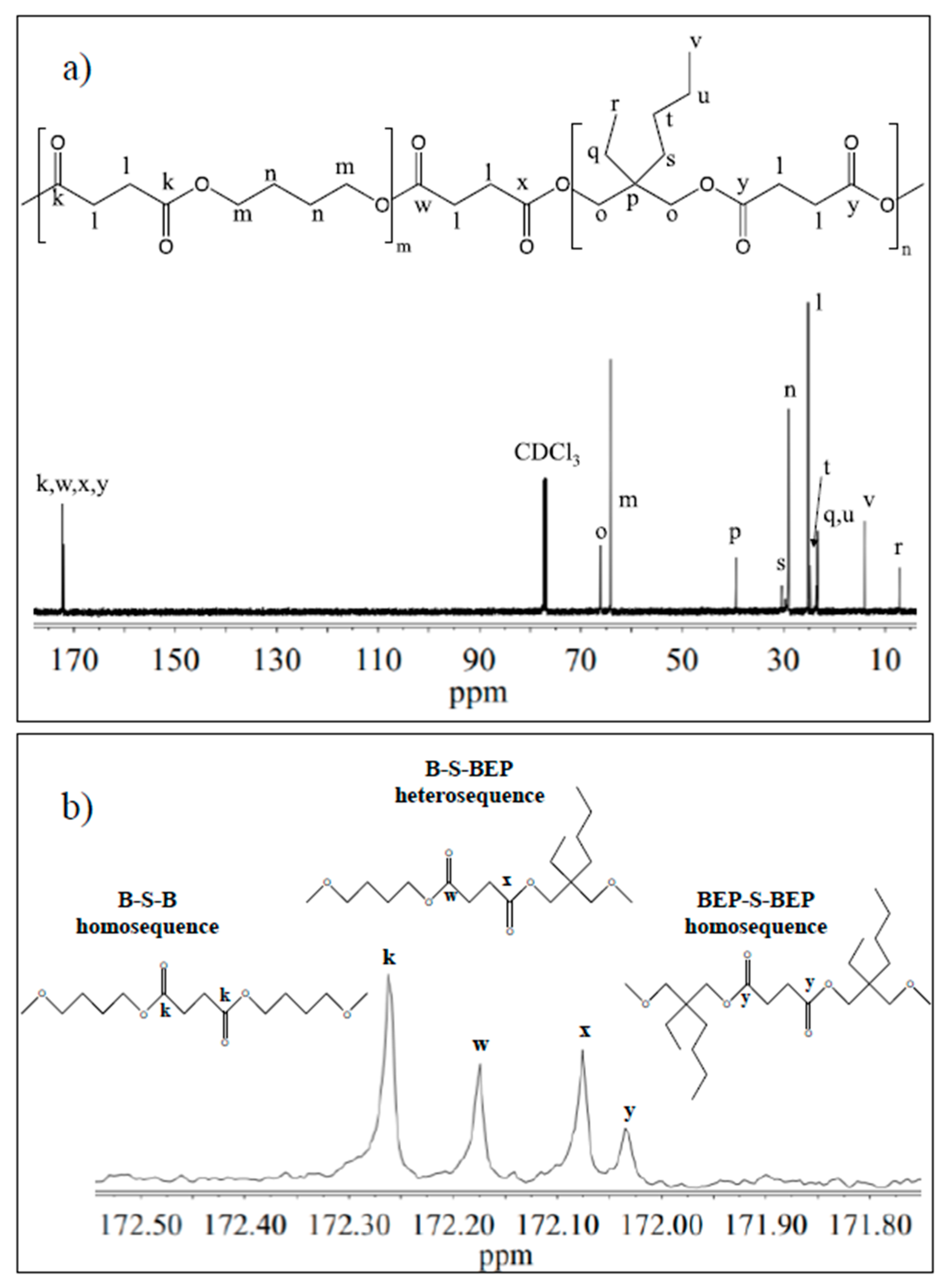
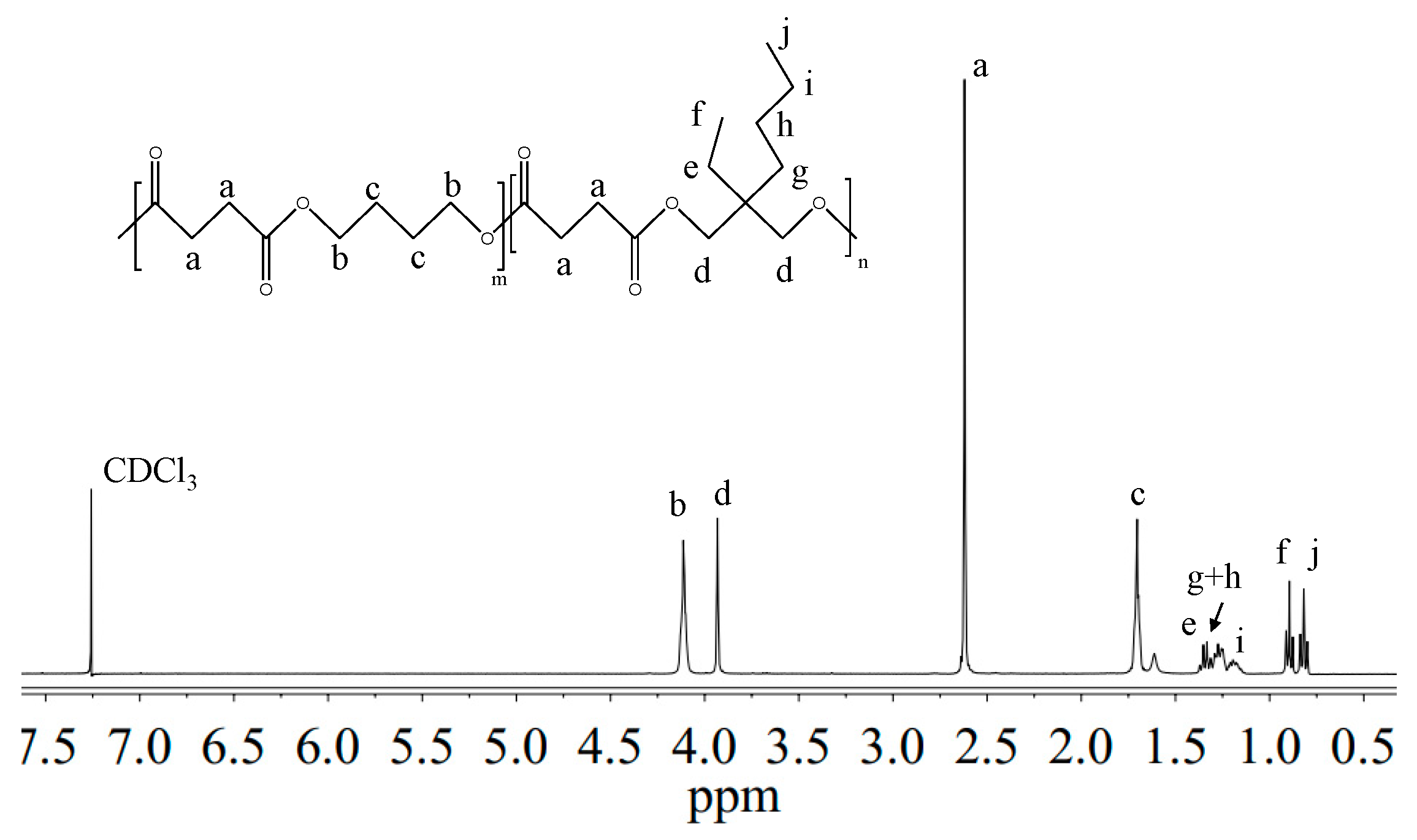
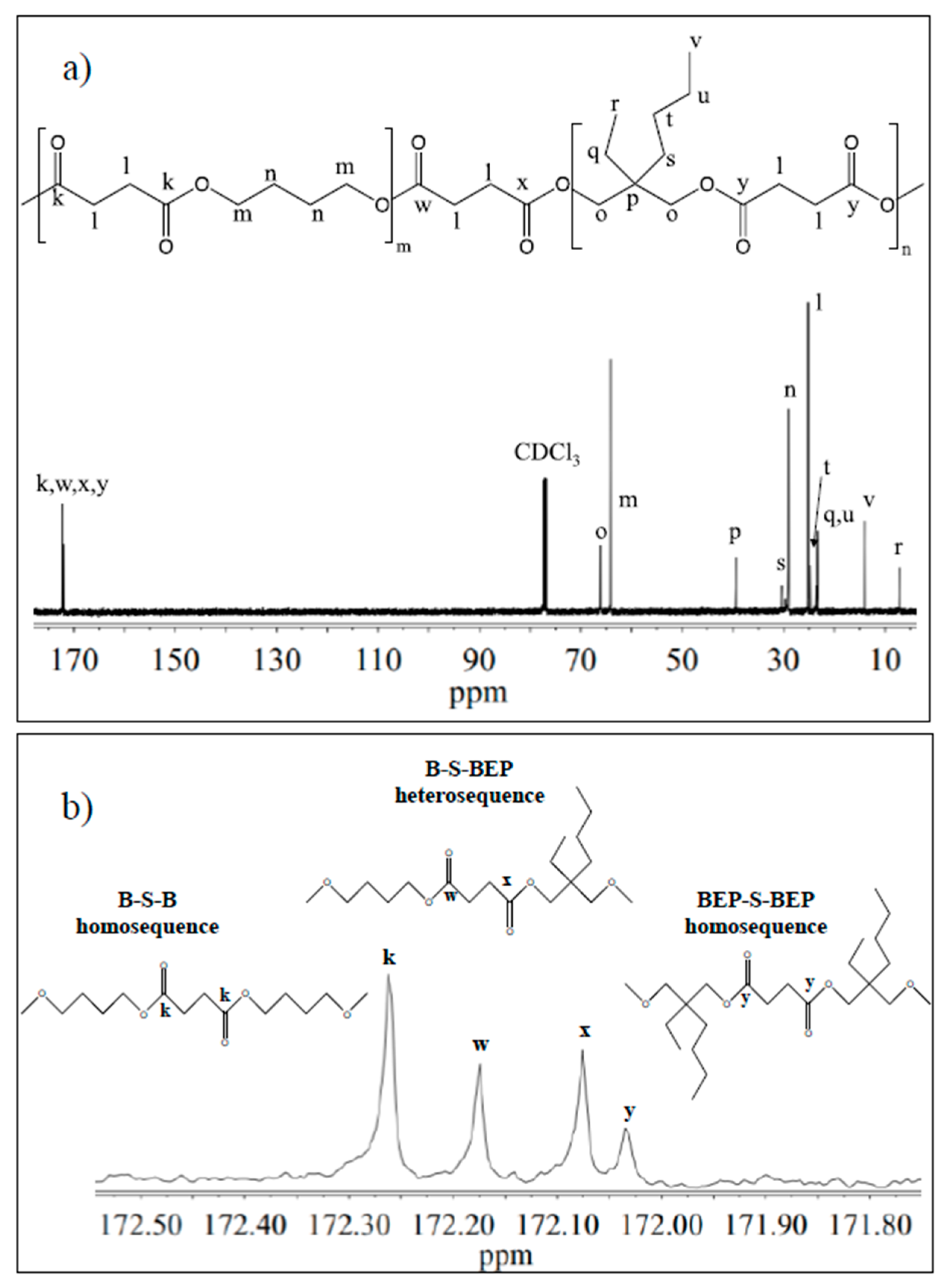
3.1. Molecular Characterization
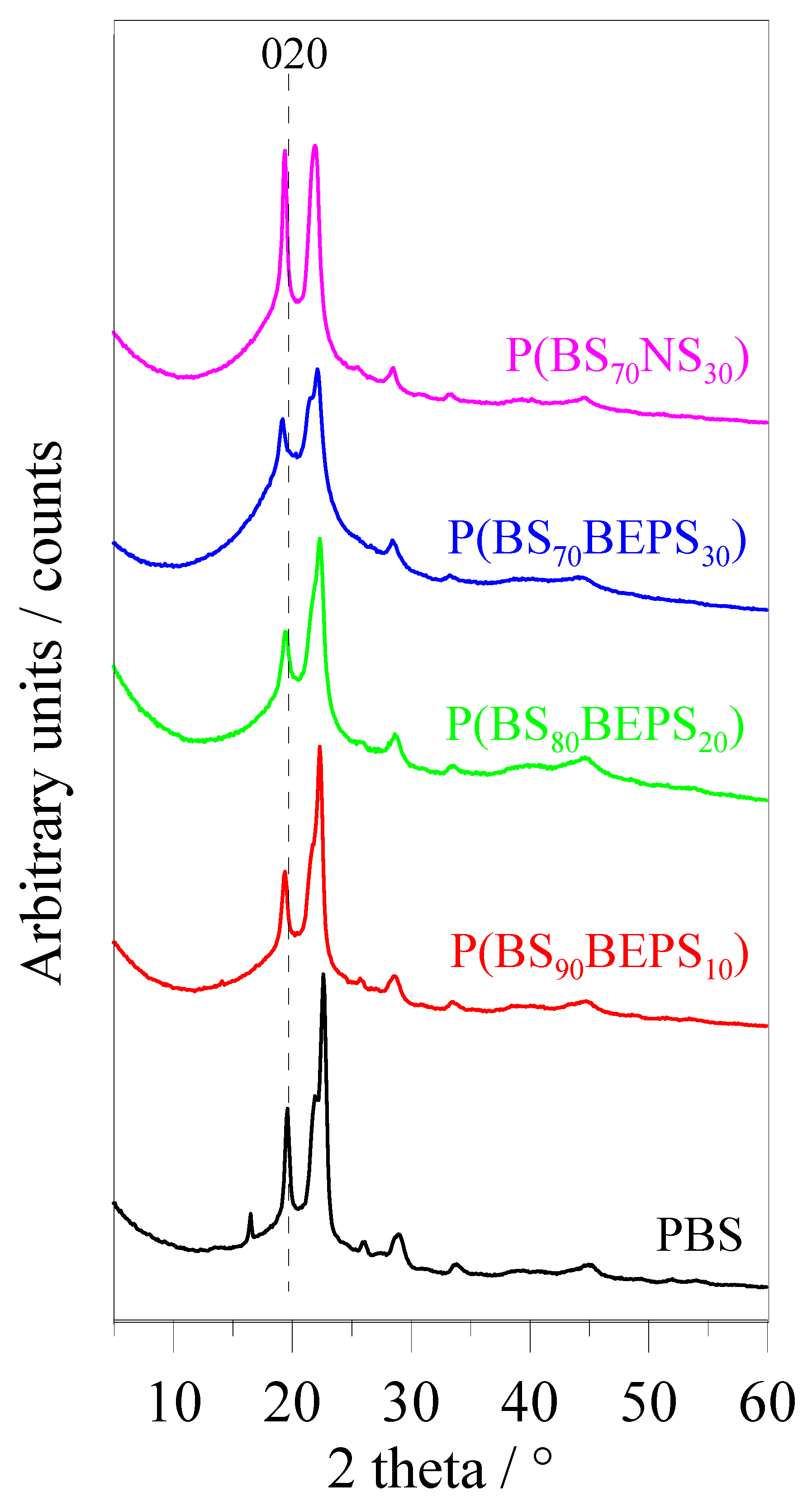
3.2. Thermal and Structural Characterization
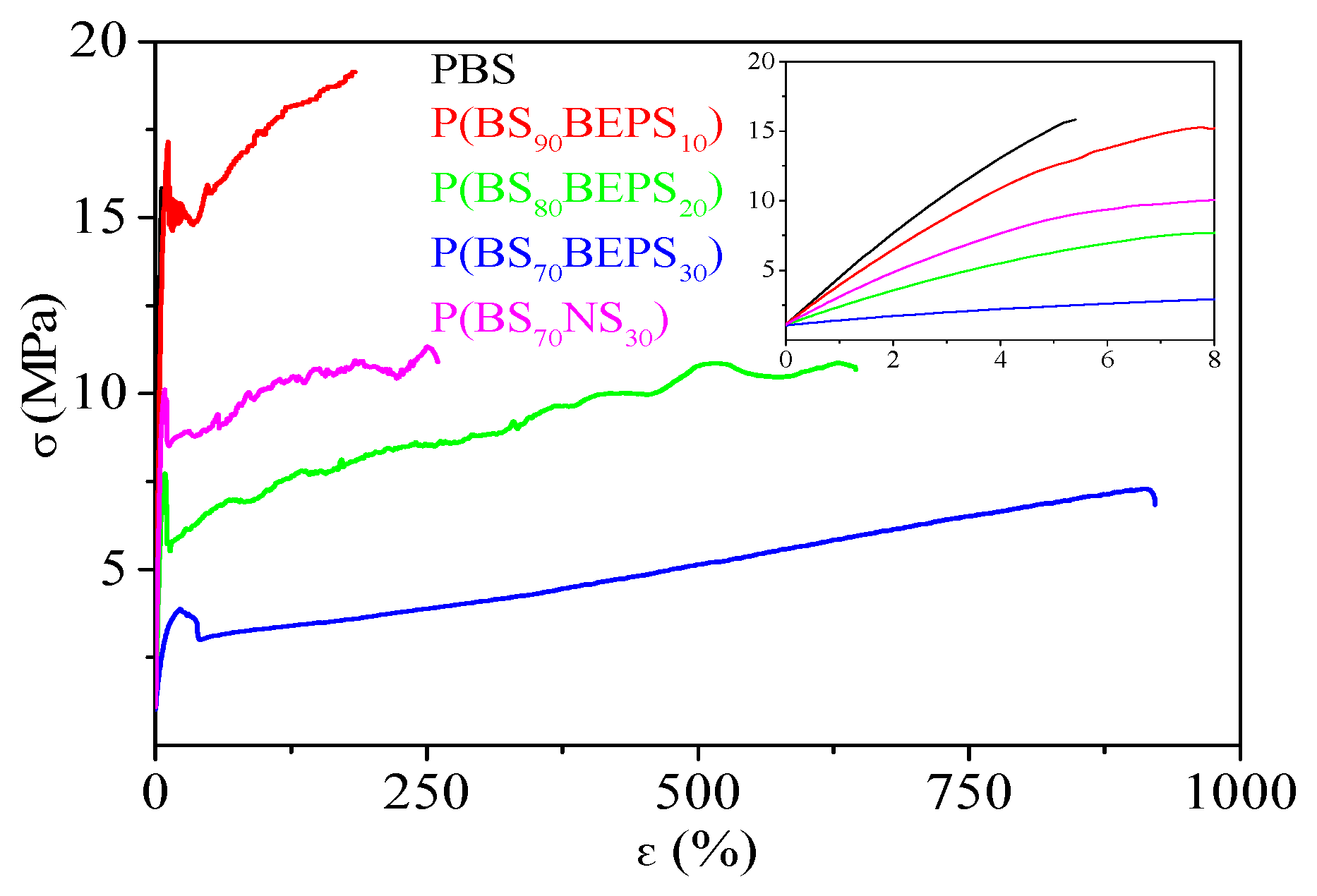
3.3. Mechanical Characterization
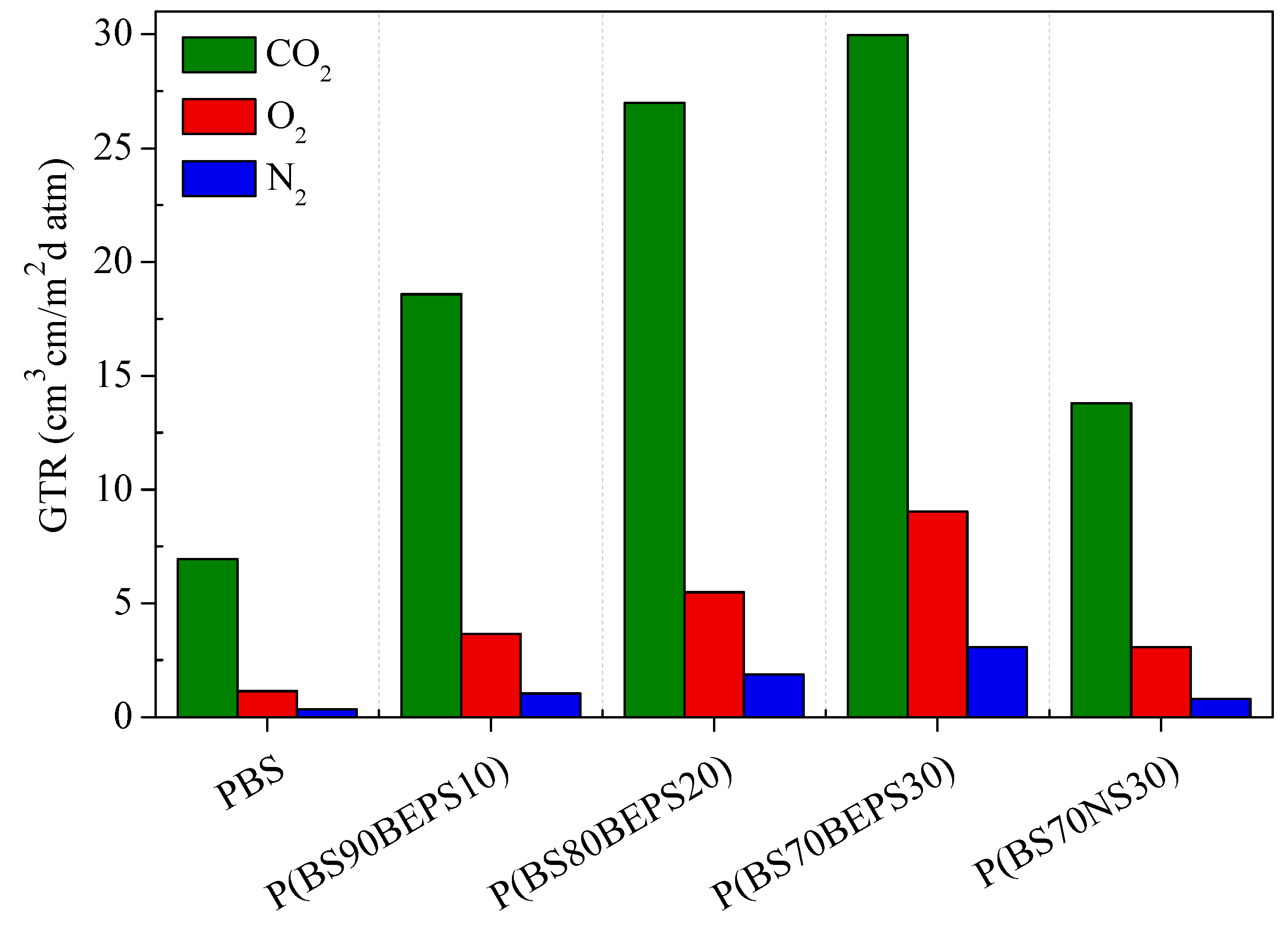
3.4. Gas Barrier Performances
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gill, M. Bioplastic: A Better Alternative to Plastics. Int. J. Res. Appl. Nat. Soc. Sci. 2014, 2, 115–120. [Google Scholar]
- Arikan, E.B.; Ozsoy, H.D. A Review: Investigation of Bioplastics. J. Civ. Eng. Arch. 2015, 9, 188–192. [Google Scholar] [CrossRef]
- Plastics Europe. Plastics—The Facts 2016. An Analysis of European Plastics Production, Demand and Waste Data. Available online: http://www.plasticseurope.org/documents/document/20161014113313-plastics_the_facts_2016_final_version.pdf (accessed on 16 December 2017).
- European Bioplastics. Bioplastics Facts and Figures. Available online: http://www.european-bioplastics.org/news/publications/ (accessed on 16 November 2017).
- Chrissafis, K.; Paraskevopoulos, K.M.; Bikiaris, D.N. Thermal degradation mechanism of poly(ethylene succinate) and poly(butylene succinate): Comparative study. Thermochim. Acta 2005, 435, 142–150. [Google Scholar] [CrossRef]
- Mochizuki, M.; Mukai, K.; Yamada, K.; Ichise, N.; Murase, S.; Iwaya, Y. Structural effects upon enzymatic hydrolysis of poly(butylene succinate-co-ethylene succinate)s. Macromolecules 1997, 30, 7403–7407. [Google Scholar] [CrossRef]
- Gan, Z.; Abe, H.; Doi, Y. Crystallization, melting, and enzymatic degradation of biodegradable poly(butylene succinate-co-14 mol % ethylene succinate) copolyester. Biomacromolecules 2001, 2, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Tserki, V.; Matzinos, P.; Pavlidou, E.; Vachliotis, D.; Panayiotou, C. Biodegradable aliphatic polyesters. Part I. Properties and biodegradation of poly(butylene succinate-co-butylene adipate). Polym. Degrad. Stab. 2006, 91, 367–376. [Google Scholar] [CrossRef]
- Papageorgiou, G.Z.; Bikiaris, D.N. Synthesis, cocrystallization, and enzymatic degradation of novel poly (butylene-co-propylene succinate) copolymers. Biomacromolecules 2007, 8, 2437–2449. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, M.N. Isolation of bacteria degrading poly (butylene succinate-co-butylene adipate) and their lip A gene. Int. Biodeterior. Biodegrad. 2010, 64, 184–190. [Google Scholar] [CrossRef]
- Tsai, Y.; Jheng, L.C.; Hung, C.Y. Synthesis, properties and enzymatic hydrolysis of biodegradable alicyclic/aliphatic copolyesters based on 1,3/1,4-cyclohexanedimethanol. Polym. Degrad. Stab. 2010, 95, 72–78. [Google Scholar] [CrossRef]
- Ding, M.; Zhang, M.; Yang, J.; Qiu, J.H. Study on the enzymatic degradation of aliphatic polyester–PBS and its copolymers. J. Appl. Polym. Sci. 2012, 124, 2902–2907. [Google Scholar] [CrossRef]
- Tan, L.; Chen, Y.; Zhou, W.; Nie, H.; Ye, S. Characterization of the mechanical properties, crystallization, and enzymatic degradation behavior of poly(butylene succinate-co-ethyleneoxide-co-dl-lactide) copolyesters. J. Appl. Polym. Sci. 2012, 123, 2272–2282. [Google Scholar] [CrossRef]
- Pepic, D.; Zagar, E.; Zigon, M.; Krzan, A.; Kunaver, M.; Djonlagic, J. Synthesis and characterization of biodegradable aliphatic copolyesters with poly(ethylene oxide) soft segments. Eur. Polym. J. 2008, 44, 904–917. [Google Scholar] [CrossRef]
- Huang, X.; Chuncheng, L.; Zheng, L.; Zhang, D.; Guan, G.; Xiao, Y. Synthesis, characterization and properties of biodegradable poly(butylene succinate)-block-poly(propylene glycol) segmented copolyesters. Polym. Int. 2009, 58, 893–899. [Google Scholar] [CrossRef]
- Zheng, L.; Li, C.; Zhang, D.; Guan, G.; Xiao, Y.; Wang, D. Multiblock copolymers composed of poly(butylene succinate) and poly(1,2-propylene succinate): Effect of molar ratio of diisocyanate to polyester-diols on crosslink densities, thermal properties, mechanical properties and biodegradability. Polym. Degrad. Stab. 2010, 95, 1743–1750. [Google Scholar] [CrossRef]
- Zheng, L.; Li, C.; Huang, X.; Huang, W.; Zhang, D.; Guan, G.; Xiao, Y.; Wang, D. Synthesis of high-impact biodegradable multiblock copolymers comprising of poly(butylene succinate) and poly(1,2-propylene succinate) with hexamethylene diisocyanate as chain extender. Polym. Adv. Technol. 2011, 22, 279–285. [Google Scholar] [CrossRef]
- Zheng, L.; Li, C.; Zhang, D.; Guan, G.; Xiao, Y.; Wang, D. Synthesis, characterization and properties of novel biodegradable multiblock copolymers comprising poly(butylene succinate) and poly(1,2-propylene terephthalate) with hexamethylene diisocyanate as a chain extender. Polym. Int. 2011, 60, 666–675. [Google Scholar] [CrossRef]
- Babu, R.P.; O’Connor, K.; Seeram, R. Current progress on bio-based polymers and their future trends. Prog. Biomater. 2013, 2, 8. [Google Scholar] [CrossRef]
- Xu, J.; Guo, B.H. Microbial Succinic Acid, Its Polymer Poly(butylene succinate), and Applications. In Plastics from Bacteria; Springer: Berlin/Heidelberg, Germany, 2010; Volume 14, pp. 347–388. ISBN 978-3-642-03286-8. [Google Scholar]
- Soccio, M.; Lotti, N.; Finelli, L.; Gazzano, M.; Munari, A. Influence of transesterification reactions on the miscibility and thermal properties of poly(butylene/diethylene succinate) copolymers. Eur. Polym. J. 2008, 44, 1722–1732. [Google Scholar] [CrossRef]
- Gigli, M.; Lotti, N.; Gazzano, M.; Finelli, L.; Munari, A. Macromolecular design of novel sulfur-containing copolyesters with promising mechanical properties. J. Appl. Polym. Sci. 2012, 126, 686–696. [Google Scholar] [CrossRef]
- Gigli, M.; Lotti, N.; Gazzano, M.; Finelli, L.; Munari, A. Novel eco-friendly random copolyesters of poly(butylene succinate) containing ether-linkages. React. Funct. Polym. 2012, 72, 303–310. [Google Scholar] [CrossRef]
- Soccio, M.; Lotti, N.; Gigli, M.; Finelli, L.; Gazzano, M.; Munari, A. Reactive blending of poly(butylene succinate) and poly(triethylenesuccinate): Characterization of the copolymers obtained. Polym. Int. 2012, 61, 1163–1169. [Google Scholar] [CrossRef]
- Soccio, M.; Lotti, N.; Gazzano, M.; Govoni, M.; Giordano, E.; Munari, A. Molecular architecture and solid-state properties of novel biocompatible PBS-based copolyesters containing sulphur atoms. React. Funct. Polym. 2012, 72, 856–867. [Google Scholar] [CrossRef]
- Gigli, M.; Lotti, N.; Gazzano, M.; Finelli, L.; Munari, A. Synthesis and characterization of novel poly(butylene succinate)-based copolyesters designed as potential candidates for soft tissue engineering. Polym. Eng. Sci. 2013, 53, 491–501. [Google Scholar] [CrossRef]
- Fabbri, M.; Gigli, M.; Gamberini, R.; Lotti, N.; Gazzano, M.; Rimini, B.; Munari, A. Hydrolysable PBS-based poly(ester urethane)s thermoplastic elastomers. Polym. Degrad. Stab. 2014, 108, 223–231. [Google Scholar] [CrossRef]
- Siracusa, V.; Lotti, N.; Munari, A.; Dalla Rosa, M. Poly(butylene succinate) and poly(butylene succinate-co-adipate) for food packaging applications: Gas barrier properties after stressed treatments. Polym. Degrad. Stab. 2015, 119, 35–45. [Google Scholar] [CrossRef]
- Genovese, L.; Lotti, N.; Gazzano, M.; Siracusa, V.; Dalla Rosa, M.; Munari, A. Novel biodegradable aliphatic copolyesters based on poly(butylene succinate) containing thioether-linkages for sustainable food packaging application. Polym. Degrad. Stab. 2016, 132, 191–201. [Google Scholar] [CrossRef]
- Siracusa, V.; Genovese, L.; Munari, A.; Lotti, N. How stress treatments influence the performance of biodegradable poly(butylene succinate)-based copolymers with thioether linkages for food packaging applications. Materials 2017, 10, 1009. [Google Scholar] [CrossRef] [PubMed]
- Genovese, L.; Soccio, M.; Lotti, N.; Gazzano, M.; Siracusa, V.; Salatelli, E.; Balestra, F.; Munari, A. Design of biobased PLLA triblock copolymers for sustainable food packaging: Thermo-mechanical properties, gas barrier ability and compostability. Eur. Polym. J. 2017, 95, 289–303. [Google Scholar] [CrossRef]
- Buchholz, V.; Agarwal, S.; Greiner, A. Synthesis and enzymatic degradation of soft aliphatic polyesters. Macromol. Biosci. 2016, 16, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Kint, D.P.R.; Martinez de Ilarduya, A.; Sansalvadò, A.; Ferrer, J.; Iribarren, J.I.; Munoz-Guerra, S. Structural characterization and thermal properties of poly(ethylene terephthalate) copolymers containing 2-Butyl-2-ethyl-1,3-propanediol. J. Appl. Polym. Sci. 2002, 86, 1077–1086. [Google Scholar] [CrossRef]
- Perrin, D.D.; Armarego, W.L.F. Purification of Laboratory Chemicals, 3rd ed.; Pergamon Press: Oxford, UK, 1966. [Google Scholar]
- Soccio, M.; Lotti, N.; Finelli, L.; Gazzano, M.; Munari, A. Neopenthyl glycol containing poly(propylene azelate)s: Synthesis and thermal properties. Eur. Polym. J. 2007, 43, 3301–3313. [Google Scholar] [CrossRef]
- Soccio, M.; Lotti, N.; Finelli, L.; Gazzano, M.; Munari, A. Neopenthyl glycol containing poly(propylene terephthalate)s: Structure–properties relationships. J. Polym. Sci. Pol. Phys. 2008, 46, 170–181. [Google Scholar] [CrossRef]
- PowdreCell 2.0 for Windows. Available online: http://www.ccp14.ac.uk/ccp/web-mirrors/powdcell/a_v/v_1/powder/details/powcell.htm (accessed on 20 November 2017).
- Siracusa, V. Food packaging permeability behaviour: A report. Int. J. Polym. Sci. 2012, 2012, 302029. [Google Scholar] [CrossRef]
- Brugger Feinmechanik GmbH. Gas Permeability Testing Manual, Registergericht Munchen HRB 77020; Brugger Feinmechanik GmbH: Munich, Germany, 2008. [Google Scholar]
- Minakov, A.A.; Mordvinsted, D.A.; Schick, C. Melting and reorganization of poly(ethylene terephthalate) on fast heating (1000 K/s). Polymer 2004, 45, 3755–3763. [Google Scholar] [CrossRef]
- Kong, Y.; Hay, J.N. Multiple melting behaviour of poly(ethylene terephthalate). Polymer 2003, 44, 623–633. [Google Scholar] [CrossRef]
- Soccio, M.; Lotti, N.; Finelli, L.; Munari, A. Effect of transesterification reactions on the crystallization behaviour and morphology of poly(butylene/diethylene succinate) block copolymers. Eur. Polym. J. 2009, 45, 171–181. [Google Scholar] [CrossRef]
- Soccio, M.; Lotti, N.; Munari, A. Influence of block length on crystallization kinetics and melting behavior of poly(butylene/thiodiethylene succinate) block copolymers. J. Therm. Anal. Calorim. 2013, 114, 677–688. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Kondo, H.; Igarashi, Y.; Noguchi, K.; Okuyama, K.; Washiyama, J. Crystal structures of α and β forms of poly(tetramethylene succinate). Polymer 2000, 41, 4719–4727. [Google Scholar] [CrossRef]
- Clegg, W. X-ray Crystallography, 2nd ed.; Oxford Chemistry Primers: Oxford, UK, 2015; ISBN 9780198700975. [Google Scholar]
- Boyer, R.F. The relation of transition temperatures to chemical structure in high polymers. Rubber Chem. Technol. 1963, 36, 1303–1421. [Google Scholar] [CrossRef]
- McMillin, K.W. Where is MAP going? A review and future potential of modified atmosphere packaging for meat. Meat Sci. 2008, 80, 43–65. [Google Scholar] [CrossRef] [PubMed]
- Caleb, O.J.; Opara, U.L.; Witthuhn, C.R. Modified atmosphere packaging of pomegranate fruit and arils: A review. Food Bioprocess. Technol. 2012, 5, 15–30. [Google Scholar] [CrossRef]
- Farber, J.M. Microbiological aspects of modified-atmosphere packaging technology—A review. J. Food Prot. 1991, 54, 58–70. [Google Scholar] [CrossRef]
- Robertson, G.L. Optical, Mechanical and Barrier Properties of Thermoplastic Polymers. In Food Packaging—Principle and Practice, 2nd ed.; Taylor & Francis Group, CRC Press: Boca Raton, FL, USA, 2013; p. 127. ISBN 978-1-4398-6242-1. [Google Scholar]
- Jamshidian, M.; Tehrany, E.A.; Cleymand, F.; Leconte, S.; Falher, T.; Desobry, S. Effects of synthetic phenol antioxidants on physical, structural, mechanical and barrier properties of poly lactic acid film. Carb. Polym. 2012, 87, 1763–1773. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | BS (mol %) Feed | BS (mol %) by 1H-NMR | BS (wt %) | Mn (g/mol) | D |
|---|---|---|---|---|---|
| PBS | 100 | 100 | 100 | 50,000 | 2 |
| P(BS90BEPS10) | 90 | 87 | 83 | 52,000 | 1.9 |
| P(BS80BEPS20) | 80 | 78 | 72 | 49,000 | 2.1 |
| P(BS70BEPS30) | 70 | 67 | 59 | 63,000 | 2.2 |
| P(BS70NS30) | 70 | 72 | 70 | 55,000 | 2.1 |
| Samples | Tonset °C | Tmax °C | I SCAN | II SCAN | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tg °C | ΔCp J/g·°C | Tm °C | ΔHm J/g | Tg °C | ΔCp J/g°C | Tcc °C | ΔHcc J/g | Tm °C | ΔHm J/g | |||
| PBS | 385 | 407 | −35 | 0.088 | 114 | 50 | −35 | 0.192 | - | - | 114 | 51 |
| P(BS90BEPS10) | 381 | 410 | −33 | 0.155 | 99 | 38 | −34 | 0.180 | 7 | 19 | 100 | 50 |
| P(BS80BEPS20) | 382 | 416 | −29 | 0.263 | 42 80 | 33 | −31 | 0.444 | 31 | 34 | 81 | 34 |
| P(BS70BEPS30) | 382 | 413 | −28 | 0.354 | 43 60 | 22 | −29 | 0.457 | - | - | - | - |
| P(BS70NS30) | 386 | 415 | −28 | 0.244 | 42 77 | 33 | −29 | 0.456 | 27 | 30 | 79 | 30 |
| Samples | Xc % | FWHM ° | a Å | b Å | c Å | β ° | V Å3 |
|---|---|---|---|---|---|---|---|
| α-PBS * | - | - | 5.23 | 9.12 | 10.90 | 123.9 | 431.5 |
| PBS | 46 | 0.43 | 5.24 | 9.09 | 10.85 | 123.7 | 429.9 |
| P(BS90BEPS10) | 38 | 0.54 | 5.29 | 9.19 | 10.72 | 123.5 | 434.6 |
| P(BS80BEPS20) | 31 | 0.66 | 5.27 | 9.22 | 10.70 | 123.3 | 437.7 |
| P(BS70BEPS30) | 18 | 0.55 | 5.29 | 9.39 | 10.70 | 122.9 | 446.1 |
| P(BS70NS30) | 31 | 0.43 | 5.31 | 9.26 | 10.87 | 122.2 | 451.3 |
| Samples | E (MPa) | σb (MPa) | εb (%) |
|---|---|---|---|
| PBS | 301 ± 25 | 16 ± 2 | 5 ± 1 |
| P(BS90BEPS10) | 282 ± 17 | 17 ± 2 | 166 ± 26 |
| P(BS80BEPS20) | 139 ± 7 | 11 ± 1 | 675 ± 27 |
| P(BS70BEPS30) | 47 ± 7 | 7 ± 1 | 1050 ± 99 |
| P(BS70NS30) | 219 ± 12 | 18 ± 1.0 | 340 ± 34 |
| Sample | Film Thickness µm | N2-GTR cm3·cm/m2·d·atm | O2-GTR cm3·cm/m2·d·atm | CO2-GTR cm3·cm/m2·d·atm |
|---|---|---|---|---|
| PBS | 112 ± 11 | 0.3473 | 1.1327 | 6.8573 |
| P(BS90BEPS10) | 102 ± 14 | 1.0286 | 3.6146 | 18.3380 |
| P(BS80BEPS20) | 109 ± 15 | 1.8600 | 5.4080 | 26.6400 |
| P(BS70BEPS30) | 137 ± 8 | 3.0444 | 8.9153 | 29.5840 |
| P(BS70NS30) | 120 ± 10 | 0.7948 | 3.0351 | 13.6101 |
| LDPE * | n.a. | n.a. | 4.4079 | 18.4210 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guidotti, G.; Soccio, M.; Siracusa, V.; Gazzano, M.; Salatelli, E.; Munari, A.; Lotti, N. Novel Random PBS-Based Copolymers Containing Aliphatic Side Chains for Sustainable Flexible Food Packaging. Polymers 2017, 9, 724. https://doi.org/10.3390/polym9120724
Guidotti G, Soccio M, Siracusa V, Gazzano M, Salatelli E, Munari A, Lotti N. Novel Random PBS-Based Copolymers Containing Aliphatic Side Chains for Sustainable Flexible Food Packaging. Polymers. 2017; 9(12):724. https://doi.org/10.3390/polym9120724
Chicago/Turabian StyleGuidotti, Giulia, Michelina Soccio, Valentina Siracusa, Massimo Gazzano, Elisabetta Salatelli, Andrea Munari, and Nadia Lotti. 2017. "Novel Random PBS-Based Copolymers Containing Aliphatic Side Chains for Sustainable Flexible Food Packaging" Polymers 9, no. 12: 724. https://doi.org/10.3390/polym9120724
APA StyleGuidotti, G., Soccio, M., Siracusa, V., Gazzano, M., Salatelli, E., Munari, A., & Lotti, N. (2017). Novel Random PBS-Based Copolymers Containing Aliphatic Side Chains for Sustainable Flexible Food Packaging. Polymers, 9(12), 724. https://doi.org/10.3390/polym9120724