In Vitro Evaluation of Essential Mechanical Properties and Cell Behaviors of a Novel Polylactic-co-Glycolic Acid (PLGA)-Based Tubular Scaffold for Small-Diameter Vascular Tissue Engineering

 ,
,
Abstract
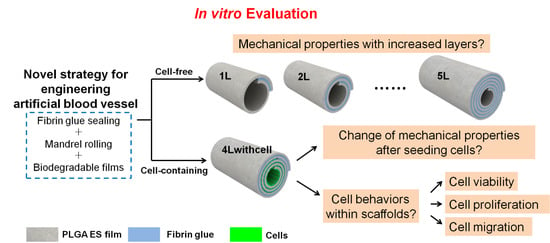
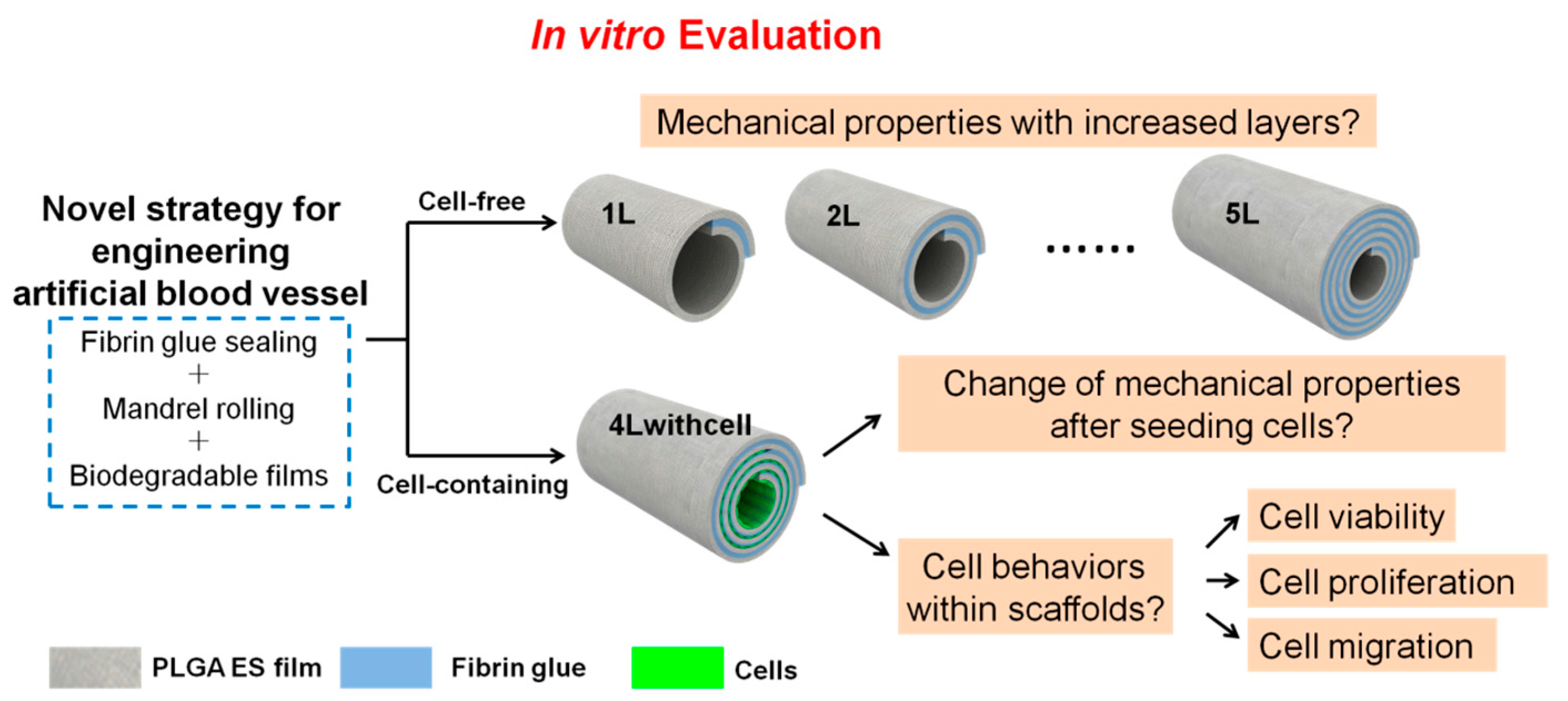
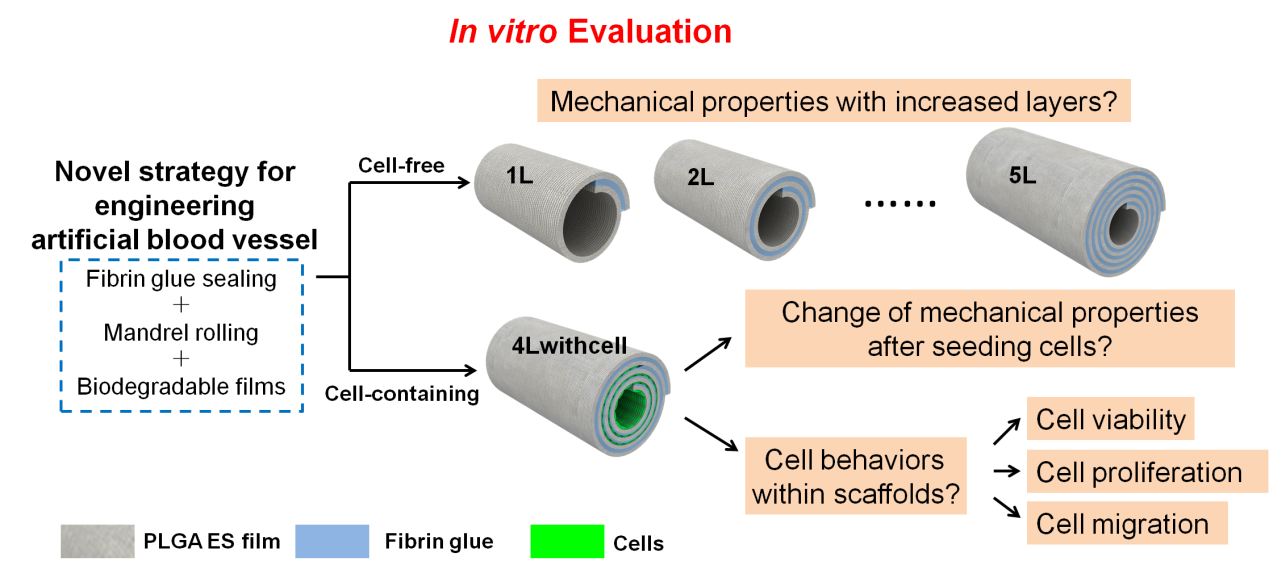{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. PLGA ES Film Preparation
2.3. PMMA Substrate and PDMS Chamber Fabrication
2.4. Cell Culture, Staining and Seeding
2.5. Fabrication of Scaffolds with or without Cells
2.6. Measurement of Mechanical Properties
2.7. Cell Viability Test
2.8. Cell Proliferation Test by a Fluorescent Assay
2.9. Cell Migration Test by a Fluorescent Assay
2.10. Cell Adhesion, Proliferation, and Migration Tests by SEM
2.11. Film Thickness, Fiber Size, Pore Size, and Cell Size Measurements
2.12. PLGA Film Degradation
2.13. Statistics
3. Results
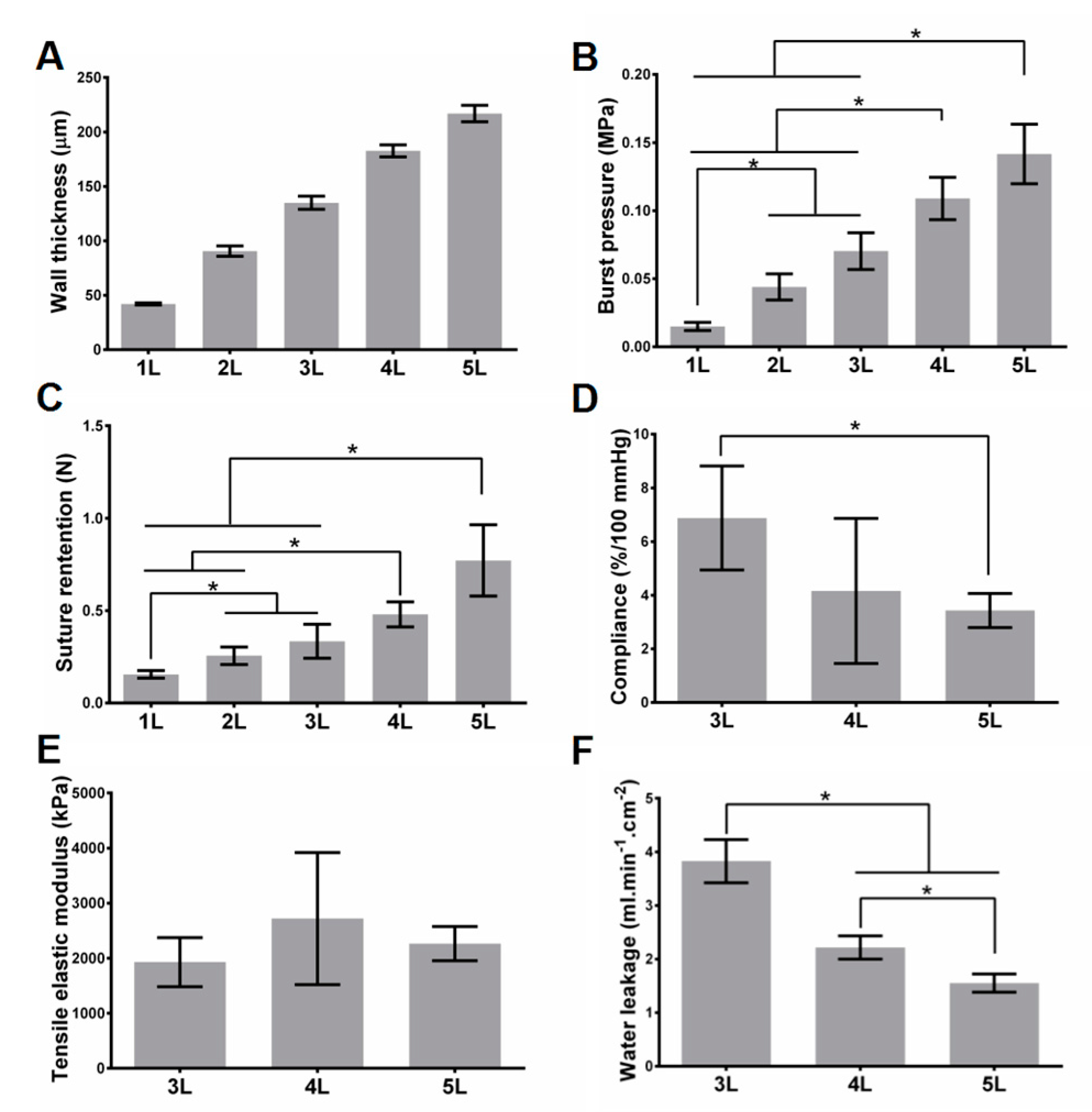
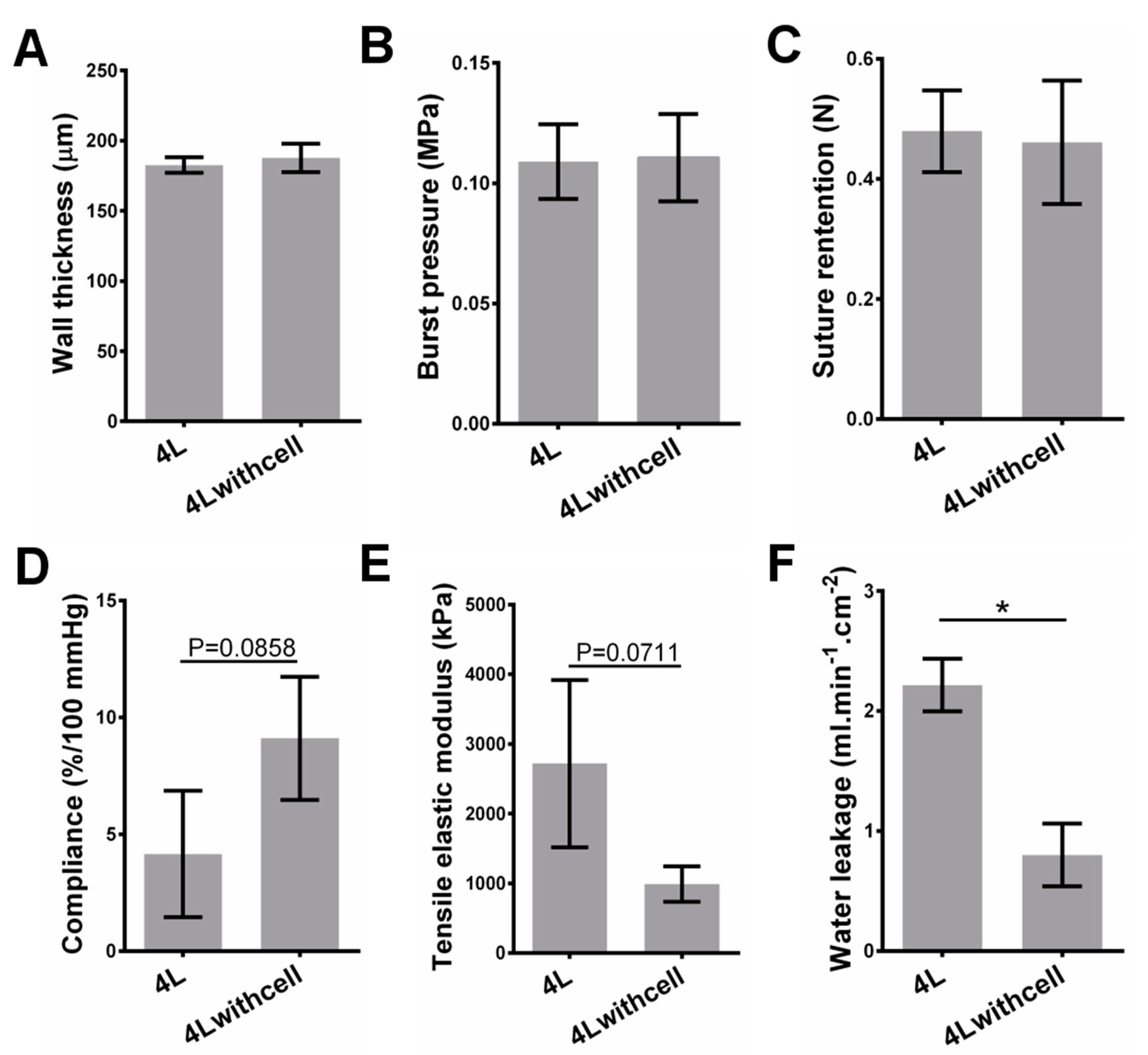
3.1. PLGA ES Film Characterization and Mechanical Properties for Scaffolds without Cells
3.2. Mechanical Properties for Scaffolds with Cells
3.3. Cell Behaviors in Cell-Containing Scaffolds
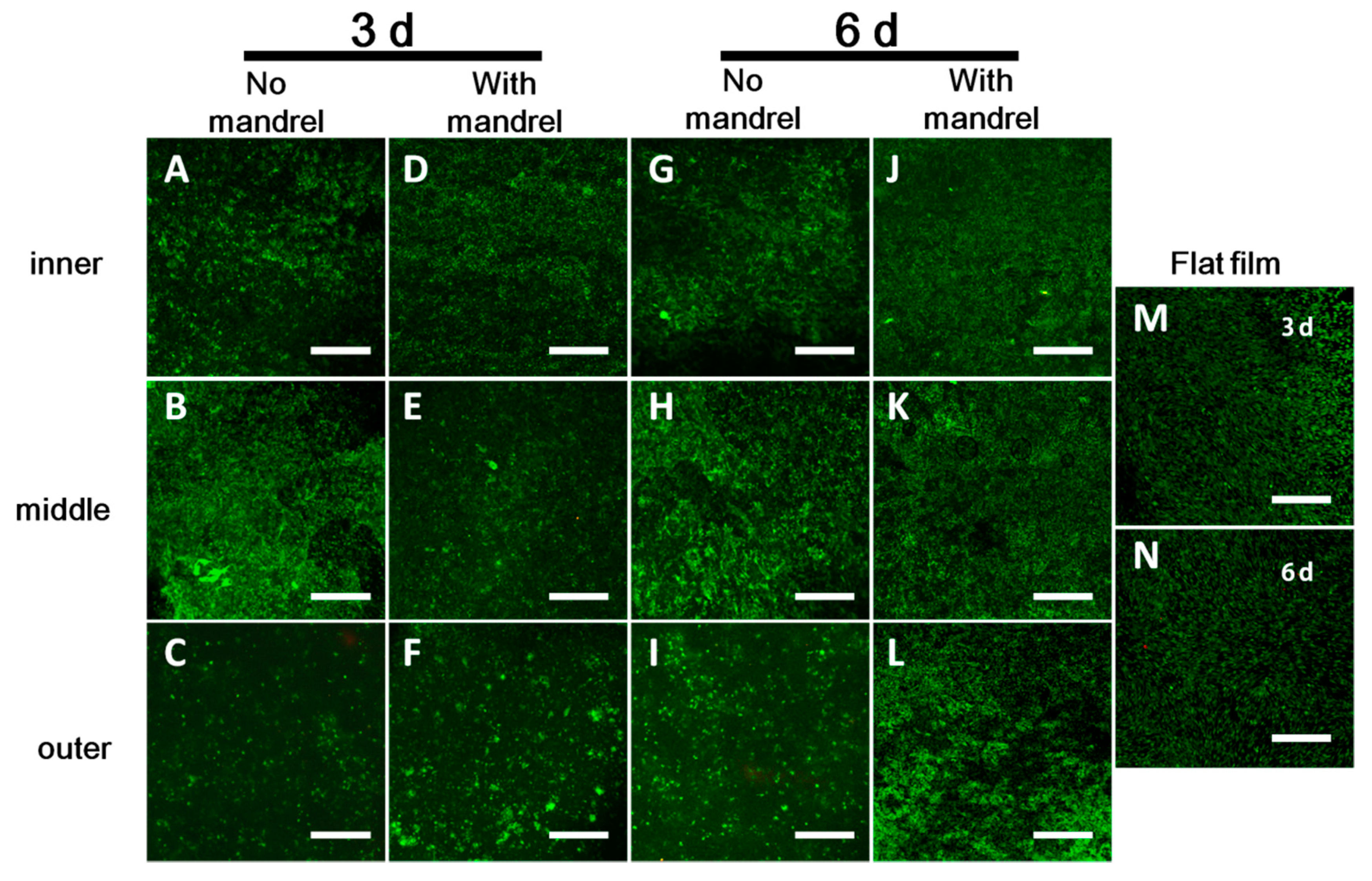
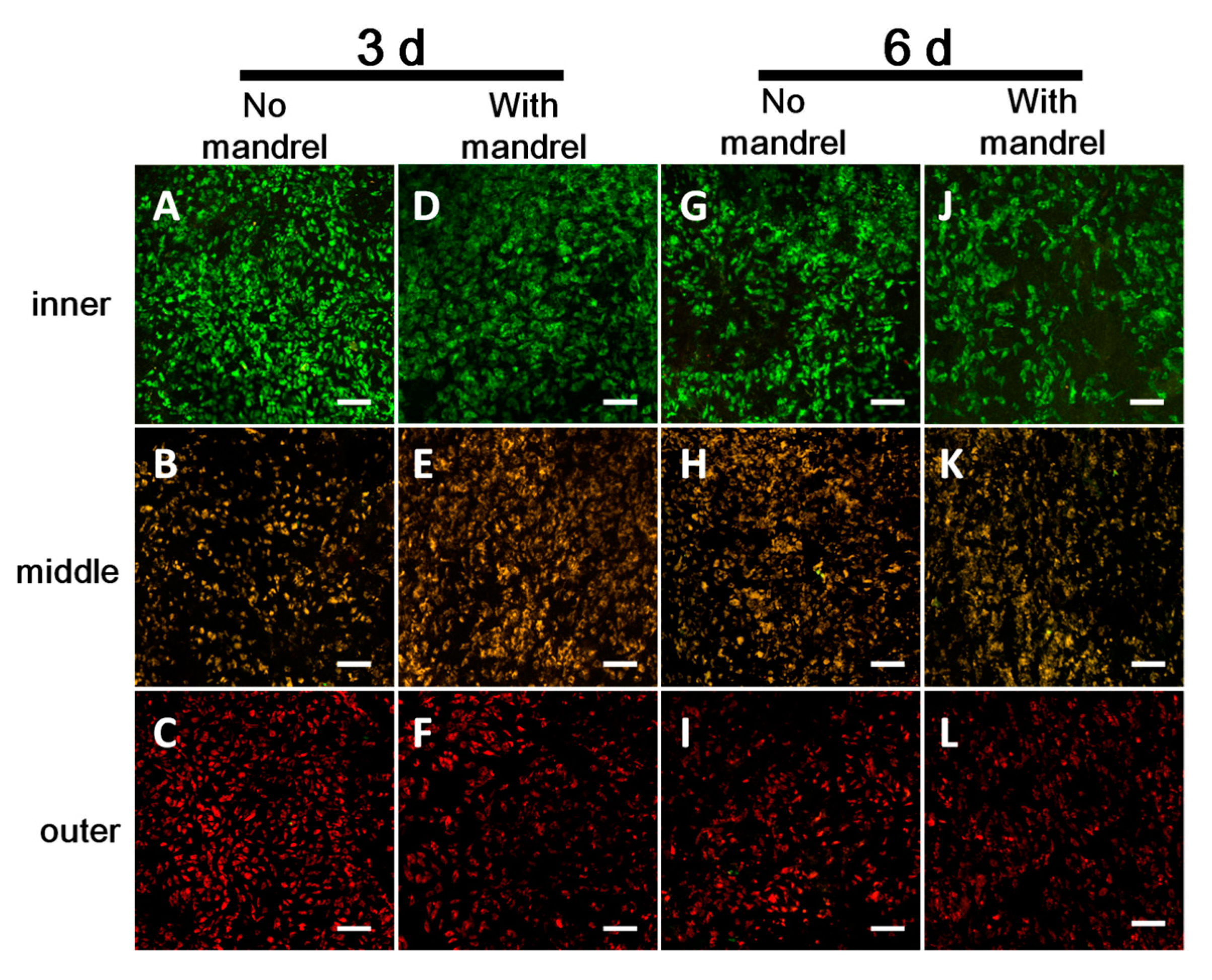
3.3.1. Cell Viability in Each Layer
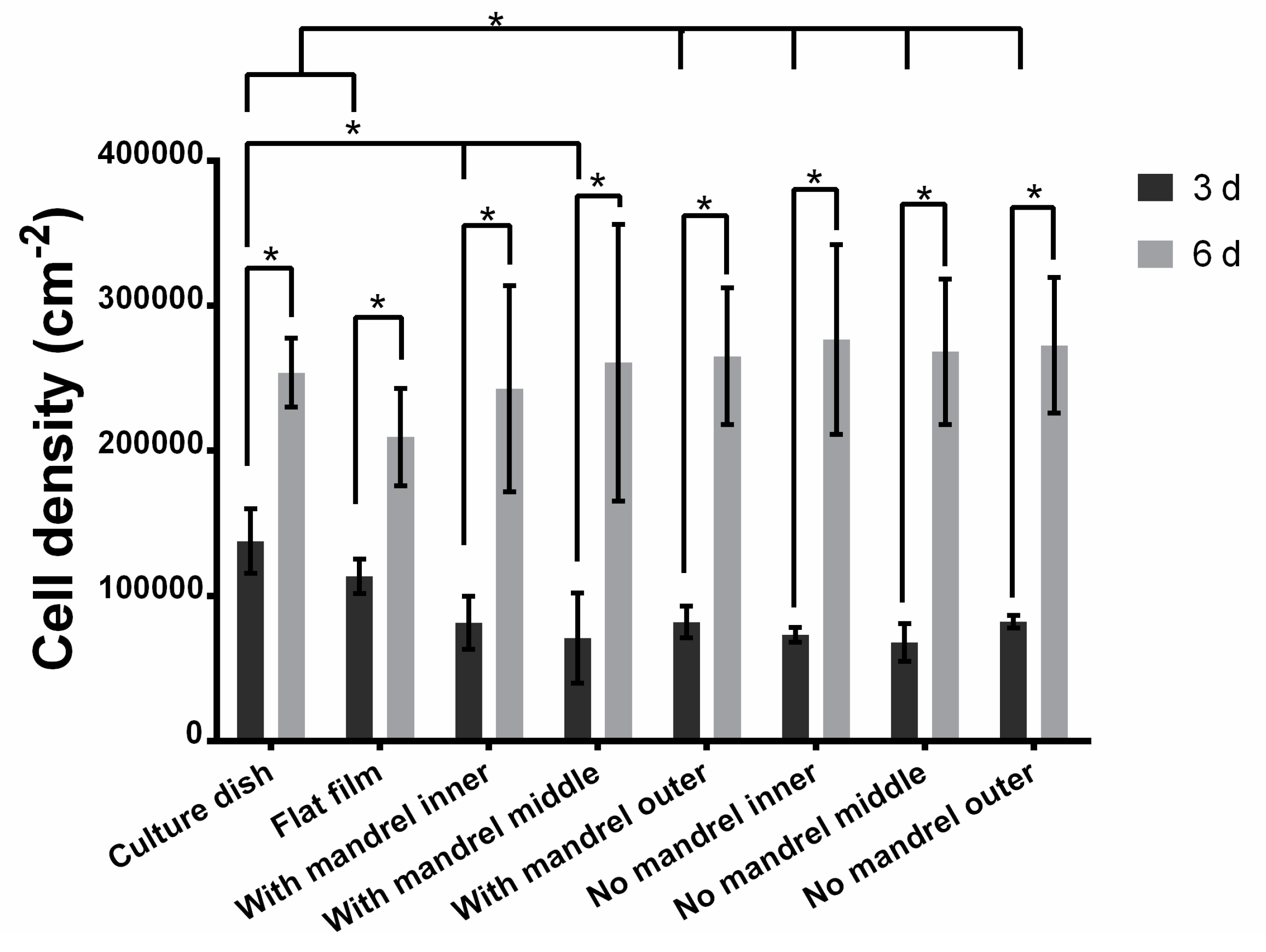
3.3.2. Cell Proliferation in Each Layer
3.3.3. Cell Migration between Layers
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rydén, L.; Grant, P.J.; Anker, S.D.; Berne, C.; Cosentino, F.; Danchin, N.; Deaton, C.; Escaned, J.; Hammes, H.-P.; Huikuri, H. ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Heart J. 2013, 34, 3035–3087. [Google Scholar] [PubMed]
- Kurobe, H.; Maxfield, M.W.; Breuer, C.K.; Shinoka, T. Concise review: Tissue-engineered vascular grafts for cardiac surgery: Past, present, and future. Stem Cells Transl. Med. 2012, 1, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Cleary, M.A.; Geiger, E.; Grady, C.; Best, C.; Naito, Y.; Breuer, C. Vascular tissue engineering: The next generation. Trends Mol. Med. 2012, 18, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Rocco, K.A.; Maxfield, M.W.; Best, C.A.; Dean, E.W.; Breuer, C.K. In vivo applications of electrospun tissue-engineered vascular grafts: A review. Tissue Eng. B Rev. 2014, 20, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Sengupta, D.; Chien, S. Vascular tissue engineering: From in vitro to in situ. Wiley Interdiscip. Rev. Syst. Biol. Med. 2014, 6, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, D.; Waldman, S.D.; Li, S. From in vitro to in situ tissue engineering. Ann. Biomed. Eng. 2014, 42, 1537–1545. [Google Scholar] [CrossRef] [PubMed]
- Chlupac, J.; Filova, E.; Bacakova, L. Blood vessel replacement: 50 years of development and tissue engineering paradigms in vascular surgery. Physiol. Res. 2009, 58, S119. [Google Scholar] [PubMed]
- Dong, C.; Lv, Y. Application of Collagen Scaffold in Tissue Engineering: Recent Advances and New Perspectives. Polymers 2016, 8, 42. [Google Scholar] [CrossRef]
- Manavitehrani, I.; Fathi, A.; Badr, H.; Daly, S.; Negahi Shirazi, A.; Dehghani, F. Biomedical Applications of Biodegradable Polyesters. Polymers 2016, 8, 20. [Google Scholar] [CrossRef]
- Yuan, B.; Jin, Y.; Sun, Y.; Wang, D.; Sun, J.; Wang, Z.; Zhang, W.; Jiang, X. A strategy for depositing different types of cells in three dimensions to mimic tubular structures in tissues. Adv. Mater. 2012, 24, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Bartalena, G.; Loosli, Y.; Zambelli, T.; Snedeker, J.G. Biomaterial surface modifications can dominate cell-substrate mechanics: The impact of PDMS plasma treatment on a quantitative assay of cell stiffness. Soft Matter 2012, 8, 673–681. [Google Scholar] [CrossRef]
- Wang, N.; Tang, L.; Zheng, W.; Peng, Y.; Cheng, S.; Lei, Y.; Zhang, L.; Hu, B.; Liu, S.; Zhang, W.; Jiang, X.Y. A strategy for rapid and facile fabrication of controlled, layered blood vessel-like structures. RSC Adv. 2016, 6, 55054–55063. [Google Scholar] [CrossRef]
- Hueper, W.C.; Martin, G.J.; Thompson, M.R. Methyl cellulose solution as a plasma substitute. Am. J. Surg. 1942, 56, 629–635. [Google Scholar] [CrossRef]
- Lee, K.-W.; Stolz, D.B.; Wang, Y. Substantial expression of mature elastin in arterial constructs. Proc. Natl. Acad. Sci. USA 2011, 108, 2705–2710. [Google Scholar] [CrossRef] [PubMed]
- Fung, Y.C.; Fronek, K.; Patitucci, P. Pseudoelasticity of arteries and the choice of its mathematical expression. Am. J. Physiol. Heart Circ. Physiol. 1979, 237, H620–H631. [Google Scholar]
- Soletti, L.; Hong, Y.; Guan, J.; Stankus, J.J.; El-Kurdi, M.S.; Wagner, W.R.; Vorp, D.A. A bilayered elastomeric scaffold for tissue engineering of small diameter vascular grafts. Acta Biomater. 2010, 6, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Burton, A.C. Relation of structure to function of the tissues of the wall of blood vessels. Physiol. Rev. 1954, 34, 610–642. [Google Scholar]
- Sarkar, S.; Lee, G.Y.; Wong, J.Y.; Desai, T.A. Development and characterization of a porous micro-patterned scaffold for vascular tissue engineering applications. Biomaterials 2006, 27, 4775–4782. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.; Grijpma, D.W.; Feijen, J. Porous polymeric structures for tissue engineering prepared by a coagulation, compression moulding and salt leaching technique. Biomaterials 2003, 24, 1937–1947. [Google Scholar] [CrossRef]
- Flemming, R.G.; Murphy, C.J.; Abrams, G.A.; Goodman, S.L.; Nealey, P.F. Effects of synthetic micro-and nano-structured surfaces on cell behavior. Biomaterials 1999, 20, 573–588. [Google Scholar] [CrossRef]
- Diban, N.; Haimi, S.; Bolhuis-Versteeg, L.; Teixeira, S.; Miettinen, S.; Poot, A.; Grijpma, D.; Stamatialis, D. Development and characterization of poly (ε-caprolactone) hollow fiber membranes for vascular tissue engineering. J. Membr. Sci. 2013, 438, 29–37. [Google Scholar] [CrossRef]
- L’Heureux, N.; Pâquet, S.; Labbé, R.; Germain, L.; Auger, F.A. A completely biological tissue-engineered human blood vessel. FASEB J. 1998, 12, 47–56. [Google Scholar] [PubMed]
- Gauvin, R.; Ahsan, T.; Larouche, D.; Lévesque, P.; Dubé, J.; Auger, F.A.; Nerem, R.M.; Germain, L. A novel single-step self-assembly approach for the fabrication of tissue-engineered vascular constructs. Tissue Eng. Part A 2010, 16, 1737–1747. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Shimizu, T.; Wada, M.; Yamato, M.; Okano, T. Automatic fabrication of 3-dimensional tissues using cell sheet manipulator technique. Biomaterials 2014, 35, 2428–2435. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Zheng, W.; Huang, Z.; Zhang, W.; Xiao, D.; Jiang, X. A strategy for the construction of controlled, three-dimensional, multilayered, tissue-like structures. Adv. Funct. Mater. 2013, 23, 42–46. [Google Scholar] [CrossRef]
- Jin, Y.; Wang, N.; Yuan, B.; Sun, J.; Li, M.; Zheng, W.; Zhang, W.; Jiang, X. Stress-Induced Self-Assembly of Complex Three Dimensional Structures by Elastic Membranes. Small 2013, 9, 2410–2414. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Shirota, T.; Kawahara, D. Fabrication Factory for Tubular Vascular Tissue Mimics based on Automated Rolling Manipulation and Thermo-Responsive Polymers. J. Tissue Sci. Eng. 2014, 5, 1–10. [Google Scholar] [CrossRef]
- Lee, Y.B.; Jun, I.; Bak, S.; Shin, Y.M.; Lim, Y.-M.; Park, H.; Shin, H. Reconstruction of Vascular Structure with Multicellular Components using Cell Transfer Printing Methods. Adv. Healthc. Mater. 2014, 3, 1465–1474. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Almeida, B. Advances in cellular and tissue engineering using layer-by-layer assembly. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2014, 6, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.L.; Li, Y.; Liu, W.W.; Zhang, D.Z.; Zhao, Y.Y.; Yuan, B.; Jiang, X.Y. Patterning mammalian cells for Mmodeling three types of naturally occurring cell-cell interactions. Angew. Chem. Int. Ed. 2009, 48, 8303–8305. [Google Scholar] [CrossRef] [PubMed]
- Palacios-Cuesta, M.; Cortajarena, A.L.; Garcia, O.; Rodriguez-Hernandez, J. Fabrication of Functional Wrinkled Interfaces from Polymer Blends: Role of the Surface Functionality on the Bacterial Adhesion. Polymers 2014, 6, 2845–2861. [Google Scholar] [CrossRef]
- Pimentel-Dominguez, R.; Velazquez-Benitez, A.M.; Rodrigo Velez-Cordero, J.; Hautefeuille, M.; Sanchez-Arevalo, F.; Hernandez-Cordero, J. Photothermal effects and applications of polydimethylsiloxane membranes with carbon nanoparticles. Polymers 2016, 8, 84. [Google Scholar] [CrossRef]
- Ren, F.; Yesildag, C.; Zhang, Z.; Lensen, C.M. Surface patterning of gold nanoparticles on PEG-based hydrogels to control cell adhesion. Polymers 2017, 9, 154. [Google Scholar] [CrossRef]
- Song, Y.; Feijen, J.; Grijpma, D.W.; Poot, A.A. Tissue engineering of small-diameter vascular grafts: A literature review. Clin. Hemorheol. Microcirc. 2011, 49, 357–374. [Google Scholar] [PubMed]
- Sorrentino, S.; Haller, H. Tissue Engineering of Blood Vessels: How to Make a Graft. Tissue Eng. 2011, 263–278. [Google Scholar]
- Peck, M.; Gebhart, D.; Dusserre, N.; McAllister, T.N.; L’Heureux, N. The evolution of vascular tissue engineering and current state of the art. Cells Tissues Organs 2011, 195, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Krawiec, J.T.; Vorp, D.A. Adult stem cell-based tissue engineered blood vessels: A review. Biomaterials 2012, 33, 3388–3400. [Google Scholar] [CrossRef] [PubMed]
- Menu, P.; Stoltz, J.F.; Kerdjoudj, H. Progress in vascular graft substitute. Clin. Hemorheol. Microcirc. 2013, 53, 117–129. [Google Scholar] [PubMed]
- Konig, G.; McAllister, T.N.; Dusserre, N.; Garrido, S.A.; Iyican, C.; Marini, A.; Fiorillo, A.; Avila, H.; Wystrychowski, W.; Zagalski, K. Mechanical properties of completely autologous human tissue engineered blood vessels compared to human saphenous vein and mammary artery. Biomaterials 2009, 30, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- L’Heureux, N.; Dusserre, N.; Marini, A.; Garrido, S.; de la Fuente, L.; McAllister, T. Technology insight: The evolution of tissue-engineered vascular grafts—From research to clinical practice. Nat. Rev. Cardiol. 2007, 4, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Bourget, J.-M.; Gauvin, R.; Larouche, D.; Lavoie, A.; Labbé, R.; Auger, F.A.; Germain, L. Human fibroblast-derived ECM as a scaffold for vascular tissue engineering. Biomaterials 2012, 33, 9205–9213. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Hillery, C.; Seifalian, A.; Hamilton, G. Critical parameter of burst pressure measurement in development of bypass grafts is highly dependent on methodology used. J. Vasc. Surg. 2006, 44, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Allen, R.A.; Wang, Y. Fast-degrading elastomer enables rapid remodeling of a cell-free synthetic graft into a neoartery. Nat. Med. 2012, 18, 1148–1153. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.A.; Wu, W.; Yao, M.; Dutta, D.; Duan, X.; Bachman, T.N.; Champion, H.C.; Stolz, D.B.; Robertson, A.M.; Kim, K. Nerve regeneration and elastin formation within poly(glycerol sebacate)-based synthetic arterial grafts one-year post-implantation in a rat model. Biomaterials 2014, 35, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Abbott, W.M.; Megerman, J.; Hasson, J.E.; L'Italien, G.; Warnock, D.F. Effect of compliance mismatch on vascular graft patency. J. Vasc. Surg. 1987, 5, 376–382. [Google Scholar] [CrossRef]
- Mehigan, D.G.; Fitzpatrick, B.; Browne, H.I.; Bouchier-Hayes, D.J. Is compliance mismatch the major cause of anastomotic arterial aneurysms? Analysis of 42 cases. J. Vasc. Surg. 1984, 26, 147–150. [Google Scholar]
- Okuhn, S.P.; Connelly, D.P.; Calakos, N.; Ferrell, L.; Man-Xiang, P.; Goldstone, J. Does compliance mismatch alone cause neointimal hyperplasia? J. Vasc. Surg. 1989, 9, 35–45. [Google Scholar] [CrossRef]
- Sarkar, S.; Salacinski, H.J.; Hamilton, G.; Seifalian, A.M. The mechanical properties of infrainguinal vascular bypass grafts: Their role in influencing patency. Eur. J. Vasc. Endovasc. Surg. 2006, 31, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Fine, B.; Sandig, M.; Mequanint, K. Elastin biosynthesis: The missing link in tissue-engineered blood vessels. Cardiovasc. Res. 2006, 71, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Wise, S.G.; Byrom, M.J.; Waterhouse, A.; Bannon, P.G.; Ng, M.K.C.; Weiss, A.S. A multilayered synthetic human elastin/polycaprolactone hybrid vascular graft with tailored mechanical properties. Acta Biomater. 2011, 7, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Gelse, K.; Pöschl, E.; Aigner, T. Collagens-structure, function, and biosynthesis. Adv. Drug Deliv. Rev. 2003, 55, 1531–1546. [Google Scholar] [CrossRef] [PubMed]
- Papenburg, B.J.; Liu, J.; Higuera, G.A.; Barradas, A.M.C.; de Boer, J.; van Blitterswijk, C.A.; Wessling, M.; Stamatialis, D. Development and analysis of multi-layer scaffolds for tissue engineering. Biomaterials 2009, 30, 6228–6239. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.C.; Milthorpe, B.K.; McFarland, C.D. Engineering thick tissues—The vascularisation problem. Eur. Cell Mater. 2007, 14, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Malda, J.; Klein, T.J.; Upton, Z. The roles of hypoxia in the in vitro engineering of tissues. Tissue Eng. 2007, 13, 2153–2162. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.M.; Haugh, M.G.; O’Brien, F.J. The effect of mean pore size on cell attachment, proliferation and migration in collagen-glycosaminoglycan scaffolds for bone tissue engineering. Biomaterials 2010, 31, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.J.; Chao, W.C.; Lee, P.Y.; Huang, C.H. Construction and characterization of an electrospun tubular scaffold for small-diameter tissue-engineered vascular grafts: A scaffold membrane approach. J. Mech. Behav. Biomed. Mater. 2012, 13, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Bettahalli, N.M.S.; Groen, N.; Steg, H.; Unadkat, H.; Boer, J.; Blitterswijk, C.A.; Wessling, M.; Stamatialis, D. Development of multilayer constructs for tissue engineering. J. Tissue Eng. Regener. Med. 2014, 8, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Martin, Y.; Vermette, P. Bioreactors for tissue mass culture: Design, characterization, and recent advances. Biomaterials 2005, 26, 7481–7503. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, C.E.; Achneck, H.E.; Reichert, W.M.; Truskey, G.A. Biological and engineering design considerations for vascular tissue engineered blood vessels (TEBVs). Curr. Opin. Chem. Eng. 2014, 3, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.A.; Spradling, C.S.; Grant, D.N.; Fox, D.B.; Jimenez, L.; Grant, D.A.; Rone, R.J. Assessment of the biocompatibility and stability of a gold nanoparticle collagen bioscaffold. J. Biomed. Mater. Res. Part A 2014, 102, 332–339. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, N.; Zheng, W.; Cheng, S.; Zhang, W.; Liu, S.; Jiang, X. In Vitro Evaluation of Essential Mechanical Properties and Cell Behaviors of a Novel Polylactic-co-Glycolic Acid (PLGA)-Based Tubular Scaffold for Small-Diameter Vascular Tissue Engineering. Polymers 2017, 9, 318. https://doi.org/10.3390/polym9080318
Wang N, Zheng W, Cheng S, Zhang W, Liu S, Jiang X. In Vitro Evaluation of Essential Mechanical Properties and Cell Behaviors of a Novel Polylactic-co-Glycolic Acid (PLGA)-Based Tubular Scaffold for Small-Diameter Vascular Tissue Engineering. Polymers. 2017; 9(8):318. https://doi.org/10.3390/polym9080318
Chicago/Turabian StyleWang, Nuoxin, Wenfu Zheng, Shiyu Cheng, Wei Zhang, Shaoqin Liu, and Xingyu Jiang. 2017. "In Vitro Evaluation of Essential Mechanical Properties and Cell Behaviors of a Novel Polylactic-co-Glycolic Acid (PLGA)-Based Tubular Scaffold for Small-Diameter Vascular Tissue Engineering" Polymers 9, no. 8: 318. https://doi.org/10.3390/polym9080318
APA StyleWang, N., Zheng, W., Cheng, S., Zhang, W., Liu, S., & Jiang, X. (2017). In Vitro Evaluation of Essential Mechanical Properties and Cell Behaviors of a Novel Polylactic-co-Glycolic Acid (PLGA)-Based Tubular Scaffold for Small-Diameter Vascular Tissue Engineering. Polymers, 9(8), 318. https://doi.org/10.3390/polym9080318