Mechanism of Base-Catalyzed Resorcinol-Formaldehyde and Phenol-Resorcinol-Formaldehyde Condensation Reactions: A Theoretical Study


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Theoretical Calculations
3. Results and Discussion
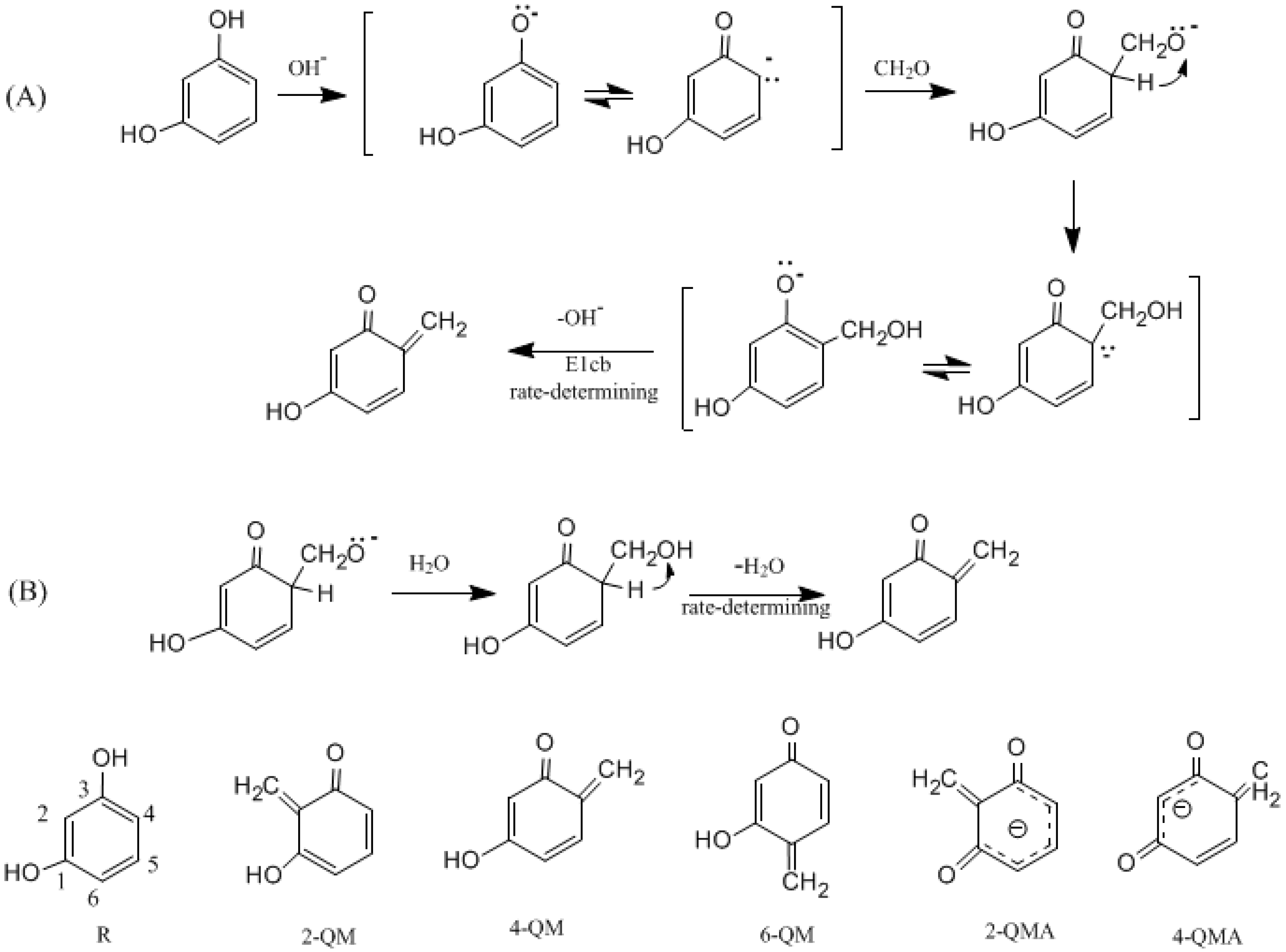
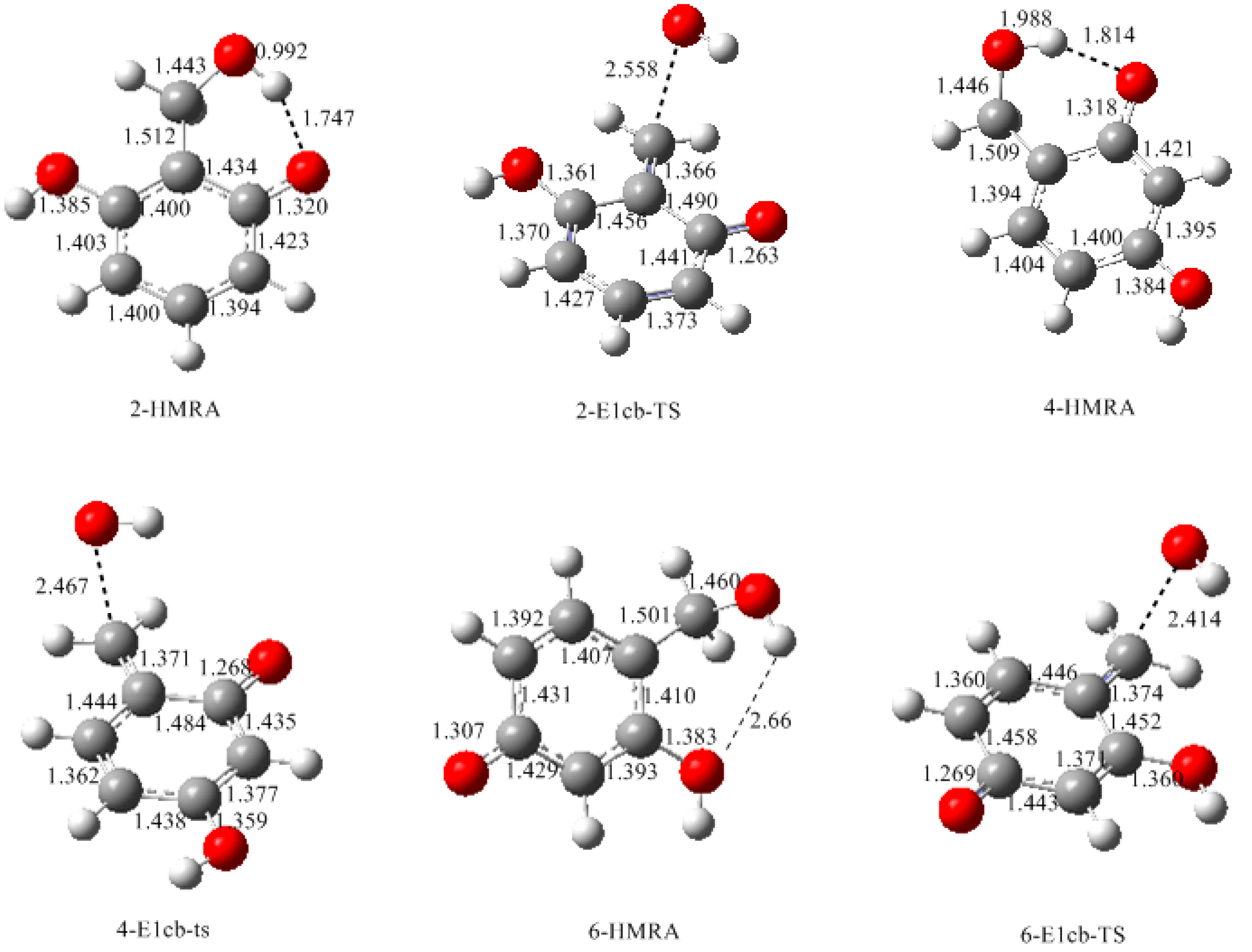
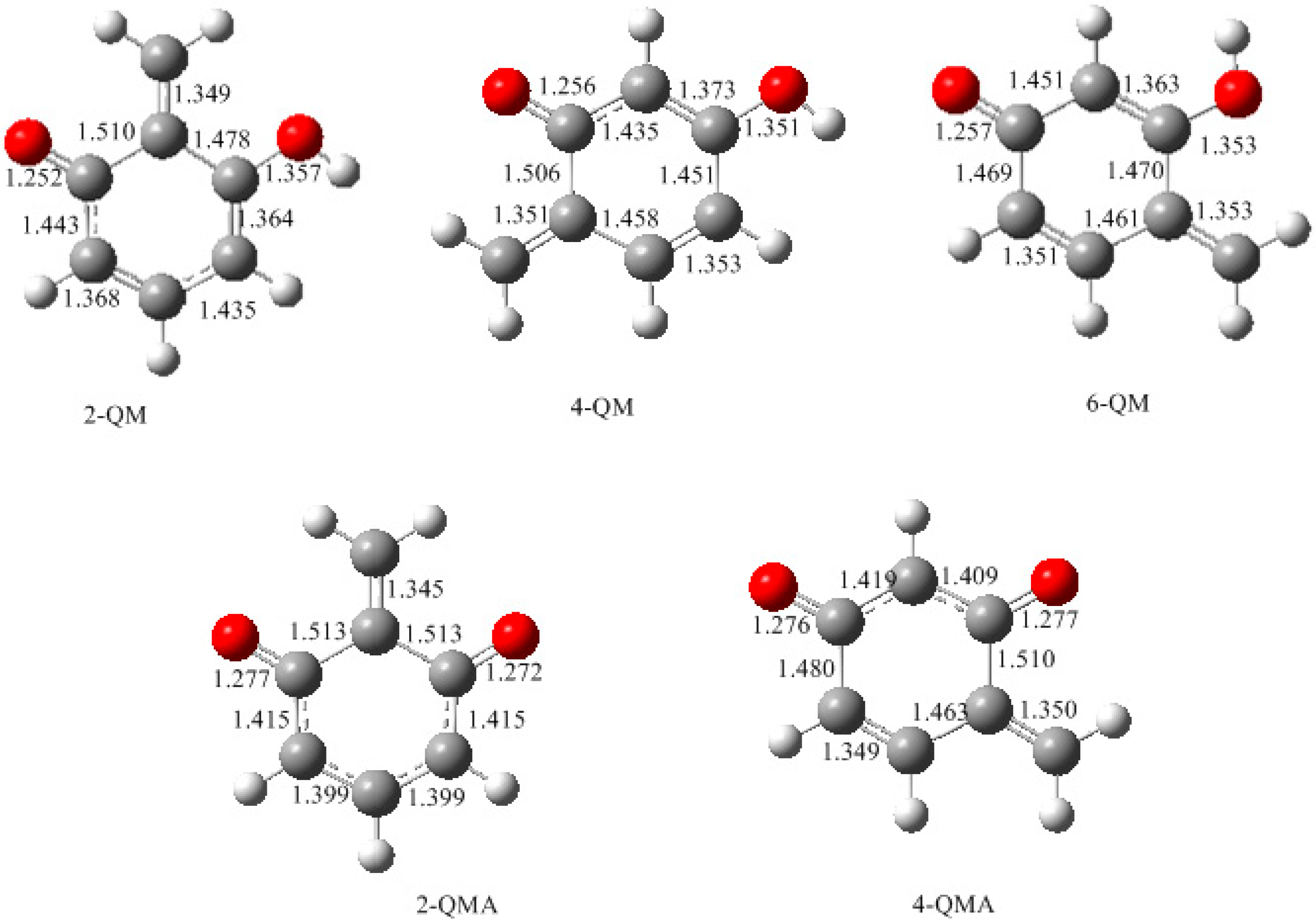
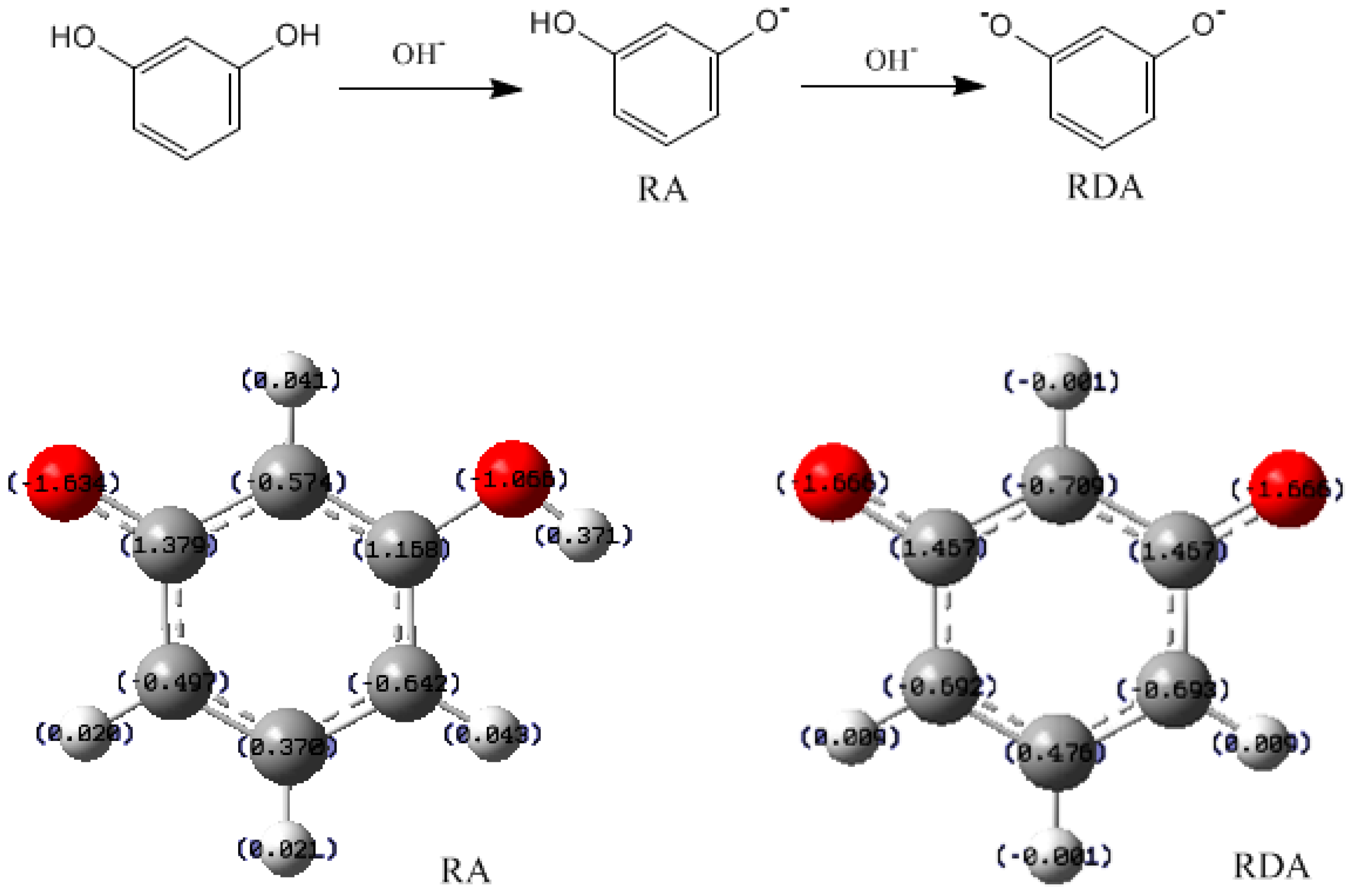
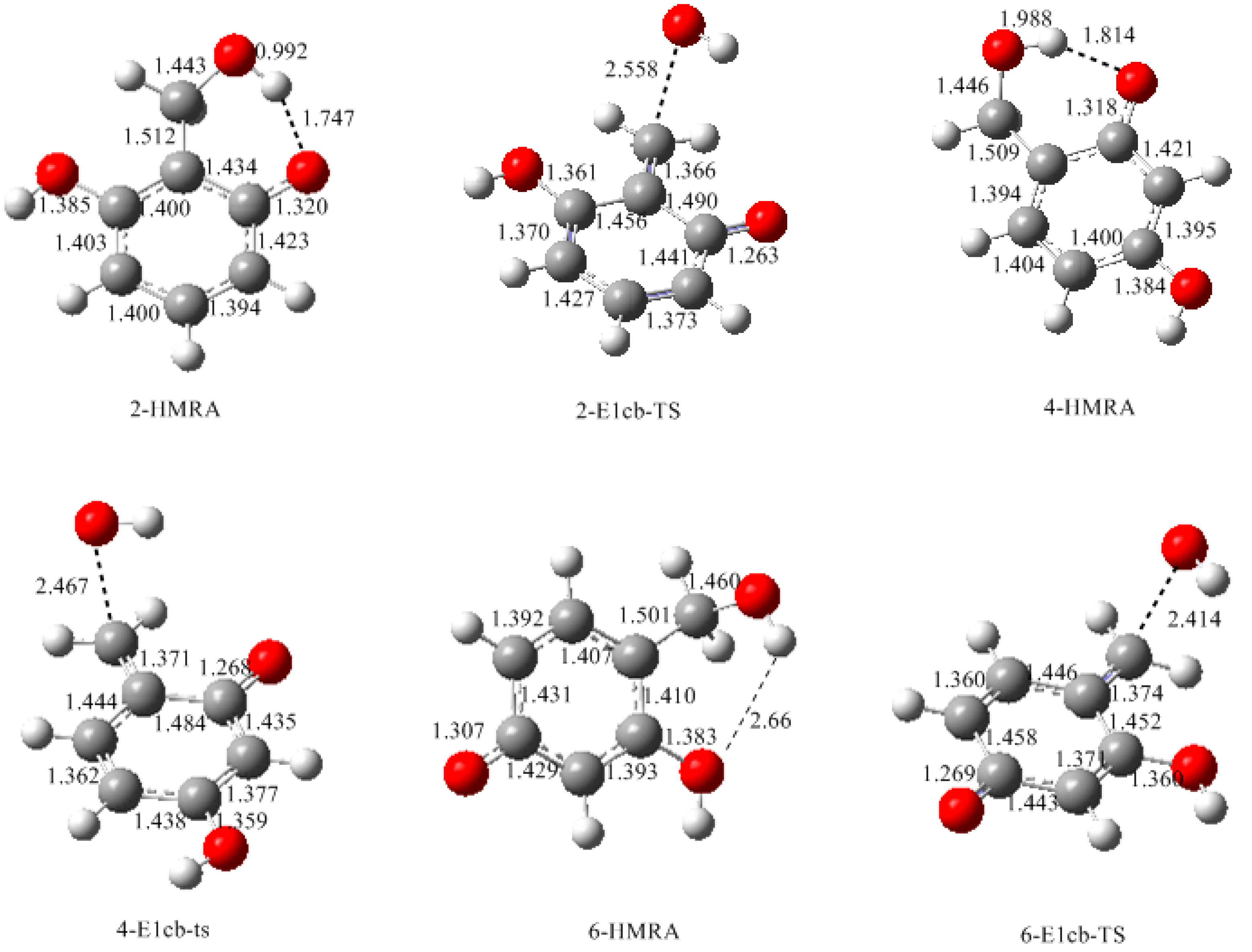
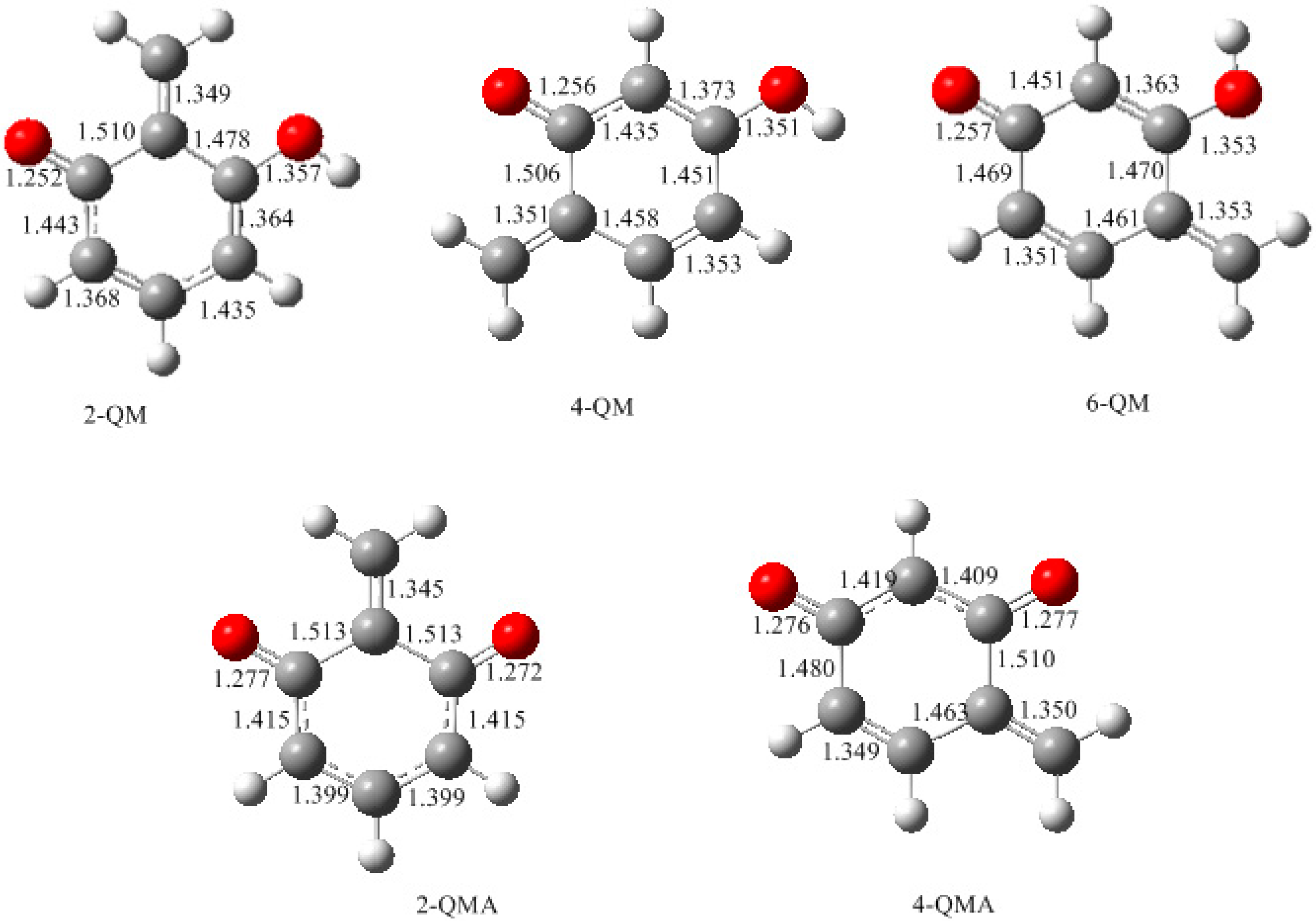
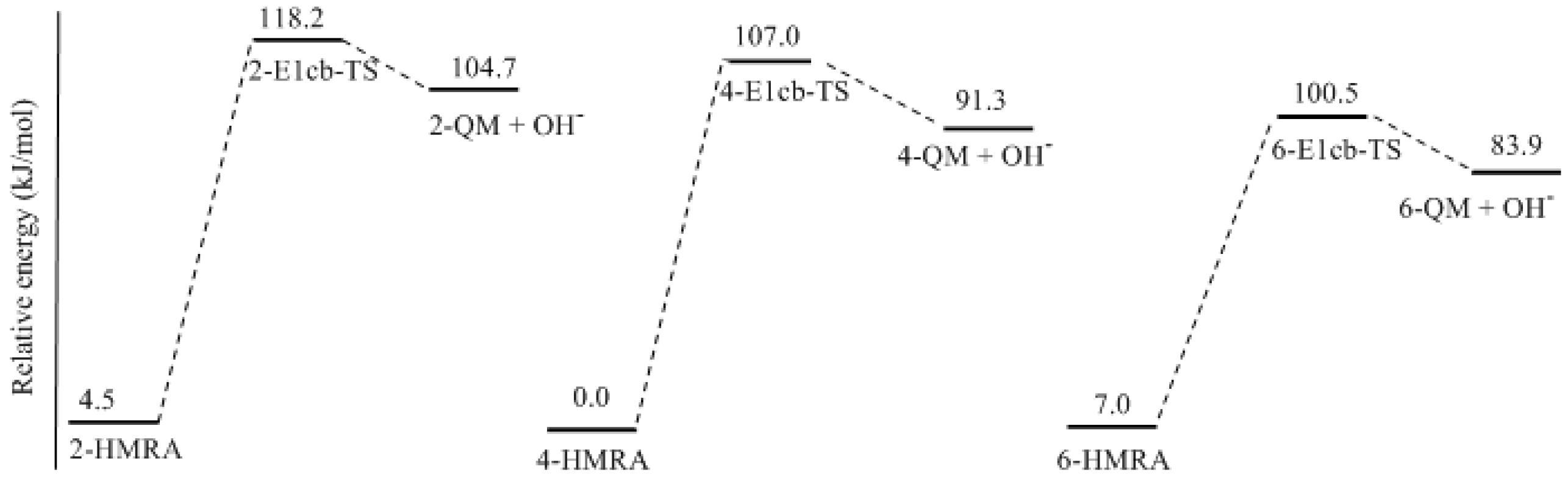
3.1. Formations of Quinine Methide Intermediates
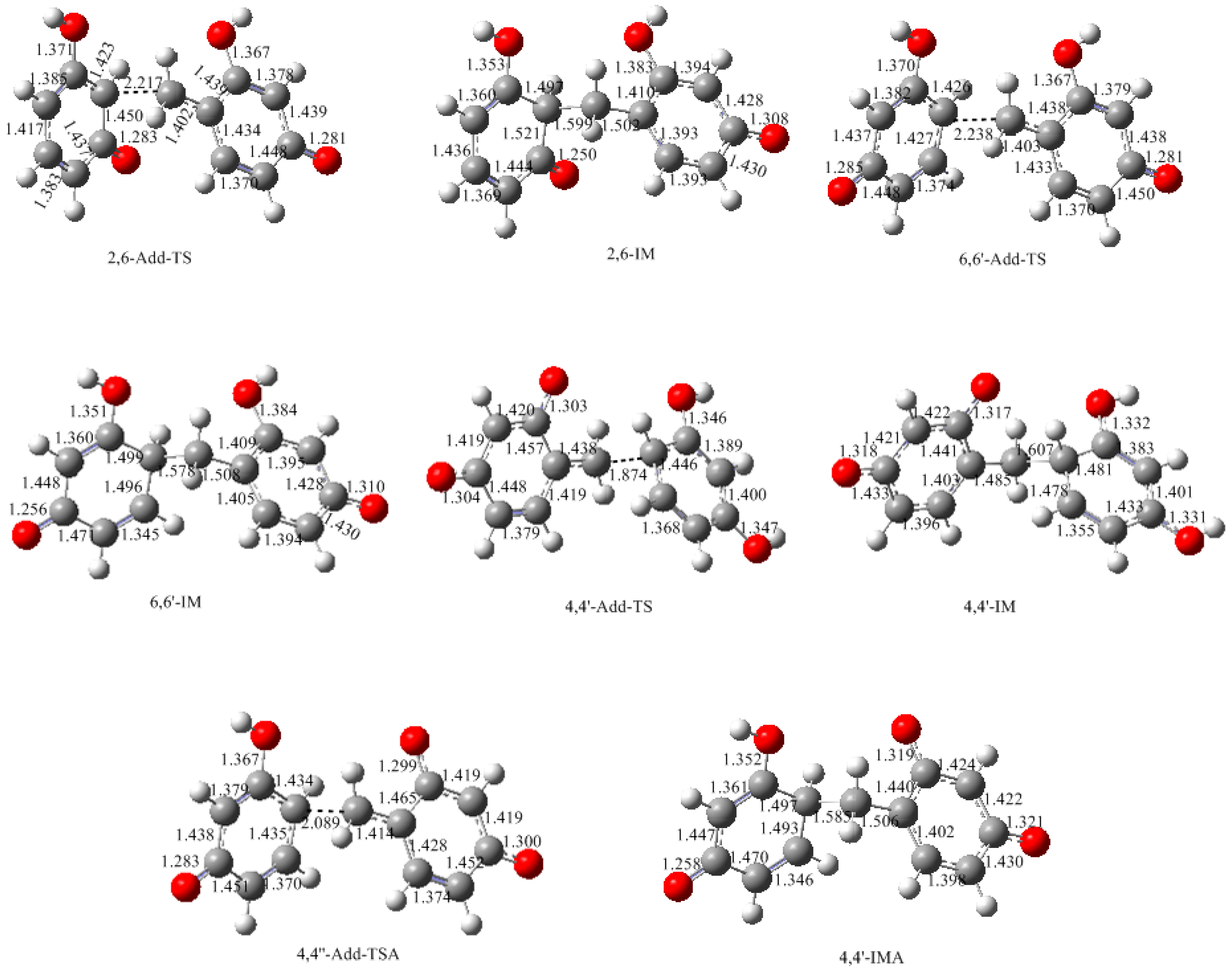
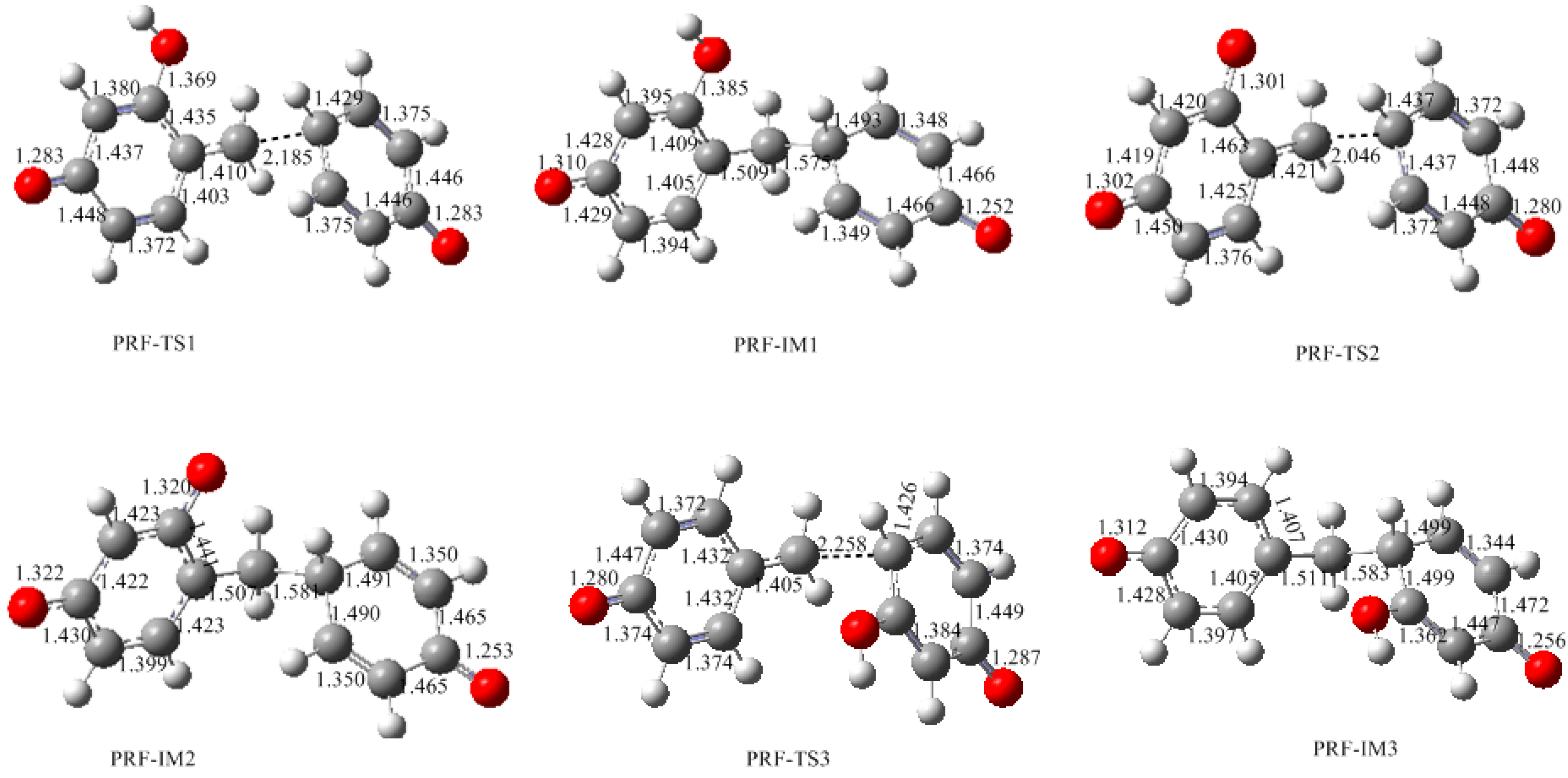
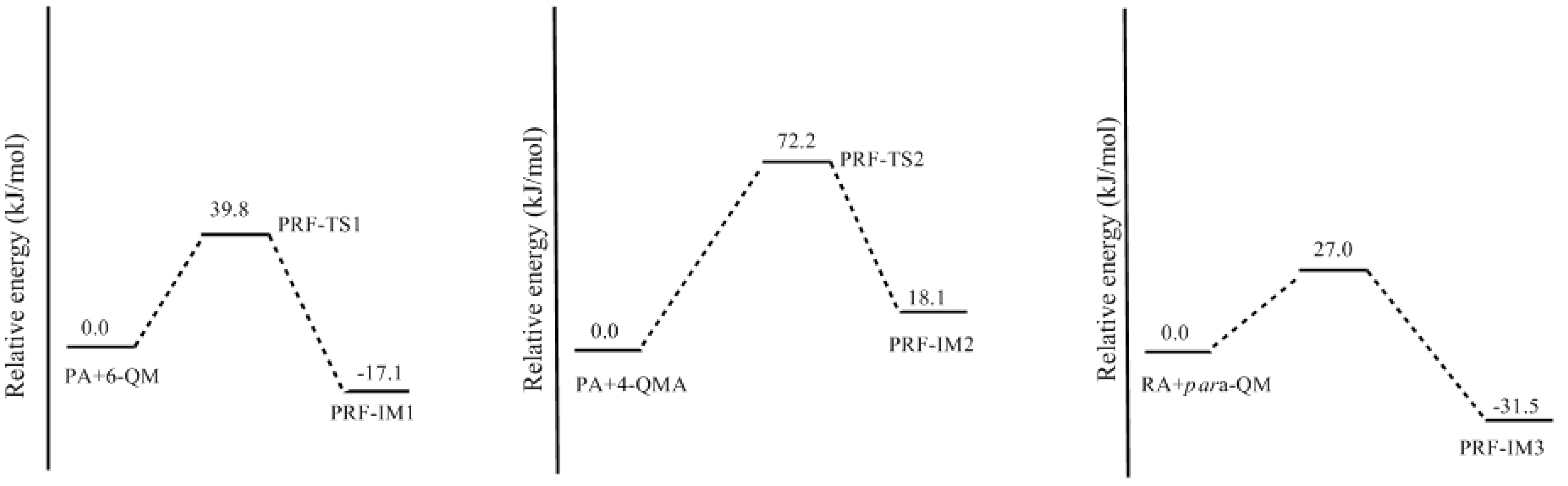
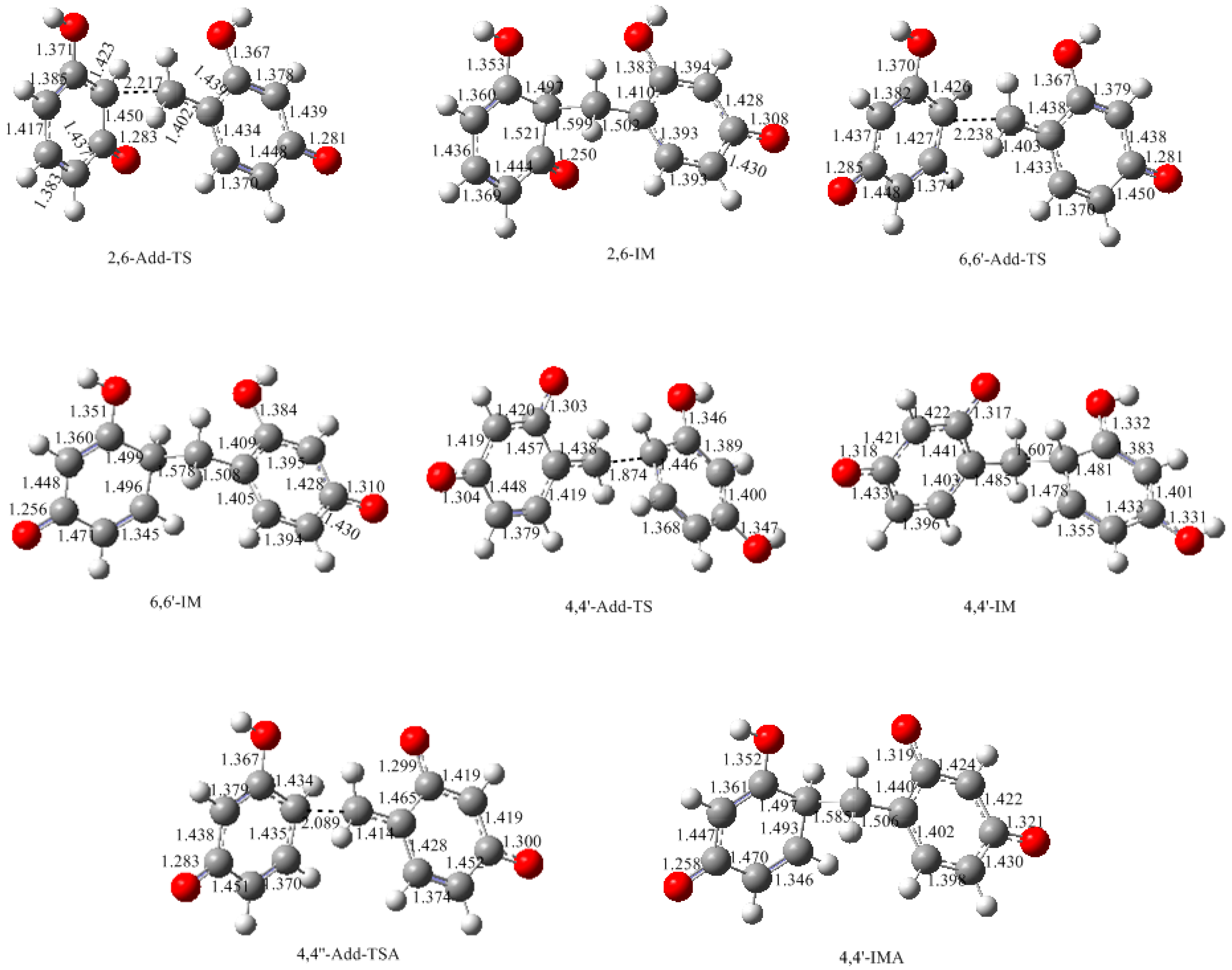
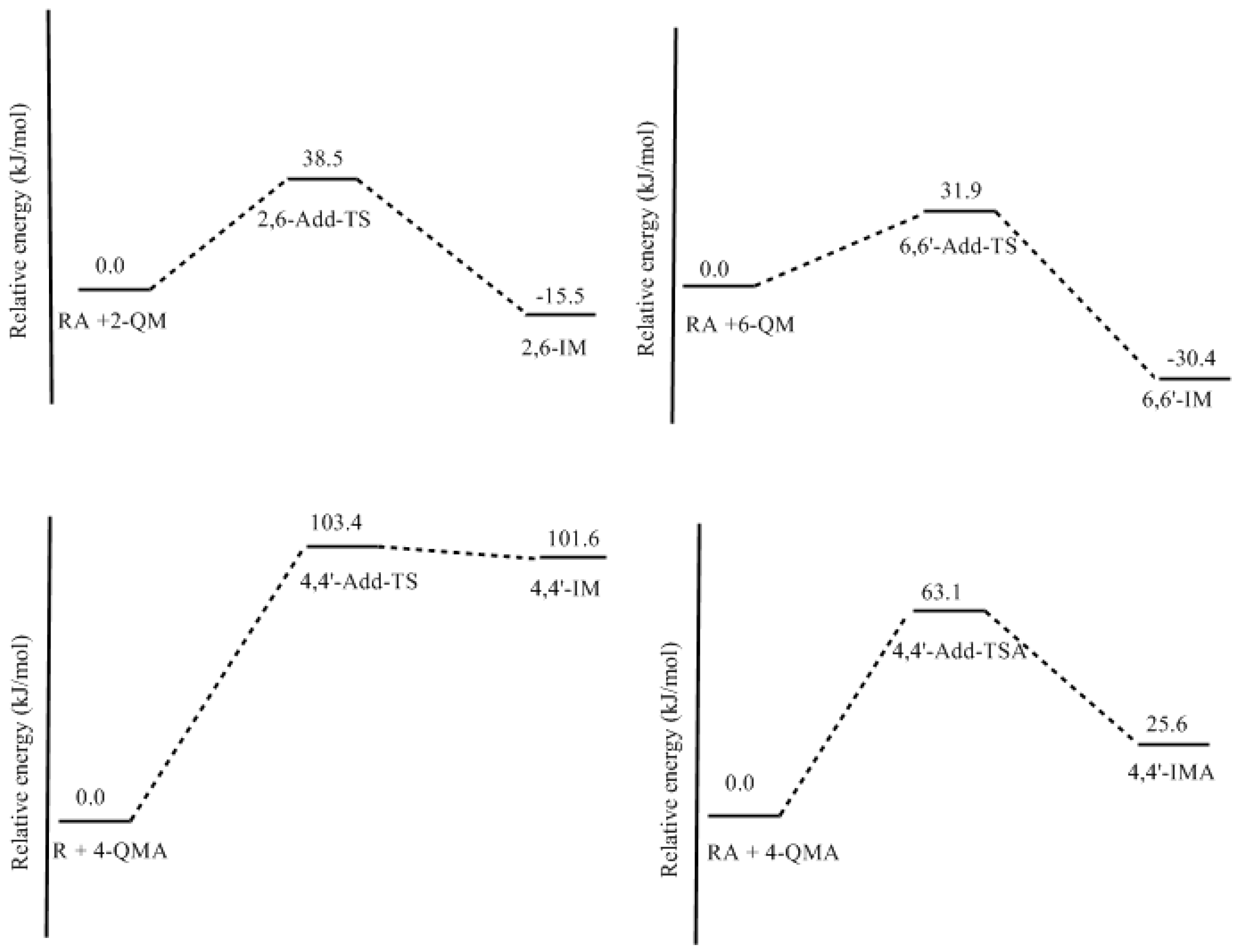
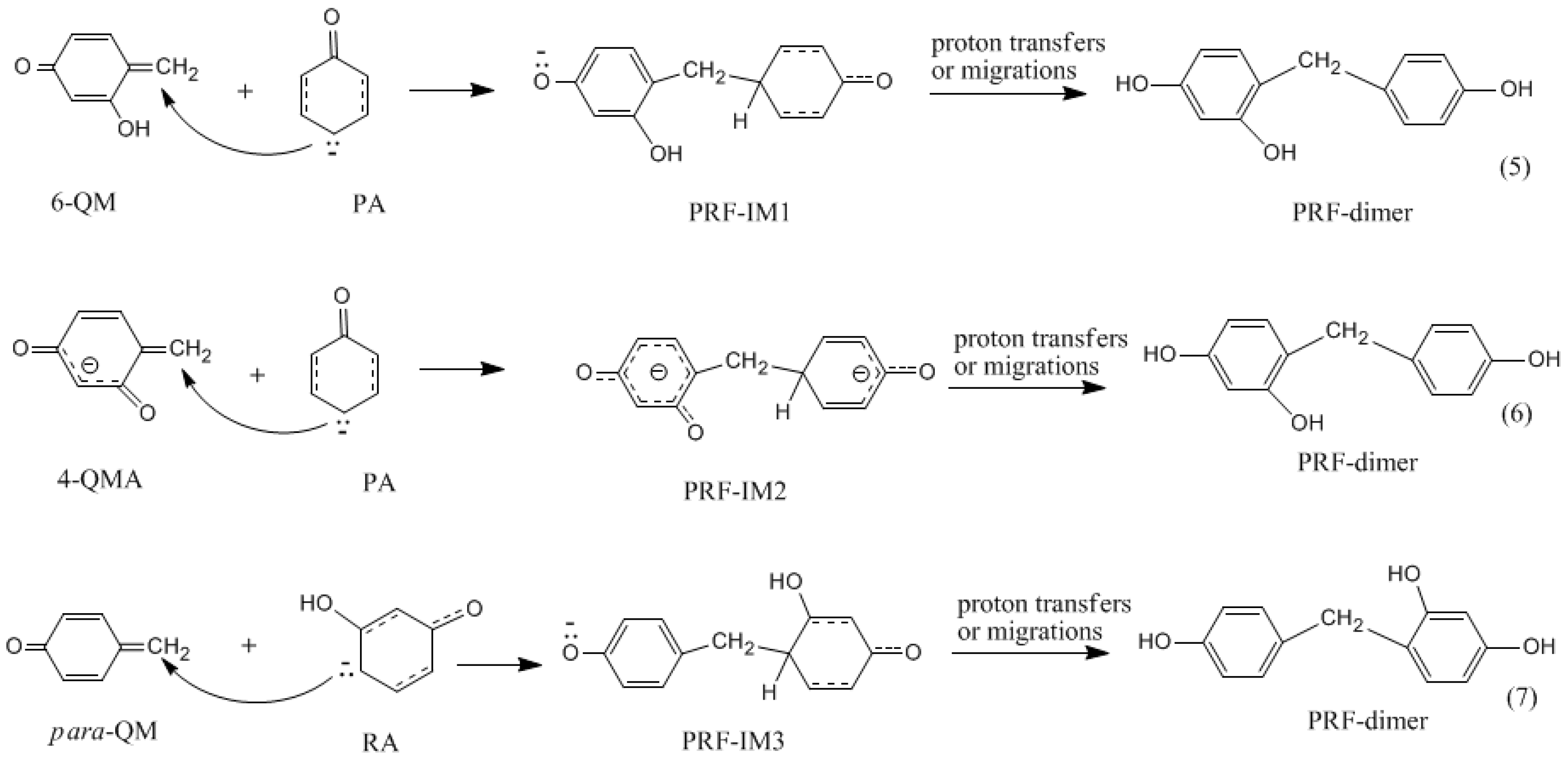
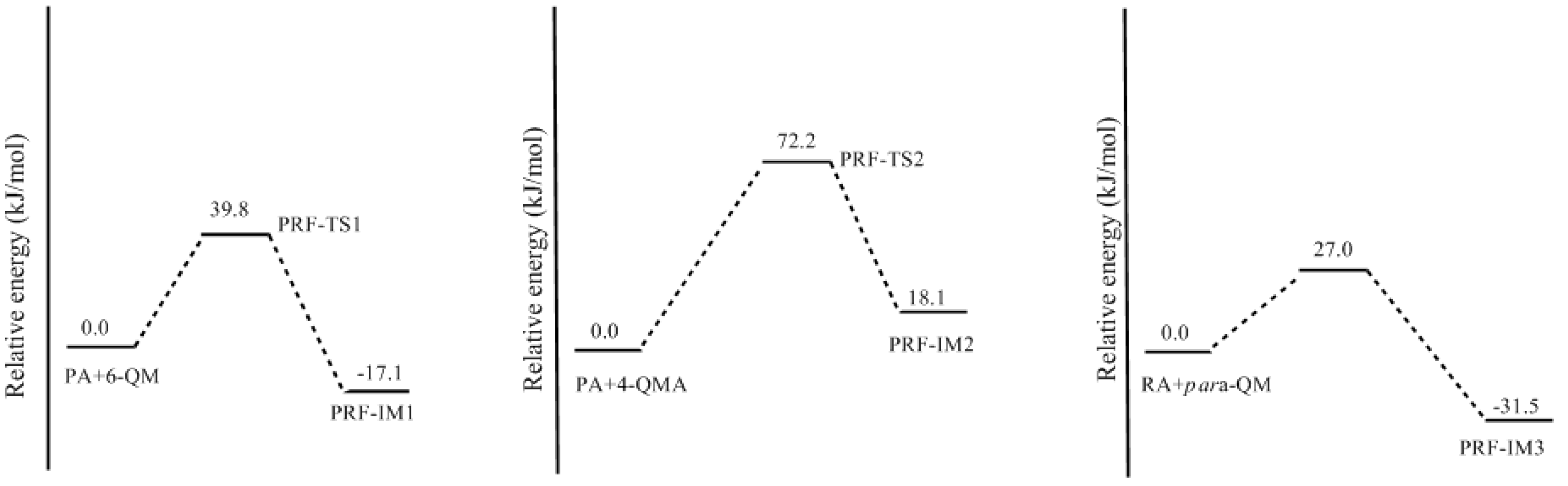
3.2. RF and PRF Polyaddtion Reactions
4. Conclusions
- (1)
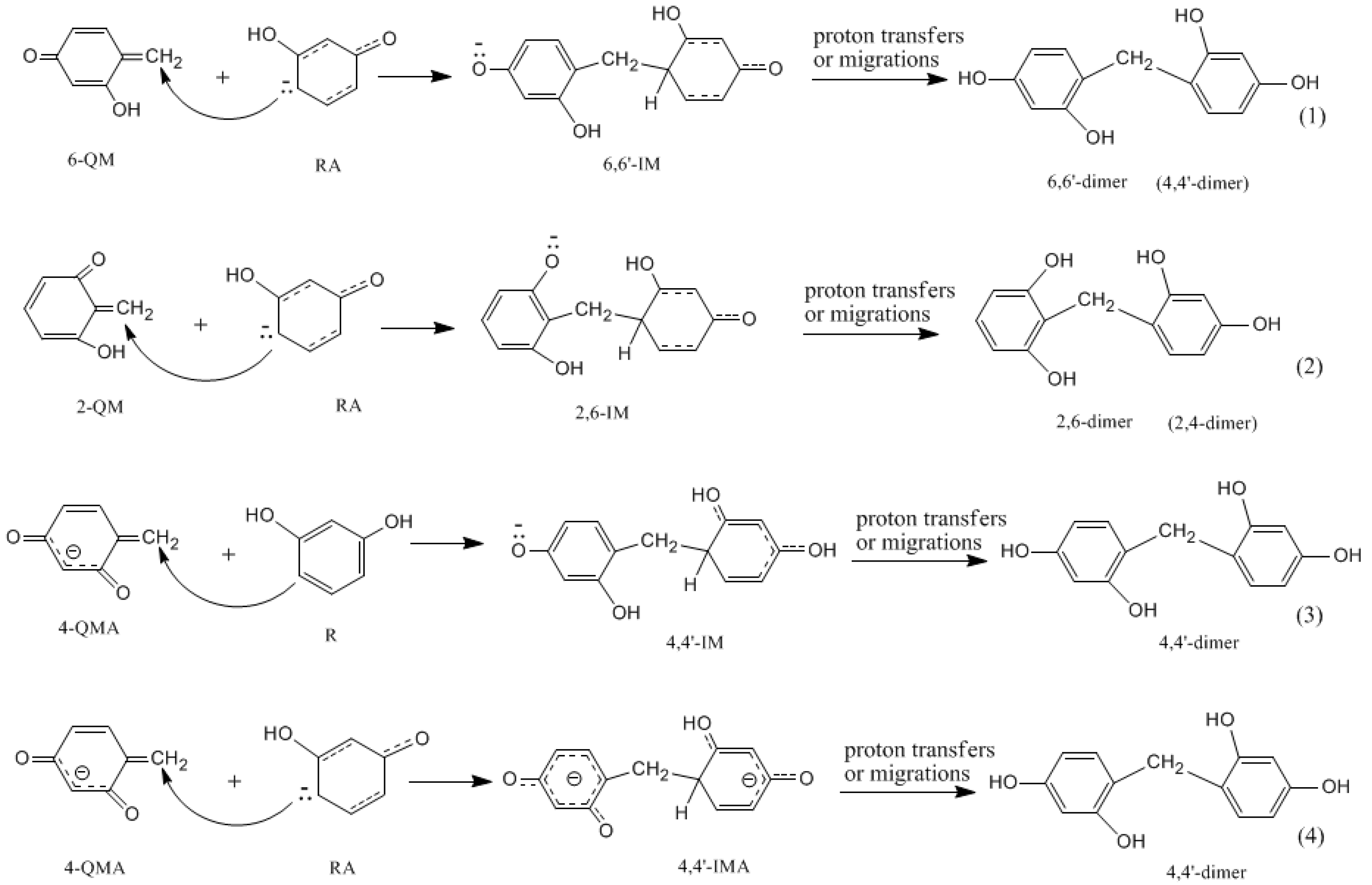
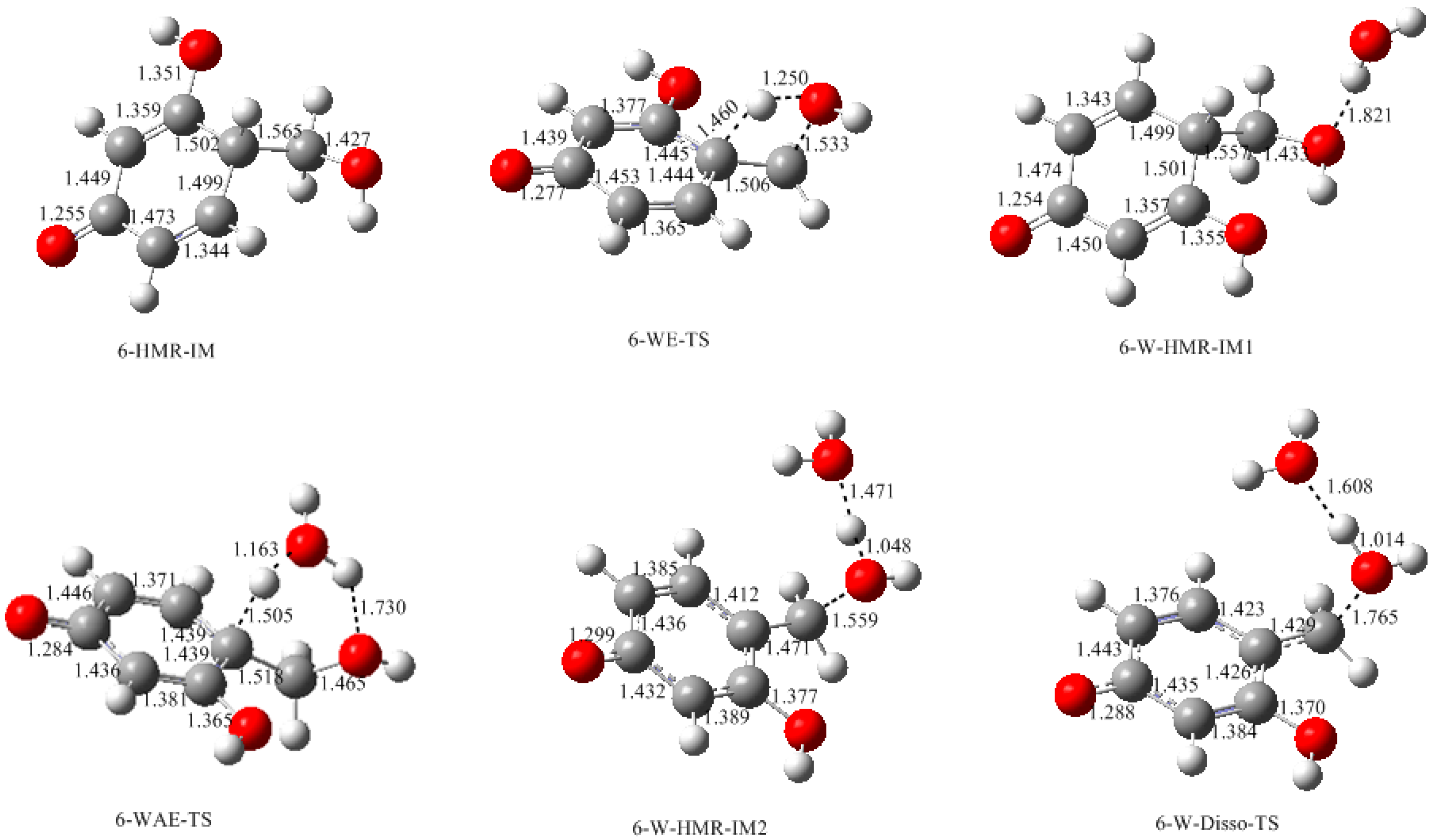
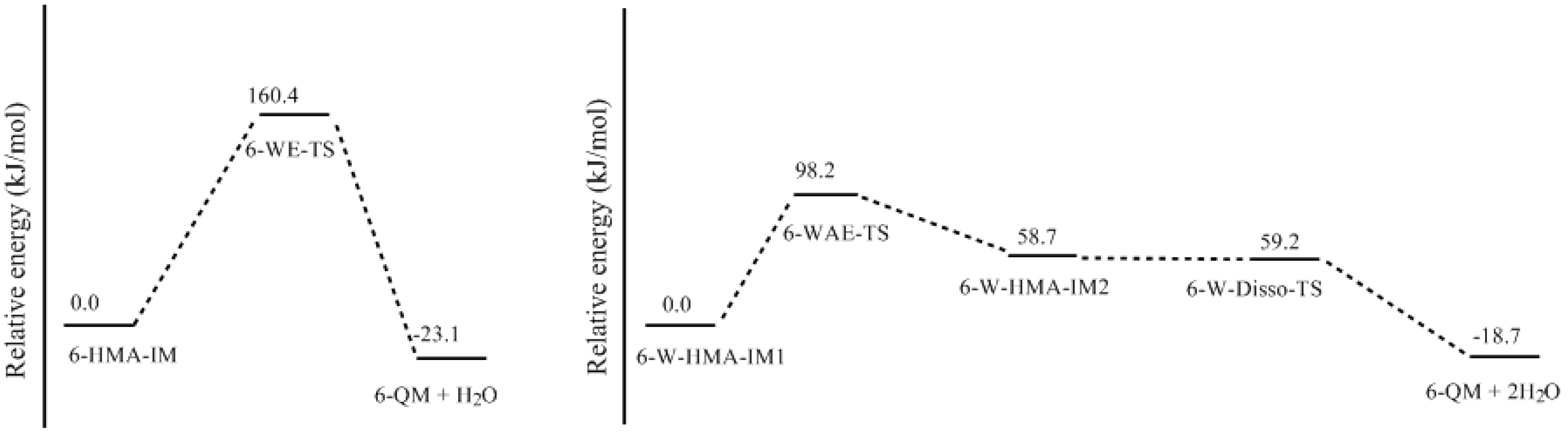
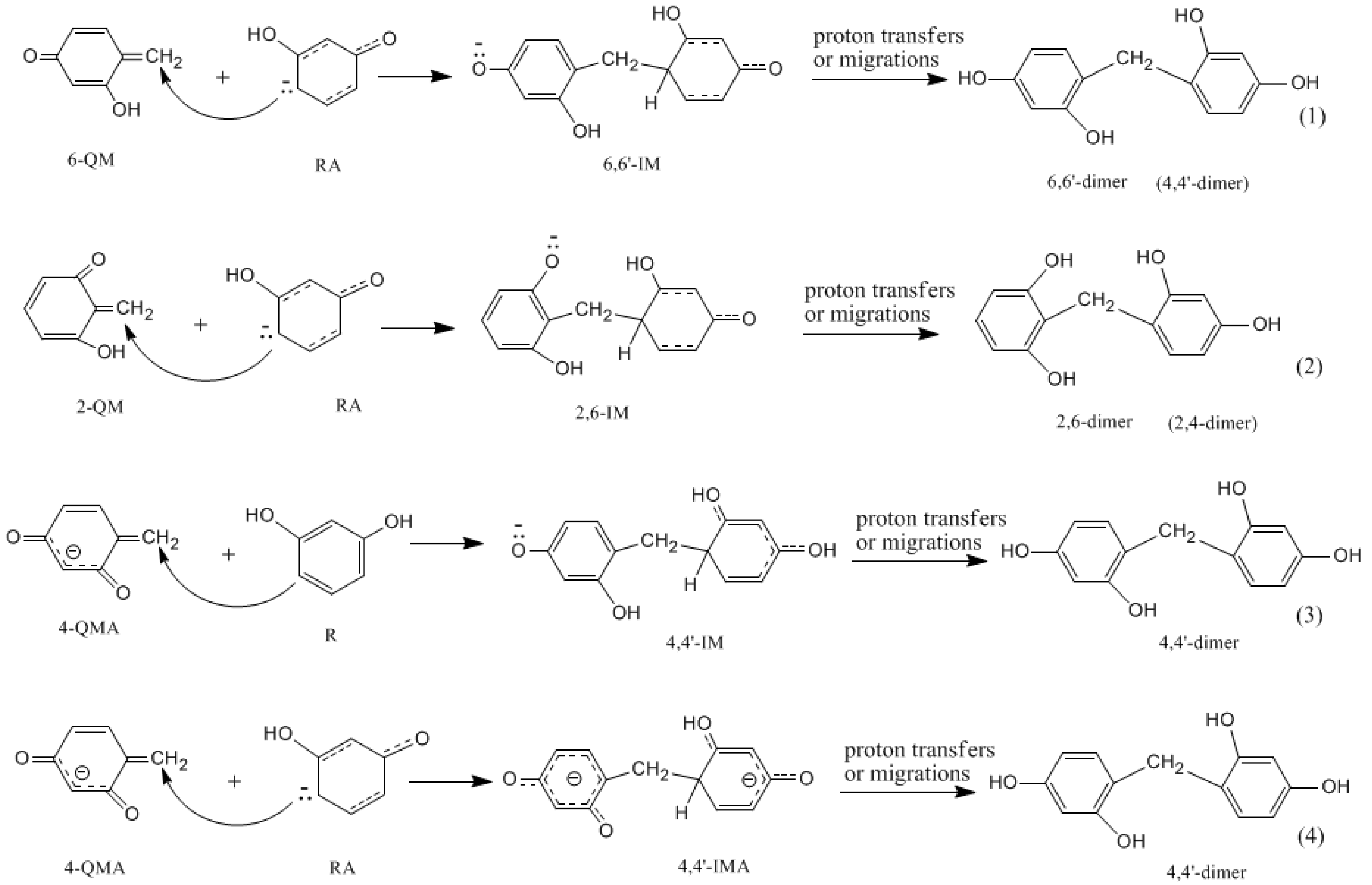
- The condensation reaction includes two steps: Formation of a quinonemethide (QM) intermediate from hydroxymethylresorcinol, followed by Michael addition between the quinonemethide and the resorcinol anion.
- (2)
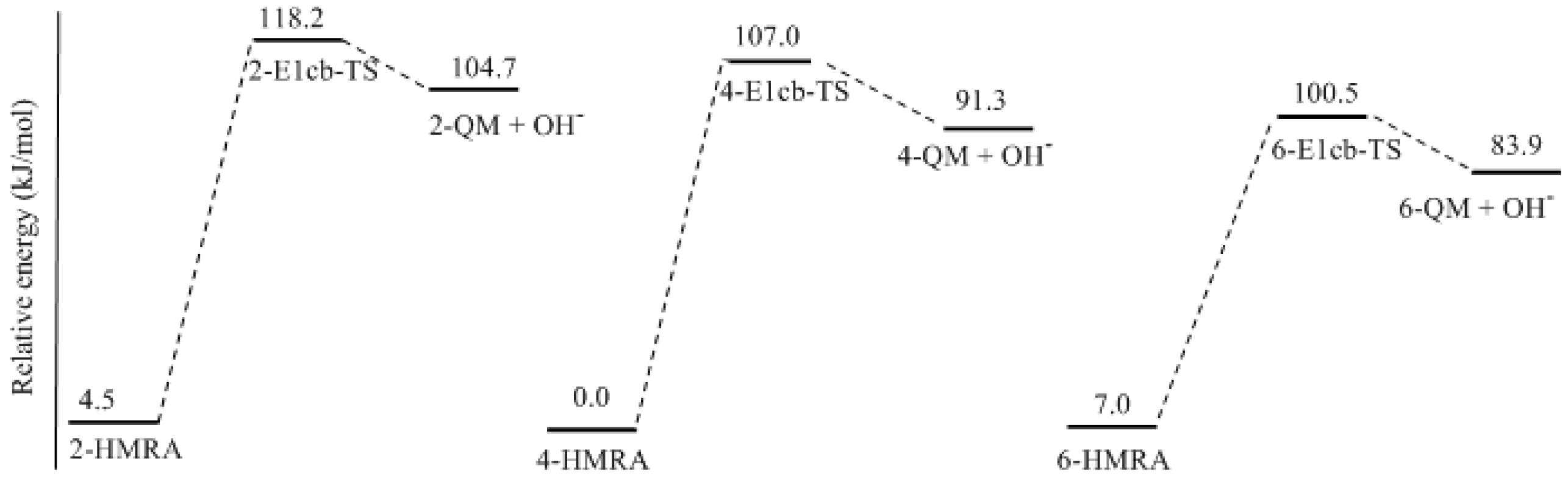
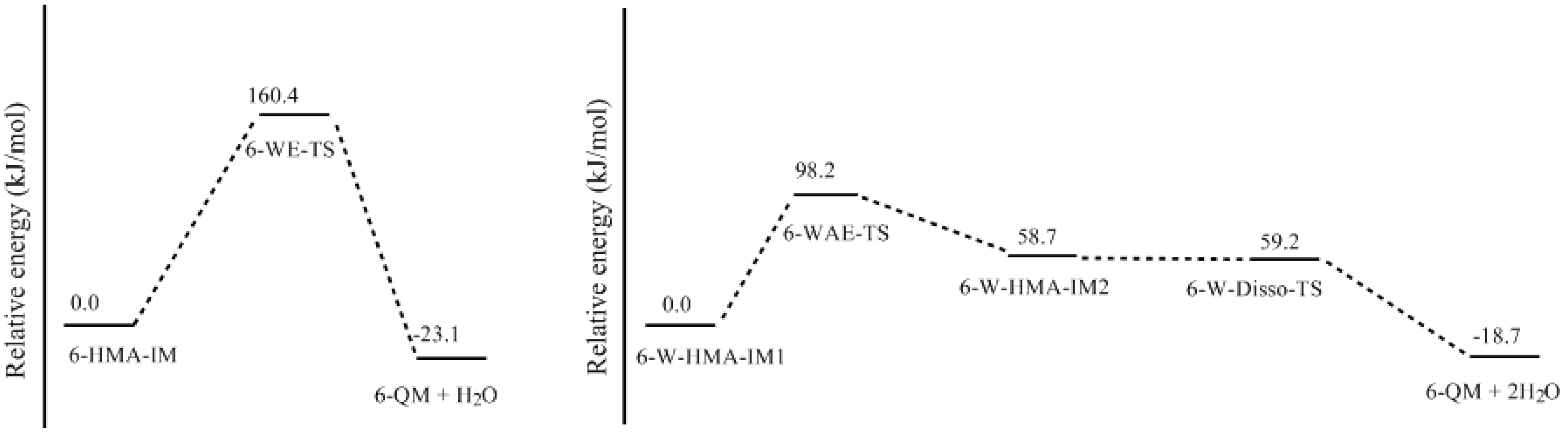
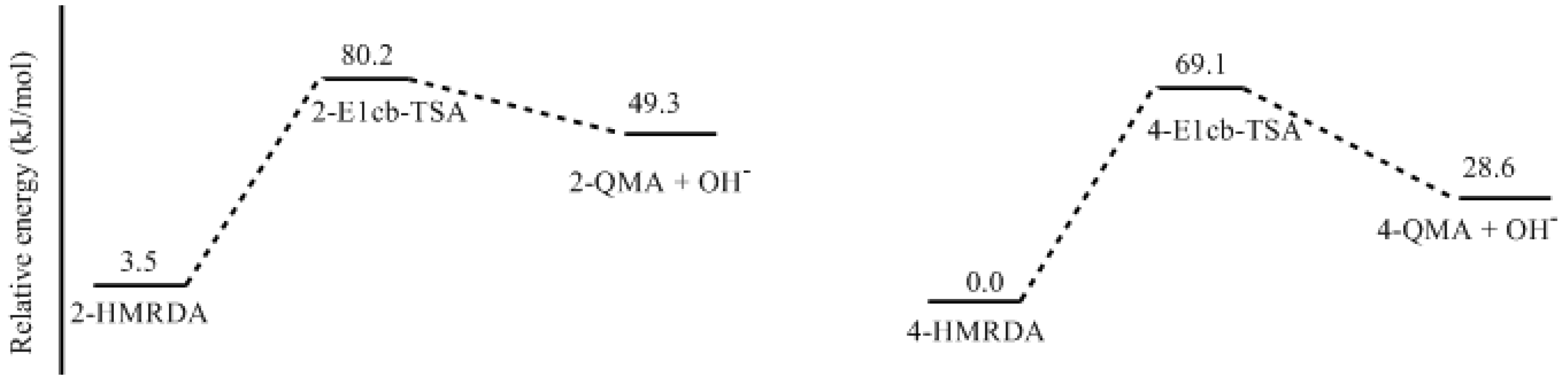
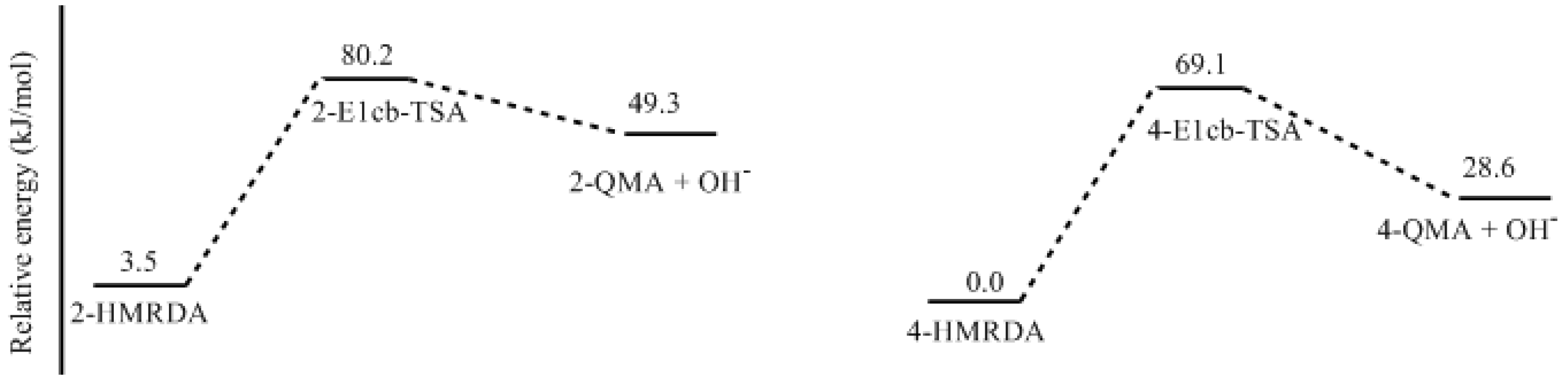
- Two mechanisms, unimolecular elimination of conjugate base (E1cb) and water-aided elimination (WAE), were identified for the formation of quinonemethide. Hydroxymethylresorcinol anion produces a neutral quinonemethide while the dianion produces a quinonemethide anion. The calculated potential energy barriers suggested that quinonemethide anion formation is much more favorable. Although resorcinol-formaldehyde and phenol-formaldehyde share common condensation mechanisms, the former would be faster if the quinonemethide anion is involved.
- (3)
- The potential energy barriers for formation of 2-QM, 4-QM, 6-QM, 2-QMA, and 4-QMA were calculated. The results show that the formation of 6-QM and 4-QMA have relatively lower energy barriers. In addition to higher reactivity of the position-6 hydroxymethylation reaction, the previous experimental observation that the 2,4-(2,6-) and 6,6′-(4,4′-) methylene linkages were dominant polycondensed structures has been rationalized.
- (4)
- The cold-setting characteristic of phenol-resorcinol-formaldehyde co-condensed resin can be attributed to participation of resorcinol quinonemethides in condensations.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chow, S.; Steiner, P.R. Determination of resorcinol content in phenol-resorcinol-formaldehyde resins by infrared spectrometry. Holzforschung 1978, 32, 120–122. [Google Scholar] [CrossRef]
- Kim, M.G.; Amos, W.L.; Barnes, E.E. Investigation of a resorcinol-formaldehyde resin by l3C-NMR spectroscopy and intrinsic viscosity measurement. J. Polym. Sci. A 1993, 31, 1871–1877. [Google Scholar] [CrossRef]
- Scopelitis, E.; Pizzi, A. The chemistry and development of branched PRF wood adhesives of low resorcinol content. J. Appl. Polym. Sci. 1993, 47, 351–360. [Google Scholar] [CrossRef]
- Christiansen, A.W. Resorcinol-formaldehyde Reactions in dilute solution observed by C13 NMR spectroscopy. J. Appl. Polym. Sci. 2000, 75, 1760–1768. [Google Scholar] [CrossRef]
- Pizzi, A.; Pasch, H.; Simon, C.; Rode, K. Structure of resorcinol, phenol, and furan resins by MALDI-TOF mass spectrometry and 13C NMR. J. Appl. Polym. Sci. 2004, 92, 2665–2674. [Google Scholar] [CrossRef]
- Yang, I.; Ahn, S.; Choi, I.; Kim, H.Y.; Oh, S. Adhesives formulated with chemically modified okara and phenol-resorcinol-formaldehyde for bonding fancy veneer onto high-density fiberboard. J. Ind. Eng. Chem. 2009, 15, 398–402. [Google Scholar] [CrossRef]
- Sauget, A.; Zhou, X.; Pizzi, A. Tannin-resorcinol-formaldehyde resin and flax fiber biocomposites. J. Renew. Mater. 2014, 2, 173–181. [Google Scholar] [CrossRef]
- Liu, J.; Chen, R.; Xu, Y.; Wang, C.P.; Chu, F. Resorcinol in high solid phenol-formaldehyde resinsfor foams production. J. Appl. Polym. Sci. 2017, 134, 44881. [Google Scholar]
- Lin, C.; Ritter, J.A. Effect of synthesis pH on the structure of carbon xerogels. Carbon 1997, 35, 1271–1278. [Google Scholar] [CrossRef]
- Liang, C.; Sha, G.; Guo, S. Resorcinol-formaldehyde aerogels prepared by supercritical acetone drying. J. Non-Cryst. Solids 2000, 271, 167–170. [Google Scholar] [CrossRef]
- Al-Muhtaseb, S.A.; Ritter, J.A. Preperation and properties of resorcinol-formaldehyde organic and carbon gels. Adv. Mater. 2003, 15, 101–114. [Google Scholar] [CrossRef]
- Yu, J.; Guo, M.; Muhammad, F.; Wang, A.; Zhang, F.; Li, Q.; Zhu, G. One-pot synthesis of highly ordered nitrogen-containing mesoporous carbon with resorcinol-urea-formaldehyde resin for CO2 capture. Carbon 2014, 69, 502–514. [Google Scholar] [CrossRef]
- Pizzi, A.; Eaton, N.J. A conformational analysis approach to phenol-formaldehyde resins adhesion to wood cellulose. J. Adhes. Sci. Technol. 1987, 1, 191–200. [Google Scholar] [CrossRef]
- Pizzi, A.; De Sousa, G. On the resolution of dihydroxydiphenylmethanes on achiral crystalline Cellulose II—Correlation of experimental and calculated results. Chem. Phys. 1992, 164, 203–216. [Google Scholar] [CrossRef]
- Pizzi, A.; Maboka, S. Calculated values of the adhesion of phenol-formaldehyde oligomers to crystalline CelluloseI. J. Adhes. Sci. Technol. 1993, 7, 81–94. [Google Scholar] [CrossRef]
- Sedano-Mendoza, M.; Alnarran-Lopez, P.; Pizzi, A. Natural lignans adhesion to cellulose: Computational vs experimental results. J. Adhes. Sci.Technol. 2010, 24, 1769–1786. [Google Scholar] [CrossRef]
- Alnarran-Lopez, P.; Pizzi, A.; Navarro-Santos, P.; Hernandes-Esparza, R.; Garza, J. Oligolignols within lignin-adhesive formulations drive their Young’s modulus: A ReaxFF-MD study. Int. J. Adhes. Adhes. 2017. [Google Scholar] [CrossRef]
- Li, T.; Cao, M.; Liang, J.; Xie, X.; Du, G. Theoretical confirmation of the quinonemethide hypothesis for the condensation reactions in phenol-formaldehyde resin synthesis. Polymers 2017, 9, 45. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation-energy density 17. functionals of Becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent molecular orbital methods. 9. Extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-consistent molecular orbital methods. 12. Further extensions of Gaussian-type basis sets for use in molecular-orbital studies of organic-molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V. Efficient diffuse function-augmented basis-sets for anion calculations. 3. The 3-21+G basis set for 1st-row elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods. 25. Supplementary functions for Gaussian Basis Sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Miertus, S.; Tomasi, J. Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. J. Chem. Phys. 1982, 65, 239–245. [Google Scholar] [CrossRef]
- Miertus, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilization of AB initio molecular potentials for the prevision of solvent effects. J. Chem. Phys. 1981, 55, 117–129. [Google Scholar]
- Cossi, M.; Barone, V.; Mennucci, B. Ab initio study of ionic solutions by a polarizable continuum dielectric model. Chem. Phys. Lett. 1998, 286, 253–260. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N. Gaussian 03, Revision B.01, Software for Quantum Chemistry Calculation; Gaussian, Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Durairaj, R.B. Resorcinol, Chemistry, Technology and Applications; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 2005. [Google Scholar]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.; Cao, M.; Liang, J.; Xie, X.; Du, G. Mechanism of Base-Catalyzed Resorcinol-Formaldehyde and Phenol-Resorcinol-Formaldehyde Condensation Reactions: A Theoretical Study. Polymers 2017, 9, 426. https://doi.org/10.3390/polym9090426
Li T, Cao M, Liang J, Xie X, Du G. Mechanism of Base-Catalyzed Resorcinol-Formaldehyde and Phenol-Resorcinol-Formaldehyde Condensation Reactions: A Theoretical Study. Polymers. 2017; 9(9):426. https://doi.org/10.3390/polym9090426
Chicago/Turabian StyleLi, Taohong, Ming Cao, Jiankun Liang, Xiaoguang Xie, and Guanben Du. 2017. "Mechanism of Base-Catalyzed Resorcinol-Formaldehyde and Phenol-Resorcinol-Formaldehyde Condensation Reactions: A Theoretical Study" Polymers 9, no. 9: 426. https://doi.org/10.3390/polym9090426
APA StyleLi, T., Cao, M., Liang, J., Xie, X., & Du, G. (2017). Mechanism of Base-Catalyzed Resorcinol-Formaldehyde and Phenol-Resorcinol-Formaldehyde Condensation Reactions: A Theoretical Study. Polymers, 9(9), 426. https://doi.org/10.3390/polym9090426