Abstract
The quality of plants is often enhanced for diverse purposes such as improved resistance to environmental pressures, better taste, and higher yields. Considering the world’s dependence on plants (nutrition, medicine, or biofuel), developing new cultivars with superior characteristics is of great importance. As part of the ‘omics’ approaches, metabolomics has been employed to investigate the large number of metabolites present in plant systems under well-defined environmental conditions. Recent advances in the metabolomics field have greatly expanded our understanding of plant metabolism, largely driven by potential application to agricultural systems. The current review presents the workflow for plant metabolome analyses, current knowledge, and future directions of such research as determinants of cultivar phenotypes. Furthermore, the value of metabolome analyses in contemporary crop science is illustrated. Here, metabolomics has provided valuable information in research on grain crops and identified significant biomarkers under different conditions and/or stressors. Moreover, the value of metabolomics has been redefined from simple biomarker identification to a tool for discovering active drivers involved in biological processes. We illustrate and conclude that the rapid advances in metabolomics are driving an explosion of information that will advance modern breeding approaches for grain crops and address problems associated with crop productivity and sustainable agriculture.
1. Introduction: The Genotype × Environment × Phenotype (G × E × P) Relationship
Progress in genomic technology has provided incomparable access to information contained in the genomes of cultivated crops (e.g., 1000 Plant Genomes Project). The metabolome can be described as the apogee of the omics trilogy, reflecting the final stage of biological information flow and determining the phenotype. It is well known that complex traits can often result from interactions between the genotype and the environment, with the former also able to prime multiple phenotypes. Moreover, the said interaction can result in different success rates in reproduction and subsequently cause alterations in the genotype, hence the triangular association (Figure 1) reiterates the interrelationship among these [1].
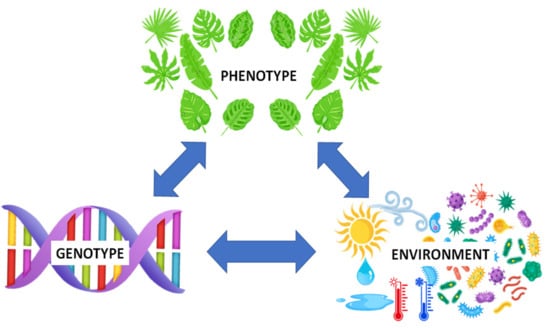
Figure 1.
Abridged illustration of the genotype-environment-phenotype (G × E × P) interaction, demonstrating the interrelationship among the various components. Growth and development of plants and phenotypic plasticity are greatly influenced by the genetic composition, environmental factors and genetic x environment interactions [2,3]. The metabolome represents the final recipient of biological information flow and determines the phenotype.
Several studies have illustrated the impact of environmental changes on the plant phenotype through remodeling of gene expression [3]. This interrelationship has been used as a basis to study complex traits and how they are influenced by environmental variations and genotype interactions [4,5]. These studies provided insights that can be applied in plant breeding during selection and in the development of cultivars with desirable traits. Although genomic selection is commonly used in plant breeding, it is important to note that multiple genetic variants linked to different traits only account for a small fraction of variety between different cultivars of the same species. Additionally, a specific, desirable agronomic trait can be multi-genic, supported by more than one gene or a network of interacting genes. Furthermore, the multi-factorial nature of stress response mechanisms, combined with the distinct influence of the environment and the intricacy of gene x environment interactions, are vital factors that need to be taken into account to understand and predict how the genotype translates into specific phenotypes. To this extent, integrative approaches have attempted to construct a full causal relationship from the genotype to the phenotype, taking into consideration possible environmental effects that might perturb this connection [6,7]. As part of these approaches, metabolomics is a fast-growing field contributing to the elucidation and understanding of plant biological processes. Metabolomics can provide a more in-depth evaluation of phenotypic variation following environmental changes and hence establish good criteria of selection for desirable traits. In the current review the use of metabolomics in crop science, with a focus on small grains, will be discussed to highlight its potential benefits and growing importance.
2. Plant Metabolomes as Responsive and Dynamic Entities
Plant metabolites are reactants, products, or intermediates of enzymatic and other chemical reactions occurring within a biological system [8]. These small molecules can be categorized as primary or secondary metabolites. Generally, the former is directly involved in plant growth, development, and reproduction; and includes sugars, lipids, amino acids, and intermediates of photosynthesis, energy sources, tricarboxylic (TCA) cycle, and glycolysis. The latter, on the other hand, is not directly implicated in plant growth, nonetheless is involved in plant x environmental interactions, and particularly in responses to biotic and abiotic stresses, as well as adaptation to environmental factors. The absence of secondary metabolites may not be fatal to the plant; however, it does jeopardize the defense system of the plant. Secondary metabolites have their origins in primary metabolic pathways and include phenylpropanoids/phenolics [9,10], glucosinolates [11], terpenes, and alkaloids [12,13]. These secondary metabolites play an important role in signaling, enzyme activation, catalytic activity, plant x environment interactions, and plant defense [14]. They are often genus- or species-specific and also responsible for phenotypical characteristics (taste, color, and aroma) of the plants [15].
Plant metabolites are frequently perturbed following interaction with environmental stresses responsible for poor plant growth, reproduction, and crop yields [16]. In fact, approximately 30% of all pre- and post-harvest yields are affected by these factors and cause production losses [17]. Abiotic stresses include heat, cold, drought, waterlogging, toxicity, alkalinity, and salinity among others [18,19], while biotic stresses comprise pathogens (fungi, bacteria, nematodes, and viruses) and various herbivores [17]. Some plants naturally develop a resistance or tolerance against these environmental factors. However, plant breeders often manipulate the production of metabolites and provide information necessary for the development of new plant cultivars, carrying resistance against abiotic and biotic factors. The pool of metabolites in plants, in comparison to other organisms, is the most abundant and diverse. This complexity is attributed to the diversity in structural compositions and the physico-chemical differences in terms of volatility, solubility, quantity, polarity, size, and stability of the compounds found therein [13,20]. Therefore, with the current methodologies and technologies, complete extraction and analysis of all metabolites in a biological system is still extremely challenging. A more holistic perspective of biological systems has been achieved by the use of ‘multi-omics’ approaches [21].
3. Potential of Metabolomics in Crop Science: Big Data, Big Expectations
Plant biology has become more explicit through the progress of ground-breaking systems biology approaches which involve the ‘omics’ technologies [22]. These technologies include genomics, epigenomics, transcriptomics, proteomics, metabolomics, and phenomics [23]. Recently, fluxomics (the study of the total set of fluxes occurring in a metabolic network) has been included and has shown great relevance in the field [24]. Among the ‘omics’ technologies, metabolomics is remarkably increasing in popularity, with the focus on quantitative and qualitative analyses of the whole set of exogenous and endogenous metabolites (metabolome) of a biological system under defined environmental conditions [13,22]. The metabolome reflects the dynamic responses of the plant to physiological, pathophysiological, and environmental stimuli (Figure 2). It has become evident that these small molecules, (metabolites, ≤1500 Da in size) affect cellular physiology through feedback modulation of other ‘omics’ levels. Metabolomics is thus a powerful platform that can provide a comprehensive understanding of the biochemical status of an organism, e.g., to inform on the processes involved in disease progression or environmental adaptation, and hence assist in monitoring gene function [25].
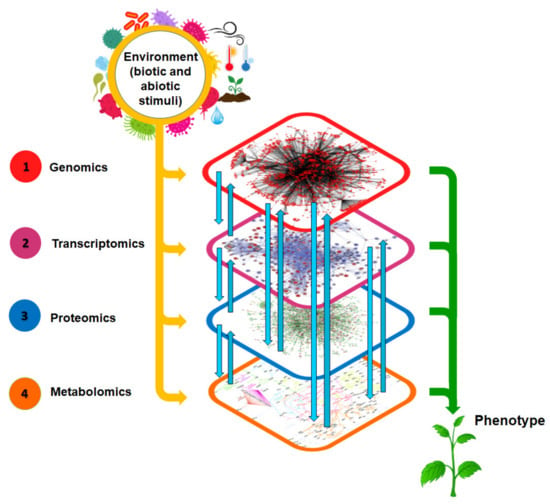
Figure 2.
A systems biology view of biological information flow from genome to phenotype. Metabolomics offers a broad view of an organism’s biochemical and physiological status, and altered metabolomes are a reflection of changes at the genome, transcriptome, and proteome levels. The metabolome is thus considered the underlying biochemical layer reflecting all information expressed and modulated throughout the other omics layers, making it the closest link to the phenotype.
Experimentally, metabolomic studies can be designated as (i) metabolite profiling, defined as the analysis (identification and quantification) of a large group of metabolites; (ii) metabolomic fingerprinting, a rapid high-throughput analysis method providing phenotype characterization and distinction between specific metabolic states [26,27,28] and (iii) metabolic foot-printing, the analysis of metabolites secreted or excreted by an organism [20,27]. These strategies can be used independently, or in combination for a broader understanding of the metabolome, as they provide a point-in-time chemical map of a biological system [20].
Furthermore, metabolomic strategies can be targeted, semi-targeted or untargeted approaches. In a targeted approach, a well-defined specific hypothesis is tested, providing deeper insights and absolute quantification of selected metabolites related to a specific metabolic reaction or pathway [20,27,29]. Thus, in this type of approach preliminary knowledge is required [30]. In semi-targeted analyses, the hypothesis is often undefined; however, the list of metabolites is predefined and quantitatively and tentatively identified [8]. Lastly, the untargeted approach profiles a multitude of metabolites in a sample and can be applied to measure relative concentrations in different conditions or across a population [8,30,31]. Although this approach offers the opportunity for finding new metabolites, one of the limitations is the correct annotation and identification of the unknown metabolites [30].
4. Experimental Designs, Workflows and Analytical Platforms Used in Plant Metabolomics
For a well-designed metabolomics study, experimental characteristics such as sample preparation, instrumental optimization and data acquisition, data analysis (data mining), and data interpretation have to be cautiously considered (Figure 3) [20,32]. The choice of the experimental design depends on the biological question; furthermore, due to the complexity of the plant metabolome, the coverage is restricted by sample preparation methods, sensitivity, and selectivity of the analytical technique. Hence, the use of combined extraction methodologies and analytical platforms to provide a comprehensive understanding of the plant metabolome is often required [12,33,34].
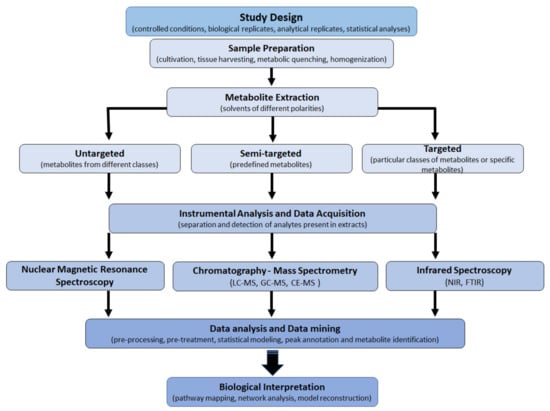
Figure 3.
Basic multi-step workflow of plant metabolomics: sample preparation, data acquisition, and data analysis form the backbone of metabolomic analysis. Analytical platforms include liquid – or gas chromatography coupled to mass spectrometery (LC/GC-MS), capillary electrophoresis coupled to MS (CE-MS), nuclear magnetic resonance (NMR) spectroscopy, and near-infrared (NIR) and Fourier transform (FTIR) spectroscopy. These steps are interrelated and funnel into biological interpretation where the goal is to analyze the intricate networks and pathways for a broad view of the metabolome.
In general, a fast, nonselective, and reproducible method is required to extract a large spectrum of plant metabolites [35]. Extraction methodologies include liquid extraction (temperature- or pressure-assisted), solid-phase extraction and microwave-assisted extraction. The choice of the sample preparation procedure is determined by the plant material used, metabolites of interest, and the chemical properties of the solvent [33,36]. Environmentally safe extractants such as natural deep eutectic solvents, pressurized hot water extraction, and aqueous two-phase extraction solvents are also available [37,38].
Following sample preparation, various analytical platforms are available for data acquisition and are often based on mass spectrometry (MS) or nuclear magnetic resonance (NMR) techniques [39]. Fourier transform infrared spectroscopy (FTIR) is also gaining popularity in the metabolomics field, due to its ability to rapidly and simply analyze and characterize complex building blocks simultaneously [40]. Again, the selection of the right platform depends on the extracted metabolite class(es). Sensitivity and selectivity are important factors to consider when selecting the ideal technique for a given experiment. For more sensitive and selective qualitative and quantitative analyses, MS in combination with liquid – and gas chromatography platforms (LC-MS and GC-MS) are commonly employed [41]. NMR spectroscopy has the advantage of providing a rapid, highly reproducible, and non-destructive high-throughput method with minimal sample preparation [41].
Although these instrumentation systems are undoubtedly main analytical platforms in metabolomics studies, there are several limitations related to their analytical capabilities. NMR, for instance, is restricted by low sensitivity and resolution affecting the number of identifiable metabolites. Such limitations have considerably been improved through developments in two-dimensional (2D) and multidimensional (nD) NMR [39,42]. Despite its longer run time, nD-NMR is able to provide insightful structural and functional information on biomolecules and even raw material. Ultra-fast (UF) 2D NMR was recently developed to reduce the long run time associated with conventional nD-NMR and has shown success in the collection of various spectra in a single scan [13,39,42]. NMR also has the advantage of being a versatile platform capable of analyzing solid, gel, and liquid samples. A new development is the comprehensive multiphase (CMP) NMR capable of analyzing the three states (solid, liquid and gels samples) simultaneously with minimal changes in the overall run time [42,43,44]. This CMP-NMR has been applied for basic structural elucidation in seeds and during plant growth [43,44].
Mass spectroscopy (MS), on the other hand, is often coupled to various chromatography systems in either one or two dimensions [45]. Ion mobility MS has gained increased popularity because of its ability to rapidly analyze samples, remove interferences, separate isomers and isobars; and its ability to identify compounds based on both ion size-to-charge and ion mass-to-charge (m/z) ratios [46]. Two dimensional liquid chromatography (LC) and gas chromatography (GC) as well as multidimensional LC/GC technologies are increasingly gaining popularity as analytical techniques in which two or more columns having different stationary phase selectivity are combined to provide greater resolution and higher peak capacities [47]. This newly established system allows for the simultaneous analysis of both the metabolome and lipidome. Furthermore, the development of hyphenated techniques such as LC-MS-NMR; 2D-LC-MS; 2D-GC-MS, 2D-GC-Q-Orbitrap-MS, among others, offers improved spectral resolution and metabolite identification capabilities [45,48].
MS-based platforms have thus been invaluable in the detection and identification of metabolites by providing spectral data, including accurate mass information and fragmentation patterns, which are essential in computing molecular formulae and structural elucidation of measured m/z ions. However, despite the technological advancements in these analytical platforms, metabolite annotation and identification still remain a bottleneck in metabolomics research. Furthermore, due to the multidimensionality and complexity of metabolome, there is no single analytical system that can cover the whole (extracted) metabolome. These are some of the limitations in (untargeted) metabolomics, which impact on the comprehensive analysis of the metabolome and biological insights generated.
5. Handling and Mining of Metabolomic Data
Data acquired from metabolomic experiments are often large-scale, complex, and require advanced data analytical tools for efficient analysis. Statistics, cheminformatics, and bioinformatics are essential for a comprehensive assessment of these datasets [30]. Data mining, a crucial step in metabolomic workflows, can be carried out in two different approaches. The first is a chemometric approach: the compounds are not firstly identified (or annotated), but their spectral patterns are statistically evaluated to extract relevant spectral features that relate to key questions of the study. The second is a targeted profiling approach: most of the metabolites are firstly annotated (or identified) and then various statistical methods are applied to extract information related to the study, changes and/or valuable biomarkers. The choice of the approach to follow would depend on the study design and availability of resources. However, it is worth pointing out that independently of the approach used, extracting information from metabolomics data is a multistep task that involves data pre-processing and pre-treatment, chemometrics, and statistical analyses, and compound annotation, and identification. Data pre-processing methods include noise filtering, peak detection, and peak alignment [49]. In addition, data pre-treatment or data correction comprises data normalization, centering, scaling, batch effect correction, and data integrity checking [20,49,50]. Both pre-processing and pre-treatment assist in data cleaning in order to emphasize only relevant biological information. These steps inevitably determine the quality and quantity of the information obtained and subsequently, the biological knowledge acquired [20,49].
Statistical analysis (either univariate or multivariate) is an important step in extracting information from the dataset obtained. Univariate data analysis (analyzing a single variable) can be applied to multidimensional data in order to independently assess the significant variation of metabolites among the different samples analyzed [49,51,52]. Examples of univariate statistical tests include Student’s t-test; analysis of variation (ANOVA) and Kruskal–Wallis test. Multivariate data analysis (MVDA) on the other hand are used to explore and extract meaningful information by analyzing multiple variables simultaneously [53,54]. MVDA can be achieved through machine learning methods, being either unsupervised or supervised [49,50,52]. Machine learning is a type of artificial intelligence enabling computers to use different algorithms in order to detect patterns and predict baseline behaviors or properties through training and observation [55]. In the case of unsupervised learning methods, underlying patterns and trends within the dataset can be identified without detailed or explicit inputs (unlabeled data) from the user [55,56,57]. Many unsupervised algorithms have been designed to uncover the complexity of multitudinous datasets. Common examples include principal component analysis (PCA), a linear method, often used to reduce the multi-dimensionality of the dataset; and hierarchical clustering (HCA) which operates based on a distance measured to group data by assessing the similarity and dissimilarity of the observations [20,57]. Other examples of unsupervised models for dimensionality reduction include locally linear embedding, isomap, and independent component analysis [58] and clustering models include K-means [59].
The supervised methods, on the other hand, are often regression-based methodologies and are applied for classification analyses to evaluate the difference between pre-defined (by the user) classes or groups. These methods include the projection to latent structures-discriminant analysis (PLS-DA), the orthogonal projection to latent structures-discriminant analysis (OPLS-DA), k nearest neighbor (KNN) clustering, and more [49,50,52,56]. Of course, the tuning and validation of these models is mandatory, evaluating performance estimates, model bias, and predictability, to ensure statistical significance, reliability, and validity of the generated models. Some of these validation procedures include cross-validation methods and permutation tests [60,61]. The detailed description of these methods is beyond the scope of this review but can be found in literature cited herein. The selection of the appropriate statistical or chemometrics methods to apply in handling and mining metabolomics data depend on the aim and design of the study, the type and size of the collected data, and to some extent, the availability of the resources. Proposed minimum reporting standards for data analysis in metabolomics [62] provide key guidelines and aspects to consider when handling metabolomics data; and detailed explanations and examples of chemometric models (e.g., PCA, HCA, and OPLS-DA) and the applications thereof can be obtained from the cited literature [9,10,11,12,20,21].
Although statistical analyses account for existing connections between variables based on their mathematical criteria, it does not take into account any pre-existing correlation originating from biological origin [30]. Thus, it is often recommended to employ several statistical or data mining techniques. Computational and bioinformatics tools as well as resources for metabolomic analysis have therefore been developed and recently improved significantly. Some of these platforms comprise licensed and non-licensed resources, such as MetaboAnalyst [63], Metabox omicsX [64], MetaCore omicsX [65], InCroMAP [66], SIMCA (soft independent modeling of class analogy) [67] and XCMS [68]. Depending on the algorithm packages in these workflows, the nature of data at hand, and intended analyses, these tools and resources can be used in combination [30,52,60].
The biological meaning and information from metabolomics data depend inevitably on assigning metabolite names or chemical formulae to measured spectral features. This step, namely, metabolite annotation and identification, is a critical step in metabolomic studies and one of the bottlenecks in maximizing the value of metabolomics data [61]. Different analytical methodologies and computational workflows have been developed and established for metabolite annotation and identification. This is an ongoing effort in the metabolomics community; and with technological advancements in analytical platforms, improvements, and exploration of machine learning and computational tools, the repertoire of annotated or identified plant metabolites is gradually expanding. An elaborated presentation and discussion of the metabolite annotation and identification procedures and methods is beyond the scope of this review. However, it is worth pointing out that annotated (or identified) metabolites and generated MVDA models or any statistical description of metabolic changes are key fundamentals in deriving information from metabolomics data. MVDA models can enable the identification of significant features or signatory markers characterizing the biological state(s) or alterations observed at a specific time point under controlled conditions [69]. Currently, there are a number of free libraries and databases often used in the annotation of compounds, such as PubChem, Massbank, Metlin, KEGG, ChEBI, MetaboID among others [70,71,72,73,74,75,76]. Thus, biological interpretation–formulating knowledge from metabolomics information–depends on annotated (or identified) metabolites and correctness of MVDA models and statistical descriptions.
6. Biological Interpretation: From Metabolite to Metabolic Pathways and Networks
One of the main objectives of metabolomics studies is to generate biological insights related to the research question, providing an understanding of the biological system under consideration based on measured (and statistically described) qualitative and quantitative metabolic changes. Thus, the biological interpretation of metabolomics data relies not only on compound identification but also on functional analysis. As part of the latter, mapping and visualization of identified metabolites on general biological networks and metabolic pathways [15] provide insight into their functions and mechanisms under stated conditions. This can be achieved manually by summarizing the information collected from each metabolite (using literature and databases) into a coherent biological explanation. The manual approach is clearly time-consuming as it focuses on each metabolite individually, and it is very limited as it lacks the computation of organized framework for a visual representation of the biochemical network of an organism. In order to overcome such limitations, computer-based approaches have been developed over the past years for data interpretation [77,78].
Recently, a chemo-enrichment analysis approach has been described [79], to overcome the setback in biological interpretation by directly predicting biological activities from spectral features and generating metabolic pathways and networks of all possible metabolite matches. The generated pathways are then compared to pinpoint possible enhanced biological processes within the plant. This approach has the potential to reduce the time and labor associated with metabolite identification and could, in turn, provide greater insight into the functioning of the plant and aid in the identification of agronomically important traits that can be used to construct models for breeding studies applied to grain crops [80]. Additionally, such innovative approaches could broaden comprehension of the plant metabolome due to the fact that current understanding stems mainly from previous studies done on a handful of model plants, which subsequently confines the knowledge to those particular pathways and limits our understanding of pathways absent in those plants [81]. The possibilities for metabolomics research, based on the current trend of advancements, are infinite. Expectedly, the future in this field could surely provide a great number of constructed metabolic pathways and a more holistic view of plant metabolome.
7. Metabolomics Applied to Cultivar/Variety Identification and Cultivar-Specific Responses
Plant breeding is the deliberate manipulation of plant attributes to enhance specific traits, it is an old technology that goes back to the domestication of the first plants; approximately 10,000 years ago [82,83]. These alterations ranged from unintentional changes at the beginning of agricultural domestication to intentional changes through the use of molecular-based tools for precision breeding. In any case, plant breeding is driven by the need to improve plant characteristics through the creation of desired genotypes and phenotypes in new cultivars. With the breakthrough of Mendel’s work in 1900 on the law of inheritance, the scientific basis of the technology was established [84]. This led to the modern era of plant breeding which has tremendously evolved since then. Advancements involved the introduction of different techniques such as double haploid technology, hybrid breeding, wild crosses, introgression of traits from wild relatives, embryo and ovule rescue, mutagenesis, protoplast, and plant cell/tissue/organs cultures and regeneration [85,86].
The integration of metabolomics with genomics, transcriptomics, proteomics, and genetic modification has been applied in studies focused on crop yield and quality improvement. Furthermore, in grain crop breeding, it has been applied for the selection of agronomically important traits, thereby aiding in the improvement of cultivars and varieties [81,87,88]. Both terms ‘varieties’ and ‘cultivars’ are sometimes interchangeably used, although having different meanings. A ‘variety’ (taxonomically ranked after subspecies) is a naturally occurring form of the same species of a plant and a ‘cultivar’ (derived from ‘cultivated variety’) refers to a plant selected for specific attributes preserved through propagation [83]. Plant varieties and cultivars resulted from the domestication of ancestral wild plant species. Since the domestication, modern plants have lost valuable traits due to the reduction of genetic diversity. The limited adaptability of crops to the ever-changing environment, climate change, and increasing food demands has resulted in growing pressure on plant breeders. Fortunately, many important traits, particularly those associated with abiotic and biotic resistance, are still conserved in wild relatives of crops such as rice, maize, wheat, barley, and oats [89,90]. Therefore, breeders are increasingly using wild relatives, with a process known as prebreeding, to reintroduce some of these traits and enhance genetic diversity. The insertion of wild relative traits into modern cultivars can be achieved by conventional breeding or by molecular breeding technologies [89,90,91].
Identification of cultivars and varieties are extremely important steps during grain crop breeding, registration, trade, inspection, and seed production. A rapid and effective fingerprinting method is required for early cultivar identification which is important for the protection of breeders’ intellectual property rights [92,93]. To date, studies in grain crop breeding have employed marker-assisted selection (MAS) for cultivar identification and crop improvement. The selection involves biochemical, morphological-, cytological- and DNA-based markers [94]. Since the development of molecular markers, key challenges in conventional crop improvement programs have been addressed [95]. The selection of desired traits in grain crop breeding is an example of such a challenge, although the presence of a gene does not always imply its full expression [96]. These studies are therefore paving the way for predicting favourable traits with the potential of creating superior hybrids in crop breeding. The plant metabolome, being a representation of the phenotype, thus allows for the application of metabolomics in crop breeding to become a valuable tool for the rapid detection of new traits, the identification of cultivars, and differentiation among cultivars [97].
The concept of applying metabolomics for grain crop breeding studies was previously explored [98]. Although metabolomics was, at the time, still an emerging field, a cost-effective integration with genome sequencing allowed its application in crop breeding programs. These earlier studies paved the way for metabolomics in plant breeding, and in recent reviews [99,100] it is apparent just how much metabolomics research has progressed to its current position. These reviews elaborated on studies that pointed out how metabolomics had enabled the selection of a greater number of desirable traits through technological advancements, allowed the construction of improved metabolic networks, and identified biomarkers for unravelling function and contribution toward improving plant yield, quality, and shelf life.
As we now know, the emergence of metabolomics is highly promising for the prediction of a variety of agronomically important phenotypes and particularly for discovering signature metabolites or metabolic markers (biomarkers) linked to traits of interest [88,101]. In plants, a metabolic marker can be described as an objectively measured characteristic, used as a predictor for plant phenotypical properties. Several studies have evaluated the predictive power and heritability of metabolic markers for the purpose of crop breeding [102]. In a study on maize hybrid crops, metabolic markers were compared to molecular markers and showed that 130 metabolites were nearly equivalent, in terms of predictive power, to 38,000 single nucleotide polymorphisms (SNPs) [103]. Additionally, metabolic inheritance patterns in various plants such as foxtail millet [104], Arabidopsis thaliana (thale cress) [105], and maize [106] have been studied with the purpose of determining the mode of inheritance (i.e., additive, non-additive, dominant and/or overdominant). For the successful implementation of biomarkers, two requirements have to be met: that the predictive ability be robust under varying environmental conditions and that the biomarker be applicable in different plant populations other than the population of origin [107,108]. Additional studies describing the use of metabolomics to identify signatory biomarkers with the aim of differentiating between plant cultivars have been reported [104,109,110,111,112,113,114,115].
The current review highlights the use of metabolomics for the purpose of small grain/cereal cultivar identification and differentiation through metabolic markers for crop improvement (Table 1). Small - or cereal grains, or simply cereals, are seeds belonging to the monocotyledonous family of Poaceae, also referred to as the Gramineae family. Cereal grains include rice, wheat, oats, maize, barley, rye, sorghum, and millet, which have been used as staple food for the world since domestication. These are essential crops for human and livestock nutrition and possess nutraceutical properties attributed to the wide array of phytochemicals therein [114,116,117]. The main phytochemicals produced in cereal grains are flavonoids [118], phenolic acids [119], phytic acid [120], coumarins [121], and terpenes [122]. These compounds are often produced by plants as part of defense response mechanisms to the environment and have health benefits for humans, including anti-oxidant, anti-inflammatory, and anti-diabetic properties to list a few [123].

Table 1.
List of plants/crops where metabolomics was applied as a tool in cultivar identification or improvement.
It is important to understand that the phytochemical content in plants depends not only on the genotype, but also on a range of factors such as biotic and abiotic influences. Additionally, phytochemical production is not only plant-specific but can also be species-specific and even variety or cultivar-specific [124]. These differences can be exploited for the identification or differentiation of cultivars and varieties. Here we have elaborated on how metabolomics was applied in crop science for the differentiation or improvement of a range of cereal crops (as outlined in Table 1). The research applications discussed below were selected to illustrate the various approaches in which metabolomics tools were utilized in order to address fundamental and applied research questions in crop science.
7.1. Rice
The world’s reliance on rice as a food crop has led to the development of a large number of new varieties/cultivars that differ genetically and phenotypically [150]. Different strategies are often employed for the improvement of rice and some examples include conventional hybridization, heterosis breeding, and genetic engineering. One example of the latter is the development of genetically modified (GM; transgenic) cultivars well illustrated in the case of “golden rice” in Asia [151]. Wild type rice does not contain vitamin A and its precursor beta-carotene. This deficiency significantly affects populations using rice as a major staple food. A multi-gene biochemical pathway was incorporated into the rice genome to express beta-carotene that can be metabolized by humans to produce vitamin A [151,152]. The argument around the development of such cultivars is often based on unintended changes that may occur due to pleiotropic effects, mutation, and inactivation of endogenous genes in the transgenic plant; resulting in an unintended difference in the phenotype [153]. However, these unintended changes may not always have a negative impact on the genome. The metabolic regulation and adaptation of “golden rice” following genetic manipulation of phytoene synthase (Psy) and phytoene desaturase (crtI) were comprehensively studied [128]. Seeds of homozygous transgenic golden rice and the non-transgenic counterpart were extracted for proteomics and metabolomics studies. HPLC results revealed high levels of carotenoids in the GM line due to the expression of the Psy and crtI genes. Using a GC-MS protocol [154] alterations in the carbohydrate metabolism pathway were detected in response to the genetic manipulation. High levels of galactose, fructo furanose, D-glucoronate, and D-sorbitol were found in the GM rice. Interestingly, proteomics data correlated with metabolomics results as higher activity of pullulanase and UDP-glucose pyrophosphorylase were observed in the transgenic line. Both enzymes play important in the carbohydrate metabolism linked to the biosynthesis of carotenoids and interconnected to diverse metabolic pathways [128,155]. Moreover, increased activity of the pyruvate, phosphate dikinase (PPDK), a key enzyme in the biosynthesis of pyruvate (a precursor in the pathway leading to carotenoid biosynthesis) was also observed in the GM lines [128]. Despite the argument around the development of golden rice, the crop has been approved in numerous countries.
Another study highlighting the role of metabolomics in assessing rice grains and leaves crop quality was conducted by [111]. Using NMR-based metabolomics, the metabolic quality of two cultivars of rice was evaluated. Distinct metabolic traits in leaves and grains of early and late maturing rice cultivars (EMC and LMC) at all growth stages were successfully identified. Cultivar-specific metabolism was observed until the milk ripe growing stage, through the over- and down-expression of sucrose in leaves of LMC and EMC respectively. It was suggested that the rapid decrease of sucrose in the EMC, probably led to the production of other metabolites namely phenylalanine, leucine, and isoleucine. In the rice grains, remarkably higher quantities of sucrose, amino acids, and fatty acids were found in EMC as compared to LMC. It was concluded that grains from the EMC rice were more nutritious than those from the LMC [111]. Relatedly, the investigation of rice bran from 17 cultivars across 7 different countries revealed a core metabolome and groups of metabolites differentiating them. An average of 411 metabolites was annotated per cultivar, and 71 metabolites were found to discriminate among them. From the cultivar-discriminating metabolites, 34 were associated with 15 metabolic pathways, linked to approximately 1500 genes in total. Gene-metabolite relationships with medicinal and nutritional importance were identified and provided information of great relevance for rice bran improvement [126]. A study was published illustrating the significant differences between cultivars of rice sub-species (Oryza sativa, spp. ‘indica’ and ‘japonica’) [127]. Among the 92 metabolites showing statistically significant variations, 66 were up-regulated in ‘japonica’- and 26 in ‘indica’ cultivars. According to the Random Forest ranking, asparagine was found to be the most discriminant metabolite with higher levels in ‘indica’. The metabolites responsible for differentiating the two sub-species were found to be linked to nitrogen metabolism, inorganic nutrition storage, translocation, and stress responses.
Abiotic stresses are major contributors to the decrease in crop production. A GC-MS metabolomics approach was used to demonstrate the effects of drought and heat on the metabolite distribution of rice cultivars and organs at different developmental stages. More than 50% of metabolites identified in the flag leaves at the flowering stage were significantly different in at least two of the three cultivars (‘Anjali’, ‘Dular’, and ‘N22′). The highest levels of these metabolites were found in the most drought- and heat susceptible cultivar ‘Anjali’. In the flowering spikelets, the highest levels of the polyols, myo-inositol and glycerol, were found in the drought- and heat-tolerant ‘N22’. In the developing seeds, while putrescine and two unknown metabolites levels were the highest in ‘N22’, compounds such as vanillic acid, arabitol, 4-hydroxy-benzoic acid, arbutin, and hydroquinone were the highest in ‘Dular’ (drought tolerant and heat sensitive) and only erythritol and myo-inositol were the highest in ‘Anjali’. Moreover, common and cultivar-specific responses to mild or severe stress were observed in different organs and at different development stages. For instance, in different developmental stages of flag leaves, nine metabolites including phenylalanine, threonine, raffinose, and others were found in all three cultivars. These metabolites were then considered to be specific to the general response to severe drought and heat pressures [113].
Seed storability is an important agronomic trait associated with seed longevity after harvest and storage [156]. This trait was investigated in two hybrid rice cultivars, ‘IIYou 998′ (low) and ‘BoYou 998′ (high) [125]. With the help of an untargeted MS-based metabolomic, it was possible to reveal the difference between the ‘IIYou 998′ and ‘BoYou 998′ cultivars as well as the difference among each cultivar before and after 24-months of storage. An increased level of soluble sugar and sugar-related compounds were found in ‘IIYou 998′ prior and post-storage. In addition, all amino acids detected were more prominent in the same cultivar suggesting their contribution to storage sensitivity. The differential occurrence of these metabolites between the two cultivars also suggested their use as discriminatory markers to distinguish rice cultivars with regard to the storability.
An example of a multi-omics and multi-platform stress response study on rice cultivars following bacterial infection is that of [129]. Prior treatment with the Xanthomonas oryzae pv. oryzae strain PXO99, the metabolic distribution of the two genotypes ‘TP309′ (the parent genotype susceptible to PXO99) and ‘TP309_XA21′ (the transgenic variety resistant to PXO99) was different in terms of TCA intermediates, miscellaneous compounds, and sugar alcohols. Combining transcriptomics and metabolomics, mechanisms affected by the challenges were highlighted. Significantly, different genes and metabolites were compared to look for possible correlation in the response. The over-expression of glutamate decarboxylase in both challenged cultivars and particularly in the resistant one, correlated with a decrease of glutamate observed on GC-MS data and an increase of GABA observed in LC-MS data. Similarly, a correlation between phenylalanine ammonia lyase (PAL) transcript levels, significantly up-regulated in the resistant cultivar, and an elevated amount of phenylalanine was observed. PAL is a stress-responsive defense-related enzyme linking primary – and secondary metabolism, leading to synthesis of phenylpropanoids. The study provided important insights into rice infection and immunity.
7.2. Barley
Barley is a fast-growing crop with great adaptation ability. Although mainly used for animal feeding and malt and beer production, barley remains an important source of high nutritional compounds in human foods [157]. As mentioned before, researchers are constantly investigating strategies for the development of cultivars resistant to environmental stresses and possessing high nutritional content. In a study [115], the effect of salinity on two Tibetan cultivars of hulless barley was investigated with a targeted metabolomics approach. The response observed varied with the stress duration and was cultivar specific. Several compounds were identified in both cultivars; however, nine metabolites including the flavonoids hesperetin and chrysoeriol, were characterized as main biomarkers correlated to salt-tolerance. The time- and cultivar-dependent response observed was in agreement with data from rice cultivars under similar stress conditions [158].
The spatial distribution of metabolites and various elements in seven varieties of barley during germination was investigated in another salt stress study [130]. Using MALDI-MS imaging (MSI), authors tentatively annotated different classes of lipids (e.g, fatty acyls, sphingolipids, and sterol lipids), in seeds of two barley varieties, Mundah and Keel. In both cultivars, the main perturbation in the lipid profile was glycerophospholipids, however, the percentage of alteration was different in each variety. In addition to the lipids, a flavonoid (gaiconin F, identified by LC-QToF-MS) was found to be a discriminant metabolite in both varieties. The study reiterated the role of flavonoids in salt-stressed plants and also the power of using multiple platforms in metabolomics studies.
Three analytical platforms (two LC-MS and NMR) were employed to detect and identify the phenolic compounds present in nine spring varieties of barley leaves [132]. A total of 152 compounds corresponding to different classes of phenolic metabolites were annotated. The study provided extensive coverage of barley metabolites and the fragmentation patterns of hordatines ions and their glycosylated forms were revealed for the first time [132]. A subsequent study [131] revealed the presence of drought-related phenolics such as derivatives of sinapic acid and ferulic acid, polyamines, hordatines and derivatives, and blumenol terpenoids. Polyamines were also found as biomarkers following metabolite profiling of two cultivars of barley, ‘Clipper’ (boron-intolerant) and ‘Sahara’ (boron-tolerant), after exposure to high concentrations of boron. This important micronutrient is capable of affecting plant development at low or high concentrations. The results suggested a link between the polyamine putrescine and boron since the metabolite tends to increase in the intolerant cultivar and decrease in the tolerant one [135].
Metabolomics and transcriptomics approaches were applied to field-grown barley genotypes. Among the four genotypes, two were transgenics (‘ChGP’ and ‘GluB’) that were developed from the parental cultivars ‘Golden Promise’ (GP) and ‘Baronesse’ (B). Alterations in the leaf metabolomes and transcriptomes resulting from the presence of transgenes, and interaction of the cultivars with arbuscular mycorrhizal fungi, were provided. The results in the study revealed cultivar-specific differences, and again highlighted the sensitivity of untargeted and targeted metabolomics employed (as compared to transcriptomics) in uncovering minor differences observed between ‘B’ and ‘GluB’. Moreover, differences resulting from the fungal infection were well-defined at a metabolic level but not evident at a transcriptomic level [134].
In a study with a nutritional focus, barley mutants of the ‘Bombi’ cultivar were investigated for the production of lysine-rich vegetable protein and for augmented β-glucan production. GC-MS metabolomics methods demonstrated a much wider impact of the mutations with unique metabolic patterns associated with the tricarboxylic acid cycle, shikimate-phenylpropanoid pathway, mevalonate, lipid and carbohydrate metabolism in mutants. Furthermore, as an example of genotype x environment x phenotype-relationships, growth temperature primarily affected shikimate-phenylpropanoid and lipid metabolism. Low-temperature markers were benzoic acid, 3-OH-benzoic acid, pyroglutamic acid and the methyl ester of hepta-2,4-dienoic acid, while high-temperature markers were glycerol and the methyl ester of 4-OH-phenylacetic acid [133].
7.3. Sorghum
Sorghum is an important food and fuel crop, and the fifth most important cereal in the world. Untargeted metabolomics was applied to characterize the biochemical variation existing between eleven lines of sorghum (grain and biomass types) and to explore the associations of the metabolome with physiological, morphological, and structural carbohydrate traits [138]. Although all metabolites were not annotated, significantly high variation was observed among genotypes. About 84% of metabolites detected from GC-MS and 76% from LC-MS fluctuated between the sorghum lines. Comparing grain and biomass types, 27% of metabolites detected with both analytical platforms exhibited considerable variation. Moreover, using univariate and multivariate methods such as Spearman’s rank correlation and two-way orthogonal projection to latent structures (O2PLS) respectively, the relationship between metabolites and morpho-physiological traits were pointed out. A positive correlation between the glycosylated flavonoids and photosynthesis-related traits was noted. Chlorogenic acids were also found to be positively correlated to photosynthesis, and negatively correlated to both growth rates and biomass. In a recent study [159], three sweet sorghum cultivars with black, red, and white seeds were investigated to reveal cultivar-specific metabolites involved in the mechanism of color change. Multivariate data analysis tools such as the OPLS-DA model using the variable importance in the projection (VIP), with the consideration of the fold change or p-value allow the selection of discriminant metabolites among the three cultivars. Different flavonoids and anthocyanins were differentially identified in sorghum cultivars. The results revealed that more flavonoids were found in dark-colored seeds in comparison to light-colored seeds, and anthocyanins were responsible for the largest variation among the cultivars. These studies reiterated the close link between the metabolome and phenotype, and the importance of metabolomics in understanding biological processes.
During agricultural production, the effect of abiotic and biotic stresses can have a devastating impact on sorghum yield and growth. The development of new cultivars capable of withstanding such pressures is of great importance and requires the understanding of the key compounds produced and mechanisms occurring during plant x environment interactions. An untargeted metabolomic approach was employed to investigate the metabolic and biochemical responses of two cultivars of sorghum (Samsorg 17 and Samsorg 40) under drought stress [160]. A variation among the two cultivars possessing different levels of drought resistance was observed in under-watered and moderate drought conditions. However, the two cultivars seemed to respond similarly in extreme drought [137]. When it comes to biotic stresses on sorghum, leaf stripe disease and anthracnose caused respectively by the bacterium Burkholderia andropogonis and the fungus Colletotrichum sublineolum, are among the most destructive diseases affecting crop yield [161]. In a recent study, an LC-MS based untargeted metabolomics approach was employed to understand the interaction of different cultivars of sorghum with B. andropogonis. Important cultivar-specific biomarkers such as metabolites from the isoflavonoid and the phenylpropanoid pathways were identified as part of the metabolic reprogramming occurring in sorghum after pathogen attack [114]. Similarly, integrated gene expression analysis was used with metabolomics to not only identify signatory biomarkers but also to reveal potential metabolic pathways associated with sorghum in response to C. sublineolum [136].
7.4. Wheat
Wheat, as a staple crop, is a close third to rice and maize in total world production and is an important source of dietary fiber, micronutrients, and protein [141]. Several studies illustrated the potential use of metabolites as markers in wheat breeding and the importance of metabolomics in identifying quantitative trait loci (QTLs) associated with specific phenotypic traits [162,163,164]. Mature kernels of 145 recombinant inbred lines were collected from the KJ-RIL population which derives from the cross between Kenong 9204 and Jing 411, two elite wheat varieties. Using an LC-MS/MS analytical platform, 1260 different metabolites were detected and quantified, out of which 351 metabolites were putatively annotated and 116 structurally annotated using authentic standards. A large number of metabolic pathways were revealed. These involved different classes of metabolites with important agronomic functions and examples are phenolamides, flavonoids, polyphenols, fatty acids, vitamins, sugar, organic acids, amino acids and derivatives, phytohormones and derivatives, and nucleic acids and derivatives. Line-specific metabolite distribution was noted, and the highest variation was observed with polyphenols and phenolamides. As mentioned before heritability is an important characteristic of metabolic markers, and a factor to consider during trait selection. In this study, the majority of annotated metabolites depicted high heritability with the highest observed being flavonoids. Interestingly, 24 candidate genes were identified directly through quantitative trait loci (QTL) mapping. One was confirmed to express UDP-glycosyltransferase (UGT) and flavonoids such as luteolin, apigenin, quercetin, and kaempferol. The study highlighted the connection between metabolomics and agronomic traits and its applications in genome-wide associations studies (GWAS) to improve functional genomics in grains crop breeding [140].
Forty-five wheat cultivars from three U.S. market classes (tetraploid durum wheat, DW; hexaploid hard wheat, HBW and soft bread wheat, SBW), were successfully distinguished in a metabolomics study [142]. The comparison of DW and bread wheat (BW) revealed 16 metabolites positively correlated with BW and 19 in DW. The identified metabolites in BW were found to be polar lipids, and in the case of DW nonpolar lipids. These metabolites included glycerolipids in BW and fatty acyls in DW and were responsible for the discrimination between the classes. This profiling of wheat cultivars provided invaluable information for the production of good bread quality as the lipid content of wheat has an indirect effect on the dough properties. Similarly, [141] profiled the metabolite composition of different BW genotypes and other wheat species types using high-throughput proton-nuclear magnetic resonance (1H-NMR) screening developed for the analysis of non-purified extracts. The method was successful to demonstrate the diversity within and between species and to quantify asparagine, glycine betaine, and choline.
Integrative biochemical networks in wheat leaves responding to water-deficient conditions were revealed by using a combination of proteomics and metabolomics approaches [139]. A clear sample grouping was visualized between drought-susceptible (‘Bahar’) and drought-tolerant (‘Kavir’) on PCA models, showing the water-deficit condition and control samples separately. A more pronounced distinction was observed between control and drought-stressed samples in ‘Bahar’ showing that the metabolites accounting for these differences are more discriminatory as compared to the ones in ‘Kavir’. In response to drought, 14 and 16 metabolites (with VIP scores > 1) were respectively selected as a discriminant in ‘Kavir’ vs. ‘Bahar’. Using metabolic pathway analysis (MetPA), interesting drought-related pathways and networks were revealed. The main pathway involved in the drought-tolerant cultivar was the metabolism of purine. Guanine and adenine were found to be upregulated in the treated tolerant ‘Kavir’ plants as compared to the controls. In the drought-sensitive ‘Bahar’ cultivar, the top nine metabolic pathways were all correlated to the metabolism of amino acids. This perturbation was characterized by the upregulation of amino acids during water-deficiency. Proline, a well-documented biomarker of drought in plants, increased intensely in both cultivars and was classified as a second variable of importance in ‘Bahar’ and fourth in ‘Kavir’. In addition, several other metabolites and pathways potentially involved in drought responses were suggested in the study, providing a foundation for future research. A similar study on heat stress of six wheat genotypes demonstrated the link between genetic variability and altered metabolic levels [112]. High-resolution LC-MS based metabolite profiling was used, and 64 known metabolites were identified to be affected by heat stress. Among these metabolites, amino acids such as tryptophan, histidine, arginine, and leucine were positively correlated; and threonine, aspartate, 4-aminobutanoic acid (GABA) and phenylalanine were negatively correlated to the stress. Elevated levels of sugar, sugar alcohols, and organic compounds were noted in plants experiencing heat stress. Furthermore, the study highlighted several compounds differentiating heat-stressed plants from controls, and which could possibly be applied as potential biomarkers for genetic improvement studies.
7.5. Maize
As described, the phenotype is strongly affected by genotype, environment, and interactions between genotype and environment. The same is true for the metabolic composition of maize as highlighted by [109], where non-genetically modified (GM) hybrids were used to metabolically characterize the complexity of the genotype, environment, and interaction in hybrid seeds. Six geographically diverse locations in North America were chosen for this purpose. Although the polar metabolite content in maize seeds is usually low (around 5%), by choosing the appropriate location and inbred line, an increase in the level of these metabolites were observed. A total of 45 different polar metabolites were reported in all samples, and their abundance across the different hybrids and locations were compared. Ultimately it was shown that the environment had a greater effect on the accumulation of polar metabolites compared to the different genotypes. Of the 45 metabolites used for comparing the different lines and regions, sucrose, glucose, fructose, and two unidentified metabolites (m/z 133 and m/z 189) contributed to most of the variation observed. Fructose stood out as a major source of variation across the different lines and showed changes influenced by both the environment and genotype.
In a non-targeted study by [143], a comprehensive metabolomic investigation of maize kernels was reported. Unlike the study by [109], [143] only compared 14 maize lines (of which 13 were inbred and 1 hybrid) that were all planted in the same region. After chemometric - and statistical analysis, out of 210 annotated metabolites, 75 metabolites contributed to the differentiation between the different lines. Of these, the main metabolites involved included dihydrokaempferol, nicotinate ribonucleoside, phosphoethanolamine, stigmasterol, and trans-4-hydroxyproline. Conversely, all eight glycolysis metabolites were similarly expressed over the different lines including sucrose and fructose. Ultimately the work provided insights into the maize kernel metabolome that would be useful for metabolic engineering or molecular breeding studies to improve maize kernel quality and yield.
A similar study, involving genotype and environmental effects of GM (Bt) maize involved four non-targeted analytical methodologies to detect unintended effects that result during genetic manipulation [144]. Data collected over three growing seasons showed distinct differences in the proteome and metabolite levels, suggesting that the effect of the environment strongly influenced their production. Among the identified metabolites, fifteen (including glucose, fructose, tocopherol, and inositol) were differentially produced throughout the three seasons. The generated data showed that the growing season had a stronger influence on the metabolome, proteome, and transcriptome of different maize genotypes compared to the genetic modification.
In a more recent study by [145], thirty genetically different inbred maize lines were studied with the aim of identifying metabolite signatures for differentiating hybrid groups based on their silage-earliness and sowing conditions. Interestingly, early sowing of the hybrid lines, compared to normal sowing, had a greater effect on the leaf composition where several metabolites associated with this response were identified. Metabolite-markers associated with sowing conditions and applicable in breeding were suggested. Among these markers were 2-hydroxy-7-methoxy-1,4-benzoxazin-3-one (HMBOA)-glucoside, caffeoylisocitrate, tricin, 2-hydroxy-4,7-dimethoxy-1,4-benzoxazin-3-one (HDMBOA)-glucoside, coumaroylquinate B, cyanidin-glucoside, and dihydroquercetin glucoside associated with chilling tolerance.
In grain crop breeding, the focus is usually on crop improvement particularly with regard to disease resistance, grain size, grain quantities, and grain quality. As previously mentioned, due to the domestication of crops, cultivars and varieties have lost some valuable traits due to intermittent selection, this obstacle has however been overcome by reintroducing these traits and creating genetic diversity through the crossing of wild relatives and cultivated varieties [89,91]. With this concept in mind, [146] set out to identify and measure the abundance of metabolites that were targeted for selection during maize domestication. Seedlings from different maize accessions, its wild relative teosinte, and maize-teosinte cross populations were screened to compare their metabolic profiles and assess the changes caused by domestication. A range of metabolites were detected among which lipids, terpenoids and alkaloids differed greatly in the teosinte and tropical maize crops. Benzoxazinoids, on the other hand, contributed to the differentiation between the tropical and temperate maize crops. Additionally, a multi-omics approach was used where the genome, transcriptome, and metabolome data were used to identify candidate genes that contributed to metabolic divergence seen in the maize-teosinte cross populations. This study has provided novel insights on the use of metabolomics in plant breeding and illustrated how the metabolome has changed during domestication in maize crops. Furthermore, it can be seen as the foundation for upcoming studies to explore metabolic divergence and how it relates to environmental influences.
Maize crops were also used for a more comprehensive understanding of the maize flavonoid pathway by combining metabolic profiling with genetic mapping and gene regulatory network analysis [147]. The authors explored flavonoid biosynthesis and various genetic influences by integrating the genomic, transcriptomic, and metabolomic data. This allowed for the detection and identification of potential candidate genes. Interestingly, when comparing the maize crops from different environments and populations, they were able to recurrently detect 25 QTL that corresponded to 23 different flavonoids. Among these, a number of flavonoids were consistently present across the six environments, many of which were apigenin, chrysoeriol, tricin, and naringenin based. This study showed potential new prospects towards fully understanding the maize flavonoid pathway by moving beyond QTL and normal association mapping.
7.6. Oats
The assessment of cultivars as germplasm for breeding purposes initially involves that the material be analyzed for characteristics that will ensure resistance to biotic and abiotic environmental factors and provide nutritional value in future cultivars. Metabolomic profiles of wild and cultivated varieties of oats were therefore investigated in a study by [110] to compare metabolic changes that occurred from the acculturation of wild varieties to cultivated varieties with respect to organic acids, fatty acids, polyhydric alcohols, and sugars. The metabolic profiles revealed the content of various identified metabolites in extracts from cultivated oats and wild varieties. From the identified metabolites, only a few indicated high variation between the cultivated and wild types. Notably, the oleic acid content of the cultivars was significantly higher than that of the wild types, while the latter had higher contents of linoleic acid and monoacylglycerol. These metabolites contributing to the differentiation of the wild and cultivated varieties are not only nutritionally important but are often associated with resistance or tolerance to abiotic factors. Overall, the study concluded that many important compounds decreased in cultivated forms due to evolution and breeding; however, an increase in the amino acid content in the cultivated varieties was a favorable outcome.
In an abiotic stress study of selected oat cultivars by [148], key metabolites and metabolic pathways were examined in order to define important processes involved in drought tolerance. Generally, the metabolic pathways involved in the production of amino acids, sugars, amines, and sugar alcohols are commonly affected in response to drought stress [165,166]. Metabolic profiles that resulted from the comparison of two oat cultivars, ‘Flega’ (susceptible) and ‘Patones’ (tolerant), after exposure to drought stress, indicated that changes in the photorespiratory pathway were sufficient in distinguishing between the two. ‘Patones’ in this regard showed an increased abundance of metabolites involved in the Calvin cycle, particularly, ribulose-1,5-bisphosphate and 2-phosphogycolate. ‘Flega’, on the other hand, had lower amounts of glyceraldehyde-3P and other components of the Calvin cycle compared to ‘Patones’. Moreover, a comparison of the glyoxylate levels indicated an earlier response to drought stress in the tolerant line and lower antioxidant capacity and photorespiratory activity in the susceptible line. Overall, the study produced models that can be used to suggest possible markers for cereal breeding.
7.7. Millet
The effect of natural variation and species-specific accumulation of primary and secondary metabolites on the metabolomes were predominant features in a study of foxtail millet and millet hybrids [104]. Metabolic analysis indicated that compounds such as flavonoids, phenolamides, hydroxycinnamoyl derivatives, vitamins, and lysophosphatidylcholines were developmentally controlled and showed natural variation in different varieties. Variation was also observed through the accumulation of secondary metabolites when millet and rice were compared. The research thus provided insight into developing predictors for hybrid performance and future analysis of the biosynthesis, and regulation, of relevant metabolic pathways in millet.
Millet is now also being explored for its potential as a biofuel source. To increase biomass production, stimulation by plant growth-promoting bacteria (PGPB) and mycorrhiza were suggested [149]. Upon metabolomic analyses, 28 metabolites were annotated in foxtail shoots after inoculation with both PGPB and mycorrhiza. A significant increase in malate and other metabolites associated with the TCA cycle was observed for PGPB treatment; additionally, in the mycorrhiza treated groups, levels of 4-aminobutyrate, succinate, and asparagine were upregulated. In contrast, downregulation of fructose and glucose was observed in most of the treated groups which the authors described as metabolic shifts toward using more complex carbohydrates during the enhancement of the biomass. Hydroxyvalerate also showed a positive correlation to foxtail height and fresh, dry biomass in groups treated with mycorrhiza only. The study ultimately showed that PGPB and mycorrhiza treatments are beneficial for the enhancement of biomass and for boosting sugar yield.
8. Conclusions and Future Perspectives
Recent reports on plant metabolomics applied in the crop sciences demonstrate the progress in using this omics strategy to understand how the phenotype correlates to the metabolome and, by extension, to reveal the active role of metabolites in normal and stress physiology. Based on the current review it is clear that metabolomics, although considered a relatively new field, is exponentially growing in application and impact in various aspects of the plant sciences. To date, metabolomics has provided valuable molecular information in research on grain crops and identified significant biomarkers under different conditions and/or stressors. However, the value of metabolomics has been redefined from simple biomarker identification to a tool for discovering active drivers involved in biological processes and, therefore, metabolomics has not yet reached its ultimate potential. Gene-based marker-assisted selection, currently a prime focus in grain crop breeding, has shown great success addressing key challenges in crop improvement. However, one of the limitations of MAS is that the presence of genes associated with disease resistance, abiotic stress resilience, or crop yields, do not always guarantee effective expression of the trait. Therefore, the use of metabolic phenotypes in genetic variation studies may provide insights into understanding crop physiology from a metabolomics point of view. The integration of metabolomics with MAS should be explored as an alternative/supplementary tool in grain crop breeding research to ensure greater coverage and confidence in the identification and differentiation of cultivars, as well as varieties in the future. Moreover, the potential of metabolomics as a field of study is continuously being improved by technical innovation of the analytical platforms, promising further perfection for a broader, faster, and more cost-effective coverage of the metabolome. The focus of grain crop breeding generally involves genotyping or phenotyping for trait selection and for cultivar differentiation. In the future, more consideration needs to be given to the use of metabolomics in understanding environmental factors (envirotyping) and how these factors contribute to changes in the genome and ultimately the phenotype and vice versa. Additionally, with the growing use of crop wild relatives to recover desirable traits and reintroduce genetic variation, the use of metabolomics should be explored to accompany current methodologies in the identification of markers for desired traits.
Author Contributions
Conceptualization, I.A.D.; Writing—original draft, C.P. and C.Y.H.D.; Writing—review and editing, L.A.P., F.T., and I.A.D. All authors have read and approved the final version of the manuscript.
Funding
This research received no external funding. The APC was funded by the University of Johannesburg.
Acknowledgments
The authors acknowledge fellowship support to CP (the South African Cultivar Technology Agency, SACTA) and to CYHD (the University of Johannesburg, South Africa).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gienapp, P.; Laine, V.N.; Mateman, A.C.; van Oers, K.; Visser, M.E. Environment-dependent genotype-phenotype associations in avian breeding time. Front. Genet. 2017, 8, 102. [Google Scholar] [CrossRef]
- Aslam, M.M.; Karanja, J.K. Genotype by environment interactions modulate sugarcane response to mechanical wounding stress. Physiol. Mol. Plant Pathol. 2020, 109, 101443. [Google Scholar] [CrossRef]
- Lande, R. Adaptation to an extraordinary environment by evolution of phenotypic plasticity and genetic assimilation. J. Evol. Biol. 2009, 22, 1435–1446. [Google Scholar] [CrossRef] [PubMed]
- Kusmec, A.; de Leon, N.; Schnable, P.S. Harnessing phenotypic plasticity to improve maize yields. Front. Plant Sci. 2018, 9, 1377. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Guo, T.; Mu, Q.; Li, X.; Yu, J. Genomic and environmental determinants and their interplay underlying phenotypic plasticity. Proc. Natl. Acad. Sci. USA 2018, 115, 6679–6684. [Google Scholar] [CrossRef] [PubMed]
- Weckwerth, W. Green systems biology—From single genomes, proteomes and metabolomes to ecosystems research and biotechnology. J. Proteom. 2011, 75, 284–305. [Google Scholar] [CrossRef]
- Nägler, M.; Nägele, T.; Gilli, C.; Fragner, L.; Korte, A.; Platzer, A.; Farlow, A.; Nordborg, M.; Weckwerth, W. Eco-metabolomics and metabolic modeling: Making the leap from model systems in the lab to native populations in the field. Front. Plant Sci. 2018, 9, 1556. [Google Scholar] [CrossRef]
- Liu, X.; Locasale, J.W. Metabolomics: A primer. Trends Biochem. Sci. 2017, 42, 274–284. [Google Scholar] [CrossRef]
- Mhlongo, M.I.; Piater, L.A.; Madala, N.E.; Steenkamp, P.A.; Dubery, I.A. Phenylpropanoid defences in Nicotiana tabacum cells: Overlapping metabolomes indicate common aspects to responses induced by lipopolysaccharides, chitosan and flagellin-22. PLoS ONE 2016, 11, e0151350. [Google Scholar] [CrossRef]
- Zeiss, D.; Mhlongo, M.; Tugizimana, F.; Steenkamp, P.; Dubery, I. Comparative metabolic phenotyping of tomato (Solanum lycopersicum) for the identification of metabolic signatures in cultivars differing in resistance to Ralstonia solanacearum. Int. J. Mol. Sci. 2018, 19, 2558. [Google Scholar] [CrossRef]
- Finnegan, T.; Steenkamp, P.A.; Piater, L.A.; Dubery, I.A. The lipopolysaccharide-induced metabolome signature in Arabidopsis thaliana reveals dynamic reprogramming of phytoalexin and phytoanticipin pathways. PLoS ONE 2016, 11, e0163572. [Google Scholar] [CrossRef] [PubMed]
- Tugizimana, F.; Steenkamp, P.A.; Piater, L.A.; Dubery, I.A. Multi-platform metabolomic analyses of ergosterol-induced dynamic changes in Nicotiana tabacum cells. PLoS ONE 2014, 9, e87846. [Google Scholar] [CrossRef] [PubMed]
- Panda, A.; Parida, A.K.; Rangani, J. Advancement of metabolomics techniques and their applications in plant science: Current scenario and future prospective. In Plant Metabolites and Regulation under Environmental Stress; Ahmad, P., Ahanger, M.A., Singh, V.P., Tripathi, D.K., Alam, P., Alyemeni, M.N., Eds.; Academic Press: London, UK, 2018; pp. 1–36. [Google Scholar]
- Tiwari, R.; Rana, C.S. Plant secondary metabolites: A review. Int. J. Eng. Res. Gen. Sic. 2015, 3, 661–670. [Google Scholar]
- Aggio, R.B.; Ruggiero, K.; Villas-Bôas, S.G. Pathway activity profiling (PAPi): From the metabolite profile to the metabolic pathway activity. Bioinformatics 2010, 26, 2969–2976. [Google Scholar] [CrossRef]
- Pandey, P.; Irulappan, V.; Bagavathiannan, M.V.; Senthil-Kumar, M. Impact of combined abiotic and biotic stresses on plant growth and avenues for crop improvement by exploiting physio-morphological traits. Front. Plant Sci. 2017, 8, 537. [Google Scholar] [CrossRef]
- Bebber, D.P.; Gurr, S.J. Crop-destroying fungal and oomycete pathogens challenge food security. Fungal Genet. Biol. 2015, 74, 62–64. [Google Scholar] [CrossRef]
- Ramegowda, V.; Senthil-Kumar, M. The interactive effects of simultaneous biotic and abiotic stresses on plants: Mechanistic understanding from drought and pathogen combination. J. Plant Physiol. 2015, 176, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Kharub, A.S.; Singh, J.; Lal, C.; Kumar, V. Abiotic stress tolerance in barley. In Abiotic Stress Management for Resilient Agriculture; Minhas, P.S., Rane, J., Pasala, R.K., Eds.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 363–374. [Google Scholar]
- Tugizimana, F.; Piater, L.; Dubery, I. Plant metabolomics: A new frontier in phytochemical analysis. S. Afr. J. Sci. 2013, 109, 1–11. [Google Scholar] [CrossRef]
- Djami-Tchatchou, A.T.; Ncube, E.N.; Steenkamp, P.A.; Dubery, I.A. Similar, but different: Structurally related azelaic acid and hexanoic acid trigger differential metabolomic and transcriptomic responses in tobacco cells. BMC Plant Biol. 2017, 17, 227. [Google Scholar] [CrossRef]
- Gorrochategui, E.; Jaumot, J.; Lacorte, S.; Tauler, R. Data analysis strategies for targeted and untargeted LC-MS metabolomic studies: Overview and workflow. Trends Anal. Chem. 2016, 82, 425–442. [Google Scholar] [CrossRef]
- Manzoni, C.; Kia, D.A.; Vandrovcova, J.; Hardy, J.; Wood, N.W.; Lewis, P.A.; Ferrari, R. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Brief. Bioinform. 2016, 19, 286–302. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.; Krömer, J.O. Fluxomics-connecting ‘omics analysis and phenotypes. Environ. Microbiol. 2013, 15, 1901–1916. [Google Scholar] [CrossRef] [PubMed]
- Ghatak, A.; Chaturvedi, P.; Weckwerth, W. Metabolomics in plant stress physiology. In Plant Genetics and Molecular Biology; Varshney, R.K., Pandey, M.K., Chitikineni, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 187–236. [Google Scholar]
- Kopka, J.; Fernie, A.; Weckwerth, W.; Gibon, Y.; Stitt, M. Metabolite profiling in plant biology: Platforms and destinations. Genome Biol. 2004, 5, 109. [Google Scholar] [CrossRef] [PubMed]
- Ellis, D.I.; Dunn, W.B.; Griffin, J.L.; Allwood, J.W.; Goodacre, R. Metabolic fingerprinting as a diagnostic tool. Pharmacogenomics 2007, 8, 1243–1266. [Google Scholar] [CrossRef]
- Llanes, A.; Andrade, A.; Alemano, S.; Luna, V. Metabolomic approach to understand plant adaptations to water and salt stress. In Plant Metabolites and Regulation under Environmental Stress; Ahmad, P., Ahanger, M.A., Singh, V.P., Tripathi, D.K., Alam, P., Alyemeni, M.N., Eds.; Academic Press: London, UK, 2018; pp. 133–144. [Google Scholar]
- Wolfender, J.L.; Marti, G.; Thomas, A.; Bertrand, S. Current approaches and challenges for the metabolite profiling of complex natural extracts. J. Chromatogr. A 2015, 1382, 136–164. [Google Scholar] [CrossRef]
- Cambiaghi, A.; Ferrario, M.; Masseroli, M. Analysis of metabolomic data: Tools, current strategies and future challenges for omics data integration. Brief. Bioinform. 2017, 18, 498–510. [Google Scholar] [CrossRef]
- Ribbenstedt, A.; Ziarrusta, H.; Benskin, J.P. Development, characterization and comparisons of targeted and non-targeted metabolomics methods. PLoS ONE 2018, 13, 0207082. [Google Scholar] [CrossRef]
- Cook, D.W.; Rutan, S.C. Chemometrics for the analysis of chromatographic data in metabolomics investigations. J. Chemom. 2014, 28, 681–687. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, S.; Cui, X.; Pan, C.; Zhang, A.; Chen, F. A review of sample preparation methods for the pesticide residue analysis in foods. Cent. Eur. J. Chem. 2012, 10, 900–925. [Google Scholar] [CrossRef]
- Mushtaq, M.Y.; Choi, Y.H.; Verpoorte, R.; Wilson, E.G. Extraction for metabolomics: Access to the metabolome. Phytochem. Anal. 2014, 25, 291–306. [Google Scholar] [CrossRef]
- Moco, S.; Vervoort, J.; Bino, R.J.; De Vos, R.C.; Bino, R. Metabolomics technologies and metabolite identification. Trends Anal. Chem. 2007, 26, 855–866. [Google Scholar] [CrossRef]
- Altemimi, A.; Lakhssassi, N.; Baharlouei, A.; Watson, D.; Lightfoot, D. Phytochemicals: Extraction, isolation, and identification of bioactive compounds from plant extracts. Plants 2017, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Khoza, B.S.; Chimuka, L.; Mukwevho, E.; Steenkamp, P.A.; Madala, N.E. The effect of temperature on pressurised hot water extraction of pharmacologically important metabolites as analysed by UPLC-qTOF-MS and PCA. Evid. Based Complement. Altern. Med. 2014, 1–9. [Google Scholar] [CrossRef]
- Hamany Djande, C.Y.; Piater, L.A.; Steenkamp, P.A.; Madala, N.E.; Dubery, I.A. Differential extraction of phytochemicals from the multipurpose tree, Moringa oleifera, using green extraction solvents. S. Afr. J. Bot. 2018, 115, 81–89. [Google Scholar] [CrossRef]
- Emwas, A.-H.; Alghrably, M.; Al-Harthi, S.; Gabriel Poulson, B.; Szczepski, K.; Chandra, K.; Jaremko, M. New Advances in Fast Methods of 2D NMR Experiments; IntechOpen Press: London, UK, 2019. [Google Scholar]
- Junot, C.; Fenaille, F. Metabolomics using Fourier transform mass spectrometry. In Fundamentals and Applications of Fourier Transform Mass Spectrometry; Schmitt-Kopplin, P., Kanawati, B., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 325–356. [Google Scholar]
- Wishart, D.S. NMR metabolomics: A look ahead. J. Magn. Reson. 2019, 306, 155–161. [Google Scholar] [CrossRef]
- Gouilleux, B.; Rouger, L.; Giraudeau, P. Ultrafast 2D NMR: Methods and Applications. Annu. Rep. NMR Spectrosc. 2018, 93, 75–144. [Google Scholar]
- Fortier-McGill, B.E.; Dutta Majumdar, R.; Lam, L.; Soong, R.; Liaghati-Mobarhan, Y.; Sutrisno, A.; de Visser, R.; Simpson, M.J.; Wheeler, H.L.; Campbell, M.; et al. Comprehensive multiphase (CMP) NMR monitoring of the structural changes and molecular flux within a growing seed. J. Agric. Food Chem. 2017, 65, 6779–6788. [Google Scholar] [CrossRef]
- Bastawrous, M.; Jenne, A.; Tabatabaei Anaraki, M.; Simpson, A.J. In-vivo NMR spectroscopy: A powerful and complimentary tool for understanding environmental toxicity. Metabolites 2018, 8, 35. [Google Scholar] [CrossRef]
- Kapoore, R.V.; Vaidyanathan, S. Towards quantitative mass spectrometry-based metabolomics in microbial and mammalian systems. Philos. Trans. Royal Soc. A 2016, 374, 20150363. [Google Scholar] [CrossRef]
- Paglia, G.; Astarita, G. Metabolomics and lipidomics using traveling-wave ion mobility mass spectrometry. Nat. Protoc. 2017, 12, 797. [Google Scholar] [CrossRef]
- Stoll, D.R.; Harmes, D.C.; Staples, G.O.; Potter, O.G.; Dammann, C.T.; Guillarme, D.; Beck, A. Development of comprehensive online two-dimensional liquid chromatography / mass spectrometry using hydrophilic interaction and reversed-phase separations for rapid and deep profiling of therapeutic antibodies. Anal. Chem. 2018, 90, 5923–5929. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.R.; Hoyt, D.W.; Walker, S.M.; Ward, J.K.; Nicora, C.D.; Bingol, K. Unambiguous metabolite identification in high-throughput metabolomics by hybrid 1D 1H NMR/ESI MS1 approach. Magn. Reson. Chem. 2016, 54, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, A.A.; da Silva, R.; Knight, R.; Lopes, N.P.; Dorrestein, P.C. Global chemical analysis of biology by mass spectrometry. Nat. Rev. Chem. 2017, 1, 54. [Google Scholar] [CrossRef]
- Saccenti, E.; Hoefsloot, H.C.; Smilde, A.K.; Westerhuis, J.A.; Hendriks, M.M. Reflections on univariate and multivariate analysis of metabolomics data. Metabolomics 2014, 10, 361–374. [Google Scholar] [CrossRef]
- Vargason, T.; Howsmon, D.; McGuinness, D.; Hahn, J. On the use of multivariate methods for analysis of data from biological networks. Processes 2017, 5, 36. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, S.; Sen, P.; Dickens, A.M.; Hyötyläinen, T.; Orešiè, M. An overview of metabolomics data analysis: Current tools and future perspectives. Comp. Anal. Chem. 2018, 82, 387–413. [Google Scholar]
- Madala, N.E.; Piater, L.A.; Steenkamp, P.A.; Dubery, I.A. Multivariate statistical models of metabolomic data reveals different metabolite distribution patterns in isonitrosoacetophenone-elicited Nicotiana tabacum and Sorghum bicolor cells. SpringerPlus 2014, 3, 254. [Google Scholar] [CrossRef]
- Schillemans, T.; Shi, L.; Liu, X.; Åkesson, A.; Landberg, R.; Brunius, C. Visualization and interpretation of multivariate associations with disease risk markers and disease risk—The Triplot. Metabolites 2019, 9, 133. [Google Scholar] [CrossRef]
- Cuperlovic-Culf, M. Machine learning methods for analysis of metabolic data and metabolic pathway modeling. Metabolites 2018, 8, 4. [Google Scholar] [CrossRef]
- Sathya, R.; Abraham, A. Comparison of supervised and unsupervised learning algorithms for pattern classification. Int. J. Adv. Res. Artif. Intell. 2013, 2, 34–38. [Google Scholar] [CrossRef]
- Maione, C.; Nelson, D.R.; Barbosa, R.M. Research on social data by means of cluster analysis. Appl. Comp. Inform. 2019, 15, 153–162. [Google Scholar] [CrossRef]
- Bunte, K.; Biehl, M.; Hammer, B. A general framework for dimensionality-reducing data visualization mapping. Neural Comput. 2012, 24, 771–804. [Google Scholar] [CrossRef]
- Capó, M.; Pérez, A.; Lozano, J.A. An efficient approximation to the K-means clustering for massive data. Knowl. Based Syst. 2017, 117, 56–69. [Google Scholar] [CrossRef]
- Wanichthanarak, K.; Fan, S.; Grapov, D.; Barupal, D.K.; Fiehn, O. Metabox: A toolbox for metabolomic data analysis, interpretation and integrative exploration. PLoS ONE 2017, 12, 0171046. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Computational approaches to metabolomics. Methods Mol. Biol. 2010, 593, 283–313. [Google Scholar]
- Sumner, L.W.; Amberg, A.; Barrett, D.A.; Beale, M.; Beger, R.; Daykin, C.; Fan, T.; Fiehn, O.; Doodacre, R.; Griffin, J.; et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef]
- MetaboAnalyst. Available online: https://www.metaboanalyst.ca/ (accessed on 4 June 2020).
- Metabox OmicsX. Available online: https://omictools.com/metabox-tool (accessed on 4 June 2020).
- MetaCore OmicsX. Available online: https://omictools.com/metabox-tool (accessed on 4 June 2020).
- InCroMAP. Available online: http://www.ra.cs.uni-tuebingen.de/software/InCroMAP/index.htm (accessed on 4 June 2020).
- SIMCA. Available online: https://umetrics.com/ (accessed on 4 June 2020).
- XCMS. Available online: https://xcmsonline.scripps.edu (accessed on 4 June 2020).
- Wolfender, J.L.; Rudaz, S.; Hae Choi, Y.; Kyong Kim, H. Plant metabolomics: From holistic data to relevant biomarkers. Curr. Med. Chem. 2013, 20, 1056–1090. [Google Scholar]
- Klassen, A.; Faccio, A.T.; Canuto, G.A.B.; da Cruz, P.L.R.; Ribeiro, H.C.; Tavares, M.F.M.; Sussulini, A. Metabolomics: Definitions and significance in systems biology. In Metabolomics: From Fundamentals to Clinical Applications; Sussulini, A., Ed.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 31–37. [Google Scholar]
- Pubchem. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 4 June 2020).
- MassBank. Available online: https://massbank.eu/MassBank/Search (accessed on 4 June 2020).
- Metlin. Available online: https://metlin.scripps.edu/ (accessed on 4 June 2020).
- KEGG. Available online: https://www.genome.jp/kegg/ (accessed on 4 June 2020).
- ChEBI. Available online: https://www.ebi.ac.uk/chebi/ (accessed on 4 June 2020).
- MetaboID. Available online: https://www.ebi.ac.uk/chebi/ (accessed on 4 June 2020).
- Cottret, L.; Frainay, C.; Chazalviel, M.; Cabanettes, F.; Gloaguen, Y.; Camenen, E.; Merlet, B.; Heux, S.; Portais, J.C.; Poupin, N.; et al. MetExplore: Collaborative edition and exploration of metabolic networks. Nucleic Acids Res. 2018, 46, 495–502. [Google Scholar] [CrossRef]
- Chong, J.; Wishart, D.S.; Xia, J. Using Metaboanalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef]
- Xia, J. Computational strategies for biological interpretation of metabolomics data. Adv. Exp. Med. Biol. 2017, 965, 191–206. [Google Scholar] [PubMed]
- Razzaq, A.; Sadia, B.; Raza, A.; Khalid Hameed, M.; Saleem, F. Metabolomics: A way forward for crop improvement. Metabolites 2019, 9, 303. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Yang, L.; Zhang, D.; Shi, J. Plant metabolomics: An indispensable system biology tool for plant science. Int. J. Mol. Sci. 2016, 17, 767. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A. Plant Breeding: A prospect in developing world. EC Microbiol. 2017, 8, 272–278. [Google Scholar]
- McCouch, S. Diversifying selection in plant breeding. PLoS Biol. 2004, 2, 347. [Google Scholar] [CrossRef]
- Duvick, D.N. Utilization of biotechnology in plant breeding for North America: A status report. In Perspektiven Nachwachsender Rohstoffe in der Chemie; Eierdanz, H., Ed.; Wiley-VCH: Weinheim, Germany, 1996; pp. 3–10. [Google Scholar] [CrossRef]
- Pilacinski, W.; Crawford, A.; Downey, R.; Harvey, B.; Huber, S.; Hunst, P.; Lahman, L.K.; MacIntosh, S.; Pohl, M.; Rickard, C.; et al. Plants with genetically modified events combined by conventional breeding: An assessment of the need for additional regulatory data. Food Chem. Toxicol. 2011, 49, 1–7. [Google Scholar] [CrossRef][Green Version]
- Lusser, M.; Parisi, C.; Plan, D.; Rodriguez-Cerezo, E. Deployment of new biotechnologies in plant breeding. Nat. Biotech. 2012, 30, 231. [Google Scholar] [CrossRef] [PubMed]
- Simó, C.; Ibáez, C.; Valdés, A.; Cifuentes, A.; García-Cañas, V. Metabolomics of genetically modified crops. Int. J. Mol. Sci. 2014, 15, 18941–18966. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Sarma, S.; Bohra, A.; Mitra, A.; Sharma, N.K.; Kumar, A. Plant metabolomics: An emerging technology for crop improvement. In New Visions in Plant Science, 1st ed.; Çelik, O., Ed.; IntechOpen: London, UK, 2018; pp. 65–79. [Google Scholar]
- Zhang, H.; Mittal, N.; Leamy, L.J.; Barazani, O.; Song, B.H. Back into the wild—Apply untapped genetic diversity of wild relatives for crop improvement. Evol. Appl. 2017, 10, 5–24. [Google Scholar] [CrossRef]
- Mammadov, J.; Buyyarapu, R.; Guttikonda, S.K.; Parliament, K.; Abdurakhmonov, I.Y.; Kumpatla, S.P. Wild relatives of maize, rice, cotton, and soybean: Treasure troves for tolerance to biotic and abiotic stresses. Front. Plant Sci. 2018, 9, 886. [Google Scholar] [CrossRef]
- Dempewolf, H.; Baute, G.; Anderson, J.; Kilian, B.; Smith, C.; Guarino, L. Past and future use of wild relatives in crop breeding. Crop Sci. 2017, 57, 1070–1082. [Google Scholar] [CrossRef]
- Xu, Y.; Crouch, J.H. Marker-assisted selection in plant breeding: From publications to practice. Crop Sci. 2008, 48, 391–407. [Google Scholar] [CrossRef]
- Korir, N.K.; Han, J.; Shangguan, L.; Wang, C.; Kayesh, E.; Zhang, Y.; Fang, J. Plant variety and cultivar identification: Advances and prospects. Crit. Rev. Biotechnol. 2013, 33, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Roychowdhury, R.; Taoutaou, A.; Hakeem, K.R.; Gawwad, M.R.A.; Tah, J. Molecular marker-assisted technologies for crop improvement. In Crop Improvement in the Era of Climate Change; Roychowdhury, R., Ed.; International Publishing House: New Delhi, India, 2013; pp. 241–258. [Google Scholar]
- Prohens, J. Plant breeding: A success story to be continued thanks to the advances in genomics. Front. Plant Sci. 2011, 2, 51. [Google Scholar] [CrossRef] [PubMed]
- Cobb, J.N.; Juma, R.U.; Biswas, P.S.; Arbelaez, J.D.; Rutkoski, J.; Atlin, G.; Hagen, T.; Quinn, M.; Ng, E.H. Enhancing the rate of genetic gain in public-sector plant breeding programs: Lessons from the breeder’s equation. Theor. Appl. Genet. 2019, 132, 627–645. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.D. Plant metabolomics: From holistic hope, to hype, to hot topic. New Phytol. 2006, 169, 453–468. [Google Scholar] [CrossRef]
- Fernie, A.R.; Schauer, N. Metabolomics-assisted breeding: A viable option for crop improvement? Trends Genet. 2009, 25, 39–48. [Google Scholar] [CrossRef]
- Kumar, R.; Bohra, A.; Pandey, A.K.; Pandey, M.K.; Kumar, A. Metabolomics for plant improvement: Status and prospects. Front. Plant Sci. 2017, 8, 302. [Google Scholar] [CrossRef]
- Alseekh, S.; Bermudez, L.; De Haro, L.A.; Fernie, A.R.; Carrari, F. Crop metabolomics: From diagnostics to assisted breeding. Metabolomics 2018, 14, 148. [Google Scholar] [CrossRef]
- Zabotina, O.A. Metabolite-based biomarkers for plant genetics and breeding. In Diagnostics in Plant Breeding; Lübberstedt, T., Varshney, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 281–309. [Google Scholar]
- Gamboa-Becerra, R.; Hernández-Hernández, M.C.; González-Ríos, Ó.; Suárez-Quiroz, M.L.; Gálvez-Ponce, E.; Ordaz-Ortiz, J.J.; Winkler, R. Metabolomic markers for the early selection of Coffea canephora plants with desirable cup quality traits. Metabolites 2019, 9, 214. [Google Scholar] [CrossRef] [PubMed]
- Riedelsheimer, C.; Czedik-Eysenberg, A.; Grieder, C.; Lisec, J.; Technow, F.; Sulpice, R.; Altmann, T.; Stitt, M.; Willmitzer, L.; Melchinger, A.E. Genomic and metabolic prediction of complex heterotic traits in hybrid maize. Nat. Genet. 2012, 44, 217–220. [Google Scholar] [CrossRef]
- Li, S.; Dong, X.; Fan, G.; Yang, Q.; Shi, J.; Wei, W.; Zhao, F.; Li, N.; Wang, X.; Wang, F.; et al. Comprehensive profiling and inheritance patterns of metabolites in foxtail millet. Front. Plant Sci. 2018, 9, 1716. [Google Scholar] [CrossRef] [PubMed]
- Cubillos, A.E.R.; Tong, H.; Alseekh, S.; Lima, F.D.A.; Yu, J.; Fernie, A.R.; Nikoloski, Z.; Laitinen, R.A. Inheritance patterns in metabolism and growth in diallel crosses of Arabidopsis thaliana from a single growth habitat. Heredity 2018, 120, 463–473. [Google Scholar] [CrossRef]
- Lisec, J.; Römisch-Margl, L.; Nikoloski, Z.; Piepho, H.P.; Giavalisco, P.; Selbig, J.; Gierl, A.; Willmitzer, L. Corn hybrids display lower metabolite variability and complex metabolite inheritance patterns. Plant J. 2011, 68, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Steinfath, M.; Strehmel, N.; Peters, R.; Schauer, N.; Groth, D.; Hummel, J.; Steup, M.; Selbig, J.; Kopka, J.; Geigenberger, P.; et al. Discovering plant metabolic biomarkers for phenotype prediction using an untargeted approach. Plant Biotechnol. J. 2010, 8, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, O.; Urrutia, M.; Bernillon, S.; Giauffret, C.; Tardieu, F.; Le Gouis, J.; Langlade, N.; Charcosset, A.; Moing, A.; Gibon, Y. Fortune telling: Metabolic markers of plant performance. Metabolomics 2016, 12, 158. [Google Scholar] [CrossRef]
- Chen, M.; Rao, R.S.P.; Zhang, Y.; Zhong, C.; Thelen, J.J. Metabolite variation in hybrid corn grain from a large-scale multisite study. Crop J. 2016, 4, 177–187. [Google Scholar] [CrossRef]
- Loskutov, I.G.; Shelenga, T.V.; Konarev, A.V.; Shavarda, A.L.; Blinova, E.V.; Dzubenko, N.I. The metabolomic approach to the comparative analysis of wild and cultivated species of oats (Avena L.). Russ. J. Genet. Appl. Res. 2017, 7, 501–508. [Google Scholar] [CrossRef]
- Song, E.H.; Jeong, J.; Park, C.Y.; Kim, H.Y.; Kim, E.H.; Bang, E.; Hong, Y.S. Metabotyping of rice (Oryza sativa L.) for understanding its intrinsic physiology and potential eating quality. Food Res. Int. 2018, 111, 20–30. [Google Scholar] [CrossRef]
- Thomason, K.; Babar, M.A.; Erickson, J.E.; Mulvaney, M.; Beecher, C.; MacDonald, G. Comparative physiological and metabolomics analysis of wheat (Triticum aestivum L.) following post-anthesis heat stress. PLoS ONE 2018, 13, 0197919. [Google Scholar] [CrossRef]
- Lawas, L.M.F.; Li, X.; Erban, A.; Kopka, J.; Jagadish, S.K.; Zuther, E.; Hincha, D.K. Metabolic responses of rice cultivars with different tolerance to combined drought and heat stress under field conditions. Gigascience 2019, 8, 50. [Google Scholar] [CrossRef]
- Mareya, C.R.; Tugizimana, F.; Piater, L.A.; Madala, N.E.; Steenkamp, P.A.; Dubery, I.A. Untargeted metabolomics reveal defensome-related metabolic reprogramming in Sorghum bicolor against infection by Burkholderia andropogonis. Metabolites 2019, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zeng, X.; Xu, Q.; Mei, X.; Yuan, H.; Jiabu, D.; Sang, Z.; Nyima, T. Metabolite profiling in two contrasting Tibetan hulless barley cultivars revealed the core salt-responsive metabolome and key salt-tolerance biomarkers. AoB Plants 2019, 11, 21. [Google Scholar] [CrossRef] [PubMed]
- Gani, A.; Wani, S.M.; Masoodi, F.A.; Hameed, G. Whole-grain cereal bioactive compounds and their health benefits: A review. Int. J. Food Process Technol. 2012, 3, 146–156. [Google Scholar] [CrossRef]
- Cooper, R. Re-discovering ancient wheat varieties as functional foods. J. Tradit. Compl. Med. 2015, 5, 138–143. [Google Scholar] [CrossRef]
- Khlestkina, E. The adaptive role of flavonoids: Emphasis on cereals. Cereal Res. Commun. 2013, 41, 185–198. [Google Scholar] [CrossRef]
- Dykes, L.; Rooney, L.W. Phenolic compounds in cereal grains and their health benefits. Cereal Foods World 2007, 52, 105–111. [Google Scholar] [CrossRef]
- Joye, I. Protein digestibility of cereal products. Foods 2019, 8, 199. [Google Scholar] [CrossRef]
- Xu, M.; Rao, J.; Chen, B. Phenolic compounds in germinated cereal and pulse seeds: Classification, transformation, and metabolic process. Crit. Rev. Food Sci. Nutr. 2020, 60, 740–759. [Google Scholar] [CrossRef]
- Sidhu, J.S.; Kabir, Y.; Huffman, F.G. Functional foods from cereal grains. Int. J. Food Prop. 2007, 10, 231–244. [Google Scholar] [CrossRef]
- Leitzmann, C. Characteristics and health benefits of phytochemicals. J. Compl. Med. Res. 2016, 23, 69–74. [Google Scholar] [CrossRef]
- Berini, J.L.; Brockman, S.A.; Hegeman, A.D.; Reich, P.B.; Muthukrishnan, R.; Montgomery, R.A.; Forester, J.D. Combinations of abiotic factors differentially alter production of plant secondary metabolites in five woody plant species in the boreal-temperate transition zone. Front. Plant Sci. 2018, 9, 1257. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Huang, W.; Gao, J.; Fu, H.; Liu, J. Comparative metabolomic analysis of seed metabolites associated with seed storability in rice (Oryza sativa L.) during natural aging. Plant Physiol. Biochem. 2018, 127, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Zarei, I.; Luna, E.; Leach, J.; McClung, A.; Vilchez, S.; Koita, O.; Ryan, E. Comparative rice bran metabolomics across diverse cultivars and functional rice gene-bran metabolite relationships. Metabolites 2018, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Shi, J.; Quan, S.; Cui, B.; Kleessen, S.; Nikoloski, Z.; Tohge, T.; Alexander, D.; Guo, L.; Lin, H.; et al. Metabolic variation between japonica and indica rice cultivars as revealed by non-targeted metabolomics. Sci. Rep. 2014, 4, 5067. [Google Scholar] [CrossRef]
- Gayen, D.; Ghosh, S.; Paul, S.; Sarkar, S.N.; Datta, S.K.; Datta, K. Metabolic regulation of carotenoid-enriched golden rice line. Front. Plant. Sci. 2016, 7, 1622. [Google Scholar] [CrossRef]
- Sana, T.R.; Fischer, S.; Wohlgemuth, G.; Katrekar, A.; Jung, K.H.; Ronald, P.C.; Fiehn, O. Metabolomic and transcriptomic analysis of the rice response to the bacterial blight pathogen Xanthomonas oryzae pv. oryzae. Metabolomics 2010, 6, 451–465. [Google Scholar] [CrossRef]
- Gupta, S.V.K.; Rupasinghe, T.W.T.; Callahan, D.L.; Natera, S.H.; Smith, P.M.; Hill, C.B.; Roessner, U.; Boughton, B.A. Spatio-temporal metabolite and elemental profiling of salt stressed barley seeds during initial stages of germination by MALDI-MSI and µ-XRF Spectrometry. Front. Plant Sci. 2019, 10, 1139. [Google Scholar] [CrossRef]
- Piasecka, A.; Sawikowska, A.; Kuczyńska, A.; Ogrodowicz, P.; Mikołajczak, K.; Krystkowiak, K.; Gudyś, K.; Guzy-Wróbelska, J.; Krajewski, P.; Kachlicki, P. Drought-related secondary metabolites of barley (Hordeum vulgare L.) leaves and their metabolomic quantitative trait loci. Plant J. 2017, 89, 898–913. [Google Scholar] [CrossRef]
- Piasecka, A.; Sawikowska, A.; Krajewski, P.; Kachlicki, P. Combined mass spectrometric and chromatographic methods for in-depth analysis of phenolic secondary metabolites in barley leaves. J. Mass Spectrom. 2015, 50, 513–532. [Google Scholar] [CrossRef]
- Khakimov, B.; Rasmussen, M.A.; Kannangara, R.M.; Jespersen, B.M.; Munck, L.; Engelsen, S.B. From metabolome to phenotype: GC-MS metabolomics of developing mutant barley seeds reveals effects of growth, temperature and genotype. Sci. Rep. 2017, 7, 8195. [Google Scholar] [CrossRef]
- Kogel, K.H.; Voll, L.M.; Schäfer, P.; Jansen, C.; Wu, Y.; Langen, G.; Imani, J.; Hofmann, J.; Schmiedl, A.; Sonnewald, S.; et al. Transcriptome and metabolome profiling of field-grown transgenic barley lack induced differences but show cultivar-specific variances. Proc. Natl. Acad. Sci. USA 2010, 107, 6198–6203. [Google Scholar] [CrossRef]
- Roessner, U.; Patterson, J.H.; Forbes, M.G.; Fincher, G.B.; Langridge, P.; Bacic, A. An investigation of boron toxicity in barley using metabolomics. Plant Physiol. 2006, 142, 1087–1101. [Google Scholar] [CrossRef] [PubMed]
- Tugizimana, F.; Djami Tchatchou, A.T.; Steenkamp, P.A.; Piater, L.A.; Dubery, I.A. Metabolomic analysis of defence-related reprogramming in Sorghum bicolor in response to Colletotrichum sublineolum infection reveals a functional metabolic web of phenylpropanoid and flavonoid pathways. Front. Plant Sci. 2019, 9, 1840. [Google Scholar] [CrossRef] [PubMed]
- Ogbaga, C.C.; Stepien, P.; Dyson, B.C.; Rattray, N.J.; Ellis, D.I.; Goodacre, R.; Johnson, G.N. Biochemical analyses of sorghum varieties reveal differential responses to drought. PLoS ONE 2016, 11, e0154423. [Google Scholar] [CrossRef]
- Turner, M.F.; Heuberger, A.L.; Kirkwood, J.S.; Collins, C.C.; Wolfrum, E.J.; Broeckling, C.D.; Prenni, J.E.; Jahn, C.E. Non-targeted metabolomics in diverse sorghum breeding lines indicates primary and secondary metabolite profiles are associated with plant biomass accumulation and photosynthesis. Front. Plant Sci. 2016, 7, 953. [Google Scholar] [CrossRef]
- Michaletti, A.; Naghavi, M.R.; Toorchi, M.; Zolla, L.; Rinalducci, S. Metabolomics and proteomics reveal drought-stress responses of leaf tissues from spring-wheat. Sci. Rep. 2018, 8, 5710. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Zhu, A.; Jia, J.; Hu, X.; Chen, J.; Liu, W.; Ren, X.; Sun, D.; Fernie, A.R.; Cui, F.; et al. Metabolomics analysis and metabolite-agronomic trait associations using kernels of wheat (Triticum aestivum) recombinant inbred lines. Plant J. 2020, (in press). [CrossRef]
- Shewry, P.R.; Corol, D.I.; Jones, H.D.; Beale, M.H.; Ward, J.L. Defining genetic and chemical diversity in wheat grain by 1H-NMR spectroscopy of polar metabolites. Mol. Nutr. Food Res. 2017, 61, 1600807. [Google Scholar] [CrossRef] [PubMed]
- Matthews, S.B.; Santra, M.; Mensack, M.M.; Wolfe, P.; Byrne, P.F.; Thompson, H.J. Metabolite profiling of a diverse collection of wheat lines using ultra performance liquid chromatography coupled with time-of-flight mass spectrometry. PLoS ONE 2012, 7, 44179. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.; Cheng, F.; Hu, C.; Quan, S.; Lin, H.; Wang, J.; Chen, G.; Zhao, X.; Alexander, D.; Guo, L.; et al. Metabolic map of mature maize kernels. Metabolomics 2014, 10, 775–787. [Google Scholar] [CrossRef]
- Barros, E.; Lezar, S.; Anttonen, M.J.; Van Dijk, J.P.; Röhlig, R.M.; Kok, E.J.; Engel, K.H. Comparison of two GM maize varieties with a near-isogenic non-GM variety using transcriptomics, proteomics and metabolomics. Plant Biotechnol. J. 2010, 8, 436–451. [Google Scholar] [CrossRef] [PubMed]
- Lamari, N.; Zhendre, V.; Urrutia, M.; Bernillon, S.; Maucourt, M.; Deborde, C.; Prodhomme, D.; Jacob, D.; Ballias, P.; Rolin, D.; et al. Metabotyping of 30 maize hybrids under early-sowing conditions reveals potential marker-metabolites for breeding. Metabolomics 2018, 14, 132. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Cao, J.; Wang, X.; Chen, Q.; Jin, W.; Li, Z.; Tian, F. Evolutionary metabolomics identifies substantial metabolic divergence between maize and its wild ancestor, teosinte. Plant Cell 2019, 31, 1990–2009. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Zhang, X.; Zhao, M.; Deng, M.; Du, Y.; Zhou, Y.; Wang, S.; Tohge, T.; Fernie, A.R.; Willmitzer, L.; et al. Integrated genomics-based mapping reveals the genetics underlying maize flavonoid biosynthesis. BMC Plant Biol. 2017, 17, 17. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martín, J.; Heald, J.; Kingston-Smith, A.; Winters, A.; Rubiales, D.; Sanz, M.; Mur, L.A.; Prats, E. A metabolomic study in oats (Avena sativa) highlights a drought tolerance mechanism based upon salicylate signalling pathways and the modulation of carbon, antioxidant and photo-oxidative metabolism. Plant Cell Environ. 2015, 38, 1434–1452. [Google Scholar] [CrossRef] [PubMed]
- Dhawi, F.; Datta, R.; Ramakrishna, W. Metabolomics, biomass and lignocellulosic total sugars analysis in foxtail millet (Setaria italica) inoculated with different combinations of plant growth promoting bacteria and mycorrhiza. Commun. Plant Sci. 2018, 8, 8–14. [Google Scholar] [CrossRef]
- Hamzelou, S.; Pascovici, D.; Kamath, K.S.; Amirkhani, A.; McKay, M.; Mirzaei, M.; Atwell, B.J.; Haynes, P.A. Proteomic responses to drought vary widely among eight diverse genotypes of rice (Oryza sativa). Int. J. Mol. Sci. 2020, 21, 363. [Google Scholar] [CrossRef]
- Khan, M.H.; Dar, Z.A.; Dar, S.A. Breeding strategies for improving rice yield-A review. Agric. Sci. 2015, 6, 467. [Google Scholar] [CrossRef]
- Baranski, M. Golden rice. In The Embryo Project Encyclopedia; Arizona State University: Tempe, AZ, USA, 2013; pp. 1940–5030. [Google Scholar]
- Matsaunyane, L.B.T.; Dubery, I.A. Genetic modification of crop plants: Molecular approaches to address intended unintended effects and substantial equivalence. In Transgenic Crops; Khan, M.S., Malik, K.A., Eds.; InTechOpen Publishers: London, UK, 2019; Chapter 7; pp. 115–131. [Google Scholar]
- Decourcelle, M.; Perez-Fons, L.; Baulande, S.; Steiger, S.; Couvelard, L.; Hem, S.; Zhu, C.; Capell, T.; Christou, P.; Fraser, P.; et al. Combined transcript, proteome, and metabolite analysis of transgenic maize seeds engineered for enhanced carotenoid synthesis reveals pleotropic effects in core metabolism. J. Exp. Bot. 2015, 66, 3141–3150. [Google Scholar] [CrossRef]
- Chen, R.; Zhao, X.; Shao, Z.; Wei, Z.; Wang, Y.; Zhu, L.; Zhao, J.; Sun, M.; He, R.; He, G. Rice UDP-glucose pyrophosphorylase1 is essential for pollen callose deposition and its consumption result in a new type of thermosensible genic male sterility. Plant Cell 2007, 19, 847–861. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hina, A.; Song, S.; Kong, J.; Bhat, J.A.; Zhao, T. Whole-genome mapping identified novel “QTL hotspots regions” for seed storability in soybean (Glycine max L.). BMC Genom. 2019, 20, 499. [Google Scholar] [CrossRef] [PubMed]
- Tricase, C.; Amicarelli, V.; Lamonaca, E.; Rana, R.L. Economic analysis of the barley market and related uses. In Grasses as Food and Feed; Tadele, Z., Ed.; IntechOpen: London, UK, 2018; pp. 25–46. [Google Scholar]
- Zhao, Y.; Ma, Q.; Jin, X.; Peng, X.; Liu, J.; Deng, L.; Yan, H.; Sheng, L.; Jiang, H.; Cheng, B. A novel maize homeodomain–leucine zipper (HD-Zip) I gene, Zmhdz10, positively regulates drought and salt tolerance in both rice and Arabidopsis. Plant Cell Physiol. 2014, 55, 1142–1156. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, Z.; Li, Y.; Li, Z.; Liu, H.; Zhou, W. Metabolite profiling of sorghum seeds of different colors from different sweet sorghum cultivars using a widely targeted metabolomics approach. Int. J. Genom. 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lamaoui, M.; Jemo, M.; Datla, R.; Bekkaoui, F. Heat and drought stresses in crops and approaches for their mitigation. Front. Chem. 2018, 6, 26. [Google Scholar] [CrossRef]
- Tesso, T.T.; Perumal, R.; Little, C.R.; Adeyanju, A.; Radwan, G.L.; Prom, L.K.; Magill, C.W. Sorghum pathology and biotechnology—A fungal disease perspective: Part II. Anthracnose, stalk rot, and downy mildew. Eur. J. Plant Sci. Biotech. 2012, 6, 31–44. [Google Scholar]
- Hill, C.B.; Taylor, J.D.; Edwards, J.; Mather, D.; Bacic, A.; Langridge, P.; Roessner, U. Whole-genome mapping of agronomic and metabolic traits to identify novel quantitative trait loci in bread wheat grown in a water-limited environment. Plant Physiol. 2013, 162, 1266–1281. [Google Scholar] [CrossRef]
- Hill, C.B.; Taylor, J.D.; Edwards, J.; Mather, D.; Langridge, P.; Bacic, A.; Roessner, U. Detection of QTL for metabolic and agronomic traits in wheat with adjustments for variation at genetic loci that affect plant phenology. Plant Sci. 2015, 233, 143–154. [Google Scholar] [CrossRef]
- Matros, A.; Liu, G.; Hartmann, A.; Jiang, Y.; Zhao, Y.; Wang, H.; Ebmeyer, E.; Korzun, V.; Schachschneider, R.; Kazman, E.; et al. Genome–metabolite associations revealed low heritability, high genetic complexity, and causal relations for leaf metabolites in winter wheat (Triticum aestivum). J. Exp. Bot. 2017, 68, 415–428. [Google Scholar] [CrossRef]
- Seki, M.; Umezawa, T.; Urano, K.; Shinozaki, K. Regulatory metabolic networks in drought stress responses. Curr. Opin. Plant Biol. 2007, 10, 296–302. [Google Scholar] [CrossRef]
- Krasensky, J.; Jonak, C. Drought, salt, and temperature stress-induced metabolic rearrangements and regulatory networks. J. Exp. Bot. 2012, 63, 1593–1608. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).