
Serial Transcriptome Analysis Reveals Genes Associated with Late Blight Resistance in Potato Cultivar Qingshu 9

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Preparation of Spore Suspension and Incubation
2.3. Collection of Samples and Microscopic Examination
2.4. RNA Extraction, Library Preparation, and Sequencing
2.5. Analysis of the Differentially Expressed Genes
2.6. GO and KEGG Enrichment Analyses of DEGs
2.7. Transcription Factor (TF) Analysis of DEGs
2.8. Comparative Analysis of Plant R Gene Database (PRGDB)
2.9. Validation of the Expression of DEGs by qRT-PCR
3. Results
3.1. Leaves at 0, 24, 48, 72 hpi Were Chosen to Prepare in Planta Materials
3.2. A Large Amount of Credible Data Were Obtained through Transcriptome Sequencing
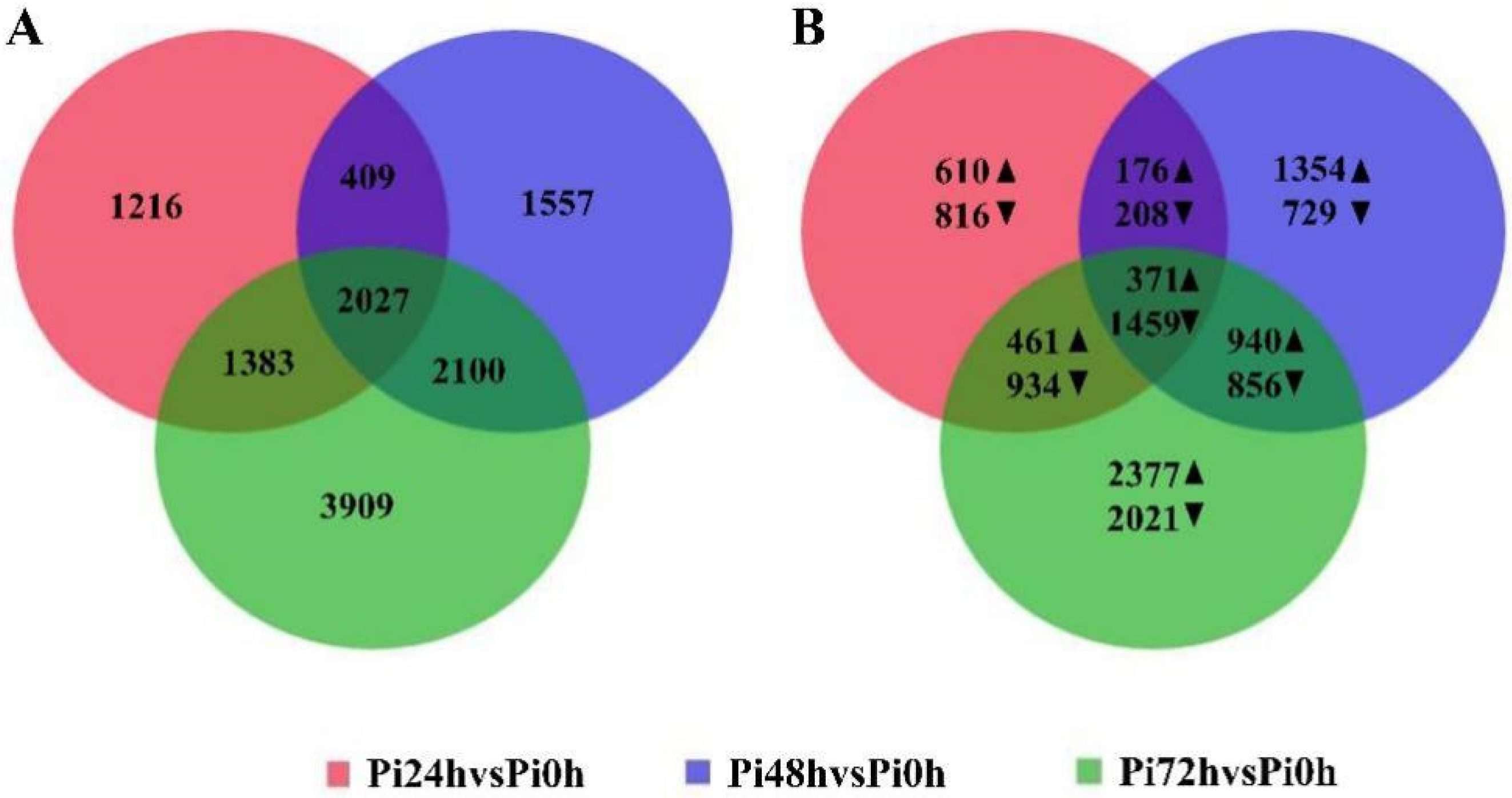
3.3. 24 hpi and 48 hpi Are Important in the Immune Response of Qingshu 9
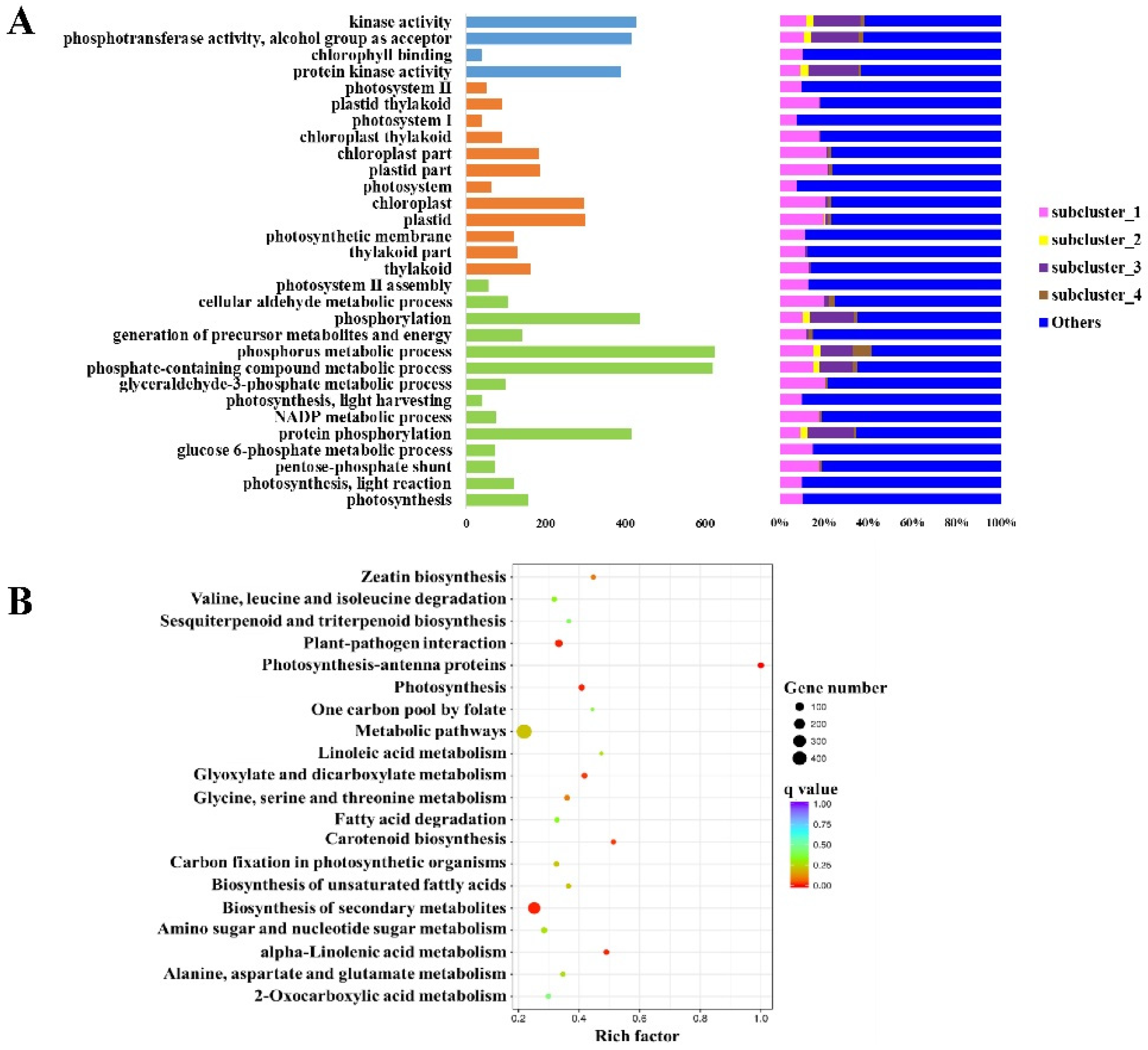
3.4. DEGs among the Groups Are Enriched in Different GO Terms and KEGG Pathways
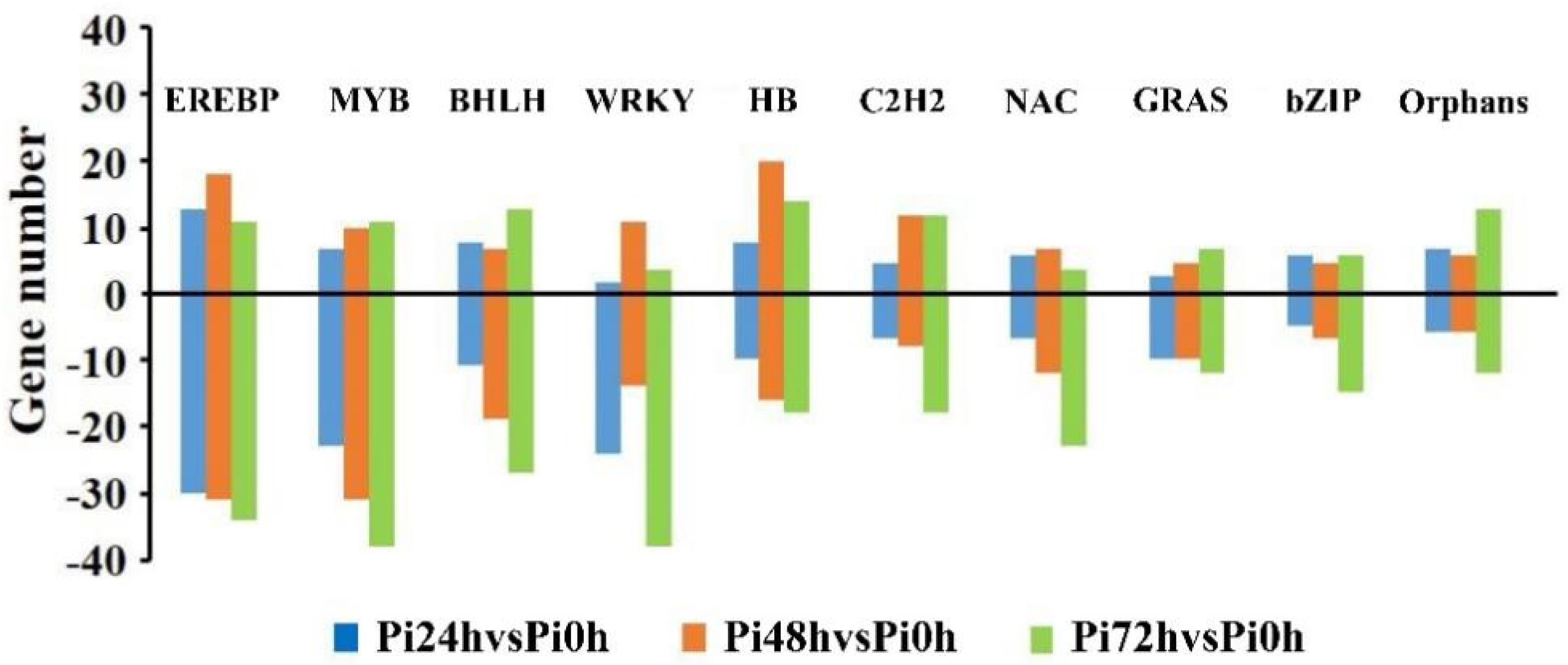
3.5. Transcription Factors Play Crucial Roles in Qingshu 9’s Resistance Response
3.6. Comparison of Results between DEGs and Plant R Gene Database
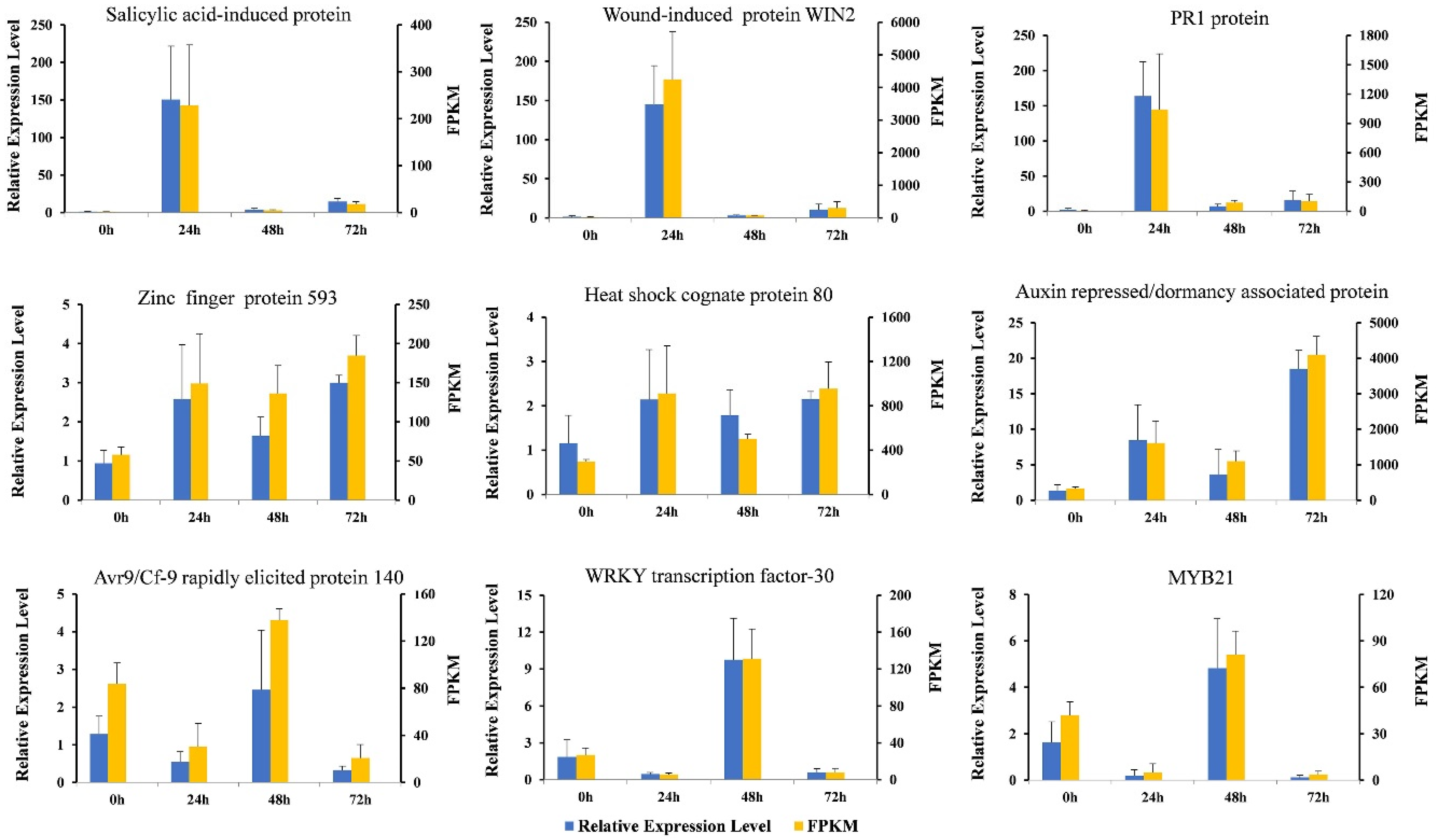
3.7. The Results of qRT-PCR and RNA-Seq Are Consistent
4. Discussion
4.1. Potato Cultivar Qingshu 9 Contains Special Resistance to P. infestans
4.2. Transcription Analysis of Qingshu 9 Infected by P. infestans
4.3. Transcription Factors May Play an Important Role in the Resistance Response to P. infestans in Qingshu 9
4.4. Protein Kinases May Participate in the Interaction between Qingshu 9 and P. infestans
4.5. Disease Resistance-Related Genes May Enhance the Disease Resistance of Qingshu 9
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Haverkort, A.J.; Hillier, J.G. Cool farm tool-potato: Model description and performance of four production systems. Potato Res. 2011, 54, 355–369. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Boevink, P.; McLellan, H.; Armstrong, M.; Bukharova, T.; Qin, Z.; Birch, P.R.J. A host KH RNA-binding protein is a susceptibility factor targeted by an RXLR effector to promote late blight disease. Mol. Plant 2015, 8, 1385–1395. [Google Scholar] [CrossRef] [Green Version]
- Boevink, P.C.; Wang, X.; McLellan, H.; He, Q.; Naqvi, S.; Armstrong, M.R.; Zhang, W.; Hein, I.; Gilroy, E.M.; Tian, Z.D.; et al. A Phytophthora infestans RXLR effector targets plant PP1c isoforms that promote late blight disease. Nat. Commun. 2016, 7, 10311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, K. Potato genetics, genomics, and applications. Breed. Sci. 2015, 65, 53–68. [Google Scholar] [CrossRef] [Green Version]
- Black, W.; Gallegly, M.E. Screening of Solanum species for resistance to physiologic races of Phytophthora infestans. Am. J. Potato Res. 1957, 34, 273–281. [Google Scholar] [CrossRef]
- Malcolmson, J.F.; Black, W. New R genes in solanum demissum lindl. and their complementary races of Phytophthora infestans (Mont.) de Bary. Euphytica 1966, 15, 199–203. [Google Scholar] [CrossRef]
- Fry, W. Phytophthora infestans: The plant (and R gene) destroyer. Mol. Plant Pathol. 2008, 9, 385–402. [Google Scholar] [CrossRef] [PubMed]
- Kwenda, S.; Motlolometsi, T.V.; Birch, P.R.J.; Moleleki, L.N. RNA-seq profiling reveals defense responses in a tolerant potato cultivar to stem infection by Pectobacterium carotovorum ssp. Brasiliense. Front. Plant Sci. 2016, 7, 1905. [Google Scholar] [CrossRef]
- Kou, Y.; Wang, S. Broad-spectrum and durability: Understanding of quantitative disease resistance. Curr. Opin. Plant Biol. 2010, 13, 181–185. [Google Scholar] [CrossRef]
- Haverkort, A.J.; Boonekamp, P.M.; Hutten, R.; Jacobsen, E.; Lotz, L.A.P.; Kessel, G.J.T.; Vossen, J.H.; Visser, R.G.F. Durable late blight resistance in potato through dynamic varieties obtained by cisgenesis: Scientific and societal advances in the durph project. Potato Res. 2016, 59, 35–66. [Google Scholar] [CrossRef] [Green Version]
- Avrova, A.O.; Stewart, H.E.; De Jong, W.; Heilbronn, J.; Lyon, G.D.; Birch, P.R.J. A cysteine protease gene is expressed early in resistant potato interactions with Phytophthora infestans. Mol. Plant-Microbe Interact. 1999, 12, 1114–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, K.; Binder, A.; Boller, T.; Collinge, M. Identification of potato genes induced during colonization by Phytophthora infestans. Mol. Plant Pathol. 2001, 2, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Collinge, M.; Boller, T. Differential induction of two potato genes, Stprx2 and StNAC, in response to infection by Phytophthora infestans and to wounding. Plant Mol. Biol. 2001, 46, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Chen, T.H.H.; Li, P.H. Expression of three osmotin-like protein genes in response to osmotic and fungal infection in potato. Plant Mol. Biol. 1995, 28, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Tyler, B.M.; Rouxel, T. Effectors of Fungi and Oomycetes: Their virulence and avirulence functions and translocation from pathogen to host cells. Mol. Plant Immun. 2012, 123–167. [Google Scholar]
- Wirthmueller, L.; Maqbool, A.; Banfield, M.J. On the front line: Structural insights into plant–pathogen interactions. Nat. Rev. Microbiol. 2013, 11, 761–776. [Google Scholar] [CrossRef]
- Birch, P.R.J.; Avrova, A.O.; Duncan, J.M.; Lyon, G.D.; Toth, R.L. Isolation of potato genes that are induced during an early stage of the hypersensitive response to Phytophthora infestans. Mol. Plant-Microbe Interact. 1999, 12, 356–361. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.W.; Hwang, B.K. Isolation, Partial sequencing, and expression of pathogenesis-related cDNA genes from pepper leaves infected by Xanthomonas campestris pv. vesicatoria. Mol. Plant-Microbe Interact. 2000, 13, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Kamoun, S.; Zody, M.C.; Jiang, R.H.Y.; Handsaker, R.E.; Cano, L.M.; Grabherr, M.; Kodira, C.D.; Raffaele, S.; Torto-Alalibo, T.; et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nat. 2009, 461, 393–398. [Google Scholar] [CrossRef]
- Potato Genome Sequencing Consortium. Genome sequence and analysis of the tuber crop potato. Nature 2011, 475, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Restrepo, S.; Myers, K.L.; del Pozo, O.; Martin, G.B.; Hart, A.L.; Buell, C.R.; Smart, C.D. Gene profiling of a compatible interaction between Phytophthora infestans and Solanum tuberosum suggests a role for carbonic anhydrase. Mol. Plant-Microbe Interact. 2005, 18, 913–922. [Google Scholar] [CrossRef] [Green Version]
- Gyetvai, G.; Sønderkær, M.; Göbel, U.; Basekow, R.; Ballvora, A.; Imhoff, M.; Kersten, B.; Nielsen, K.; Gebhardt, C. The transcriptome of compatible and incompatible interactions of potato (Solanum tuberosum) with Phytophthora infestans revealed by DeepSAGE analysis. PLoS ONE 2012, 7, e31526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Draffehn, A.M.; Li, L.; Krezdorn, N.; Ding, J.; Lübeck, J.; Strahwald, J.; Muktar, M.S.; Walkemeier, B.; Rotter, B.; Gebhardt, C. Comparative transcript profiling by SuperSAGE identifies novel candidate genes for controlling potato quantitative resistance to late blight not compromised by late maturity. Front. Plant Sci. 2013, 4, 423. [Google Scholar] [CrossRef] [Green Version]
- Barakat, A.; DiLoreto, D.S.; Zhang, Y.; Smith, C.; Baier, K.; Powell, W.A.; Wheeler, N.; Sederoff, R.; Carlson, J.E. Comparison of the transcriptomes of American chestnut (Castanea dentata) and Chinese chestnut (Castanea mollissima) in response to the chestnut blight infection. BMC Plant Biol. 2009, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.L.; Tu, Z.J.; Millett, B.P.; Bradeen, J.M. Insights into organ-specifific pathogen defense responses in plants: RNA-seq analysis of potato tuber-Phytophthora infestans interactions. BMC Genom. 2013, 14, 340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, A.; Alexandersson, E.; Sandin, M.; Resjö, S.; Lenman, M.; Hedley, P.; Levander, F.; Andreasson, E. Quantitative proteomics and transcriptomics of potato in response to Phytophthora infestans in compatible and incompatible interactions. BMC Genom. 2014, 15, 497. [Google Scholar] [CrossRef] [Green Version]
- Ballester, A.; Norelli, J.; Burchard, E.; Abdelfattah, A.; Levin, E.; González-Candelas, L.; Dorby, S.; Wisniewski, M. Transcriptomic Response of Resistant (PI613981-Malus sieversii) and Susceptible (“Royal Gala”) Genotypes of Apple to Blue Mold (Penicillium expansum) Infection. Front. Plant Sci. 2017, 8, 1981. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Guo, X.; Yang, Y.; Ye, P.; Xiong, X.; Liu, J.; Dong, D.; Li, G. Gene profiling in late blight resistance in potato genotype SD20. Int. J. Mol. Sci. 2018, 19, 1728. [Google Scholar] [CrossRef] [Green Version]
- Frades, I.; Abreha, K.B.; Proux-Wéra, E.; Lankinen, Å.; Andreasson, E.; Alexandersson, E. A novel workflow correlating RNA-seq data to Phythophthora infestans resistance levels in wild Solanum species and potato clones. Front. Plant Sci. 2015, 6, 718. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.L. Evaluation of Major Potato Varieties in Northwestern China for Late Blight Resistance and Identification of Candidate Core RXLR Effector Genes from Phytophthora Infestans; Northwest A&F University: Xianyang, China, 2017. [Google Scholar]
- Bolser, D.; Staines, D.M.; Pritchard, E.; Kersey, P. Ensembl Plants: Integrating tools for visualizing, mining, and analyzing plant genomics data. Methods Mol. Biol. 2016, 115–140. [Google Scholar]
- Kim, D.; Langmead, B.; Salzberg, S.L. Hisat: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, 316–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Rodriguez, P.; Riano-Pachon, D.M.; Correa, L.G.G.; Rensing, S.A.; Kersten, B.; Mueller-Roeber, B. PlnTFDB: Updated content and new features of the plant transcription factor database. Nucleic Acids Res. 2010, 38, 822–827. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Zhang, H.; Kong, L.; Gao, G.; Luo, J. PlantTFDB 3.0: A portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 2014, 42, 1182–1187. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-Time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Chang, C.; Yu, D.; Jiao, J.; Jing, S.; Schulze-Lefert, P.; Shen, Q.H. Barley MLA immune receptors directly interfere with antagonistically acting transcription factors to initiate disease resistance signaling. Plant Cell 2013, 25, 1158–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, S.P.; Somssich, I.E. The role of WRKY transcription factors in plant immunity. Plant Physiology 2009, 150, 1648–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Dang, F.; Liu, Z.; Wang, X.; Eulgem, T.; Lai, Y.; Yu, L.; She, J.; Shi, Y.; Lin, J.; et al. CaWRKY58, encoding a group I WRKY transcription factor of capsicum annuum, negatively regulates resistance to ralstonia solanacearum infection. Mol. Plant Pathol. 2013, 14, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Xu, P.; Meng, J.; Li, J.; Jiang, N.; Luan, Y. Transcriptome signatures of tomato leaf induced by Phytophthora infestans and functional identification of transcription factor SpWRKY3. Theor. Appl. Genet. 2017, 131, 787–800. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Bradeen, J.M.; Sek-Man, W. Contrasting potato foliage and tuber defense mechanisms against the late blight pathogen Phytophthora infestans. PLoS ONE 2016, 11, e0159969. [Google Scholar] [CrossRef] [PubMed]
- Massa, A.N.; Childs, K.L.; Lin, H.; Bryan, G.J.; Giuliano, G.; Buell, C.R. The transcriptome of the reference potato genome Solanum tuberosum group phureja clone DM1-3 516R44. PLoS ONE 2011, 6, e26801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eulgem, T.; Rushton, P.; Robatzek, S.; Somssich, I. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000, 5, 199–206. [Google Scholar] [CrossRef]
- Jakoby, M.; Weisshaar, B.; Dröge-Laser, W.; Vicente-Carbajosa, J.; Tiedemann, J.; Kroj, T.; Parcy, F. bZIP transcription factors in Arabidopsis. Trends Plant Sci. 2002, 7, 106–111. [Google Scholar] [CrossRef]
- Singh, K.B.; Foley, R.C.; Oñate-Sánchez, L. Transcription factors in plant defense and stress responses. Curr. Opin. Plant Biol. 2002, 5, 430–436. [Google Scholar] [CrossRef]
- Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. AP2/ERF family transcription factors in plant abiotic stress responses. BBA-Gene Regul. Mech. 2012, 1819, 86–96. [Google Scholar] [CrossRef]
- Maruyama, Y.; Yamoto, N.; Suzuki, Y.; Chiba, Y.; Yamazaki, K.; Sato, T.; Yamaguchi, J. The Arabidopsis transcriptional repressor ERF9 participates in resistance against necrotrophic fungi. Plant Sci. 2013, 213, 79–87. [Google Scholar] [CrossRef]
- Tian, Z.; He, Q.; Wang, H.; Liu, Y.; Zhang, Y.; Shao, F.; Xie, C. The potato ERF transcription factor StERF3 negatively regulates resistance to Phytophthora infestans and salt tolerance in potato. Plant Cell Physiol. 2015, 56, 992–1005. [Google Scholar] [CrossRef] [Green Version]
- Alves, M.; Dadalto, S.; Gonçalves, A.; de Souza, G.; Barros, V.; Fietto, L. Transcription factor functional protein-protein interactions in plant defense responses. Proteomes 2014, 2, 85–106. [Google Scholar] [CrossRef] [Green Version]
- Yogendra, K.N.; Kumar, A.; Sarkar, K.; Li, Y.; Pushpa, D.; Mosa, K.A.; Duggavathi, R.; Kushalappa, A.C. Transcription factor StWRKY1 regulates phenylpropanoid metabolites conferring late blight resistance in potato. J. Exp. Bot. 2015, 66, 7377–7389. [Google Scholar] [CrossRef] [Green Version]
- Yogendra, K.N.; Dhokane, D.; Kushalappa, A.C.; Sarmiento, F.; Rodriguez, E.; Mosquera, T. StWRKY8 transcription factor regulates benzylisoquinoline alkaloid pathway in potato conferring resistance to late blight. Plant Sci. 2017, 256, 208–216. [Google Scholar] [CrossRef]
- Kalenahalli, Y.N.; Sarkar, K.; Kage, U.; Kushalappa, A.C. Potato NAC43 and MYB8 mediated transcriptional regulation of secondary cell wall biosynthesis to contain Phytophthora infestans Infection. Plant Mol. Biol. Report. 2017, 35, 519–533. [Google Scholar]
- Chinchilla, D.; Zipfel, C.; Robatzek, S.; Kemmerling, B.; Nürnberger, T.; Jones, J.D.G.; Boller, T. A flagellin-induced complex of the receptor FLS2 and BAK1 initiates plant defence. Nature 2007, 448, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Gromadka, R.; Cieśla, J.; Olszak, K.; Szczegielniak, J.; Muszyńska, G.; Polkowska-Kowalczyk, L. Genome-wide analysis and expression profiling of calcium-dependent protein kinases in potato (Solanum tuberosum). Plant Growth Regul. 2017, 84, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Ohura, I.; Kawakita, K.; Yokota, N.; Fujiwara, M.; Shimamoto, K.; Yoshioka, H. Calcium-dependent protein kinases regulate the production of reactive oxygen species by potato NADPH oxidase. Plant Cell Online 2007, 19, 1065–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elisa, F.; María, E.S.; Franco, S.; Federico, G.M.; Rita, M.U. Analysis of the potato calcium-dependent protein kinase family and characterization of StCDPK7, a member induced upon infection with phytophthora infestans. Plant Cell Rep. 2017, 36, 1137–1157. [Google Scholar]
- Kobayashi, M.; Yoshioka, M.; Asai, S.; Nomura, H.; Kuchimura, K.; Mori, H.; Doke, N.; Yoshioka, H. StCDPK5 confers resistance to late blight pathogen but increases susceptibility to early blight pathogen in potato via reactive oxygen species burst. New Phytol. 2012, 196, 223–237. [Google Scholar] [CrossRef]
- Glazebrook, J. Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annu. Rev. Phytopathol. 2005, 43, 205–227. [Google Scholar] [CrossRef] [PubMed]
- Robb, J.; Lee, B.; Nazar, R.N. Gene suppression in a tolerant tomato vascular pathogen interaction. Planta 2007, 226, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Spoel, S.H.; Johnson, J.S.; Dong, X. Regulation of tradeoffs between plant defense against pathogens with different lifestyles. Proc. Natl. Acad. Sci. USA 2007, 104, 18842–18847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Jiang, H.; Zhu, X.; Wang, W.; He, X.; Shi, Y.; Yuan, Y.; Du, X.; Cai, Y. Analysis of sea-island cotton and upland cotton in response to Verticillium dahliae infection by RNA sequencing. BMC Genom. 2013, 14, 852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, G.; Liu, K.; Kang, J.; Xu, K.; Zhang, Y.; Hu, L.; Zhang, J.; Li, C. Transcriptome analysis of the compatible interaction of tomato with verticillium dahliae using RNA-sequencing. Front. Plant Sci. 2015, 6, 428. [Google Scholar] [CrossRef] [Green Version]
- Friend, J.; Reynolds, S.B.; Aveyard, M.A. Phenylalanine ammonia lyase, chlorogenic acid and lignin in potato tuber tissue inoculated with Phytophthora infestans. Physiol. Plant Pathol. 1973, 3, 495–507. [Google Scholar] [CrossRef]
- Henderson, S.J.; Friend, J. Increases in phenylalanine ammonia-lyase activity and lignin-like compounds as race-specific-resistance responses of potato tubers to Phytophthora infestans. Biochem. Soc. Trans. 1978, 6, 393. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; El Hadrami, A.; Adam, L.R.; Daayf, F. Differential activation and suppression of potato defence responses by Phytophthora infestans isolates representing US-1 and US-8 genotypes. Plant Pathol. 2010, 57, 1026–1037. [Google Scholar] [CrossRef]
- Gayoso, C.; Pomar, F.; Novo-Uzal, E.; Merino, F.; Martínez de Ilárduya, Ó. The Ve-mediated resistance response of the tomato to Verticillium dahliae involves H2O2, peroxidase and lignins and drives PAL gene expression. BMC Plant Biol. 2010, 10, 232. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| PGSC0003DMG400003863 (Zinc finger protein 593) | TCAAGACGAAGAAGCACAGGAAGC | AGCAGCTAAGTCGGCATCAAGTTG |
| PGSC0003DMG400005113 (PR1 protein) | GCGGCTGGTGCTGTGAAGATG | ACCTGACTCTAGCACAACCGAGAC |
| PGSC0003DMG400022159 (Heat shock cognate protein 80) | AGCTGAGAAGGTTGTTGTGACCAC | CAGGTCCTTGAGCCTACGTTCTTC |
| PGSC0003DMG400029732 (Auxin repressed/dormancy associated protein) | TACCAGAGGTCTCTGTCGATGCC | GTAGGAGCGTTAGGATGAGAAGGC |
| PGSC0003DMG400019435 (Wound-induced protein WIN2) | CAACTTGTGTTGCAGCCAATTCGG | CAGCGGCTCTGGCAACCTTG |
| PGSC0003DMG400010173 (Avr9/Cf-9 rapidly elicited protein 140) | GGTCCAATTGCTGGGATTCT | CGTCACTGGAATAACCACCTTAT |
| PGSC0003DMG400005835 (WRKY transcription factor-30) | ACATACCGAGGCTCCCATAA | TTGCTGCCCTGTTGGATAAA |
| PGSC0003DMG400008337 (MYB21) | GGCATCTGGAATTGGAAACAG | CTAACATCAGGGCGGAGATAAT |
| PGSC0003DMG400022134 (Salicylic acid-induced protein 19) | GAAATTCCTGAAGTGGCATGTG | GATACGGTGGCTCTGTTTGT |
| GenBank No. X83206 | AGATGCTTACGCTGGATGGAATGC | TTCCGGTGTGGTTGGATTCTGTTC |
| Sample | Raw Reads | Clean Reads | Uniquely Mapped Reads | Average Mapped Rate | Average Raw Reads | Average Gene Number |
|---|---|---|---|---|---|---|
| Pi0h_1 | 45,925,462 | 45,011,852 | 34,474,929 | 77.58% | 47,753,443 | 24,251 |
| Pi0h_2 | 54,412,460 | 52,408,268 | 42,404,666 | |||
| Pi0h_3 | 42,922,408 | 41,532,392 | 31,252,486 | |||
| Pi24h_1 | 44,129,574 | 42,199,746 | 33,418,531 | 77.82% | 50,630,987 | 23,579 |
| Pi24h_2 | 61,477,952 | 57,247,998 | 46,896,846 | |||
| Pi24h_3 | 46,285,436 | 44,600,984 | 32,263,042 | |||
| Pi48h_1 | 47,926,188 | 45,964,682 | 36,934,989 | 75.99% | 49,302,732 | 23,412 |
| Pi48h_2 | 48,552,120 | 46,549,340 | 33,115,129 | |||
| Pi48h_3 | 51,429,890 | 48,463,464 | 37,059,231 | |||
| Pi72h_1 | 44,484,052 | 42,866,804 | 32,606,650 | 74.79% | 48,372,645 | 23,736 |
| Pi72h_2 | 54,973,320 | 53,716,862 | 42,717,598 | |||
| Pi72h_3 | 45,660,564 | 44,668,070 | 30,717,314 |
| Sample | Pi0h_1 | Pi0h_2 | Pi0h_3 | Pi24h_1 | Pi24h_2 | Pi24h_3 | Pi48h_1 | Pi48h_2 | Pi48h_3 | Pi72h_1 | Pi72h_2 | Pi72h_3 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pi0h_1 | 1 | |||||||||||
| Pi0h_2 | 0.99 | 1 | ||||||||||
| Pi0h_3 | 0.99 | 0.99 | 1 | |||||||||
| Pi24h_1 | 0.49 | 0.51 | 0.54 | 1 | ||||||||
| Pi24h_2 | 0.57 | 0.57 | 0.60 | 0.90 | 1 | |||||||
| Pi24h_3 | 0.44 | 0.45 | 0.49 | 0.84 | 0.73 | 1 | ||||||
| Pi48h_1 | 0.88 | 0.89 | 0.89 | 0.54 | 0.60 | 0.49 | 1 | |||||
| Pi48h_2 | 0.73 | 0.73 | 0.76 | 0.55 | 0.60 | 0.58 | 0.90 | 1 | ||||
| Pi48h_3 | 0.89 | 0.88 | 0.88 | 0.42 | 0.51 | 0.39 | 0.95 | 0.82 | 1 | |||
| Pi72h_1 | 0.83 | 0.84 | 0.84 | 0.66 | 0.74 | 0.55 | 0.87 | 0.74 | 0.84 | 1 | ||
| Pi72h_2 | 0.87 | 0.88 | 0.86 | 0.59 | 0.66 | 0.49 | 0.88 | 0.69 | 0.88 | 0.96 | 1 | |
| Pi72h_3 | 0.84 | 0.85 | 0.85 | 0.69 | 0.75 | 0.54 | 0.87 | 0.72 | 0.83 | 0.98 | 0.96 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, M.; Zhou, Y.; Ye, G.; Zheng, J.; Meng, Y.; Wang, J.; Shan, W. Serial Transcriptome Analysis Reveals Genes Associated with Late Blight Resistance in Potato Cultivar Qingshu 9. Agronomy 2021, 11, 1919. https://doi.org/10.3390/agronomy11101919
He M, Zhou Y, Ye G, Zheng J, Meng Y, Wang J, Shan W. Serial Transcriptome Analysis Reveals Genes Associated with Late Blight Resistance in Potato Cultivar Qingshu 9. Agronomy. 2021; 11(10):1919. https://doi.org/10.3390/agronomy11101919
Chicago/Turabian StyleHe, Miaomiao, Yun Zhou, Guangji Ye, Jie Zheng, Yuling Meng, Jian Wang, and Weixing Shan. 2021. "Serial Transcriptome Analysis Reveals Genes Associated with Late Blight Resistance in Potato Cultivar Qingshu 9" Agronomy 11, no. 10: 1919. https://doi.org/10.3390/agronomy11101919