Abstract
Daisychain is an interactive graph visualisation and search tool for custom-built gene homology databases. The main goal of Daisychain is to allow researchers working with specific genes to identify homologs in other annotation releases. The gene-centric representation includes local gene neighborhood to distinguish orthologs and paralogs by local synteny. The software supports genome sequences in FASTA format and GFF3 formatted annotation files, and the process of building the homology database requires a minimum amount of user interaction. Daisychain includes an integrated web viewer that can be used for both data analysis and data publishing. The web interface extends KnetMaps.js and is based on JavaScript.
1. Introduction
The decline in sequencing costs and the increasing performance of genome assembly methods provide the scientific community with continued growth in the number of reference genome and pangenome assemblies. A range of comparative genomics tools and databases have been developed in recent years. One of their key features remains the search for gene and protein homologies. Public resources applied for this purpose include NCBIs HomoloGene [1], OrthoDB [2], Inparanoid [3], SHOOT [4] and eggNOG [5]. However, these databases are built from a preselected set of assemblies, and their analysis tools and visualisation cannot easily be applied for private or unpublished data. Homology clustering of individual datasets can be accomplished using software packages such as OrthoMCL [6], InParanoid [3], UCLUST [7], CD-Hit [8], GET_HOMOLOGUES(-EST) [9,10] or OrthoFinder [11]. However, a drawback of these tools is their lack of an intuitive and interactive representation of the clustering results.
These tools have become necessary with an ever-growing amount of high-quality genome sequences. One example is Brassica napus in plants. The first reference genome for B. napus was published in 2014 [12], followed by two other cultivars’ genome assemblies in 2017 [13,14], then eight high-quality genomes in 2020 [15] and two high-quality versions of earlier published genomes in 2021 [16,17]. The number of available bacterial genomes has exploded; for Escherichia coli alone, there are now more than 10,000 available genome assemblies [18]. These available genomes have led to the possibility of pangenomes representing the entire gene content of a species [19,20]. The concept originated in bacterial genomics [21]. Due to the large number of available genomes, pangenomes are now a standard approach in plant genomics, too. Pangenomes have been published for B. oleracea [22], B. napus [23], soybean [24,25], pigeon pea [26], sesame [27], rice [28,29], banana [30], wheat [31] and many more.
With the increasing number of genome assemblies, there is a requirement for a flexible tool to analyse and visualise the genic relationships between different assemblies of the same or similar individuals, which combines homology clustering of assemblies and a user-friendly representation of clustering results.
Here we present Daisychain, a tool to find and visualise homologies in genome data sets. Daisychain first builds homology databases from input genome assemblies and is therefore not limited to published data but can be used with private datasets. Homology relations are shown in a graph structure with genes represented as nodes. The visualisation includes the genomic context and an estimate of the conservation for each homology relationship. Daisychain was developed to support a server/client infrastructure but can also be run as a standalone tool. It supports multicore processing for CPU-intensive tasks and is implemented using the graph database Neo4j (www.neo4j.com (accessed on 21 August 2021)) to allow efficient storage and browsing. We demonstrate Daisychain’s features using public E. coli and B. napus assemblies and show how Daisychain can be used to explore homologies and how results can be exported for subsequent analysis or publication.
2. Materials and Methods
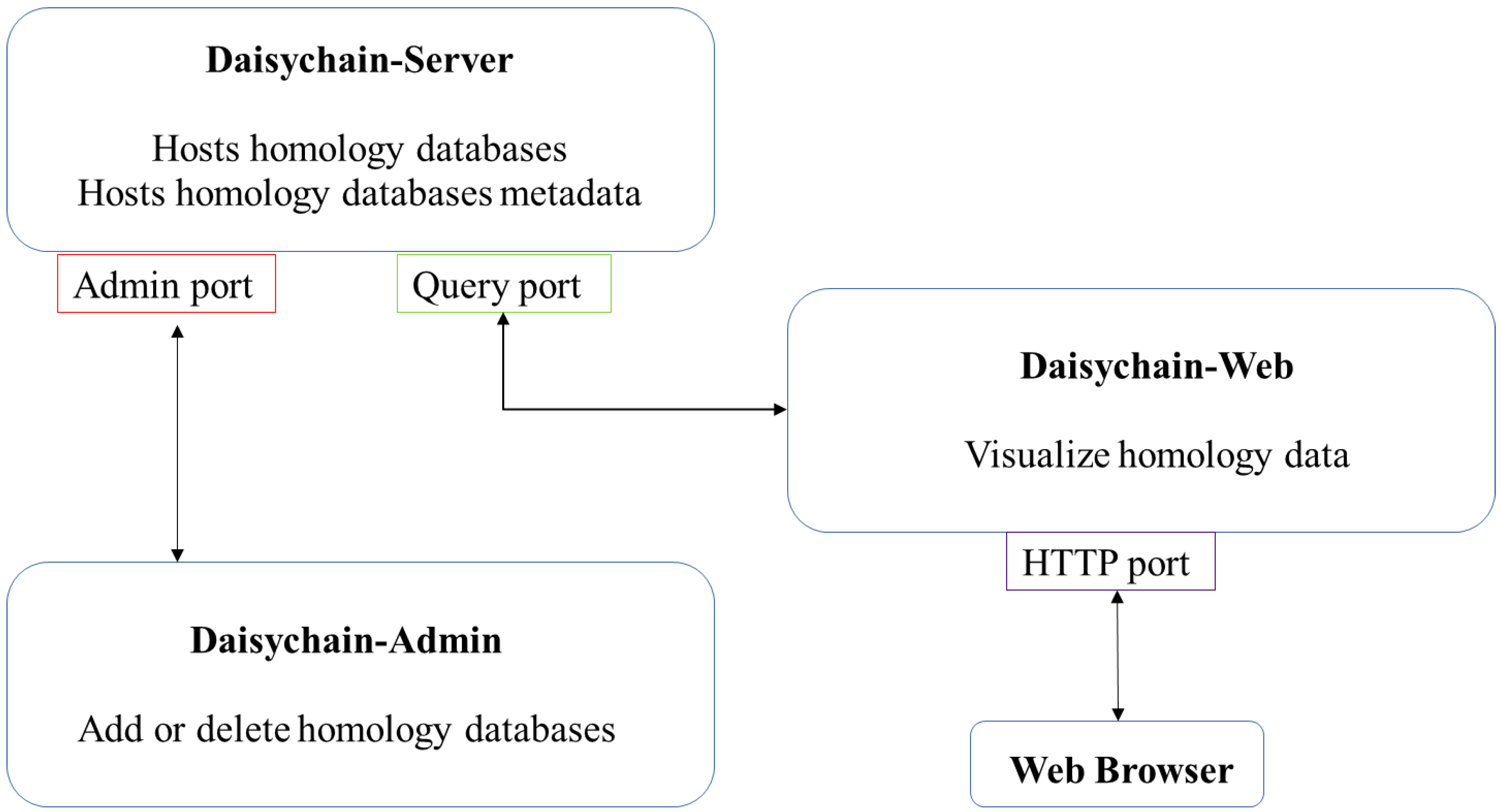
Daisychain consists of three modules: Daisychain-Server, Daisychain-Admin and Daisychain-Web (Figure 1). All modules are written in Python 3.5. Daisychain-Server hosts the server routines and all data. Daisychain-Admin is used to administrate the server from the local computer or remotely, while Daisychain-Web provides the web service used for database querying and data visualisation. Daisychain can host multiple independent homology databases, with data and metadata stored in Neo4j graph databases. The web application is built upon KnetMaps.js [32], (https://github.com/Rothamsted/knetmaps.js (accessed on 21 August 2021)) which uses cytoscape.JS 2.4.0 [33] and jQuery 1.11.2 (https://jquery.com/ (accessed on 21 August 2021)). Data is represented in graph format, with genes or proteins as nodes and homology and gene neighbourhood relations as edges. Queries are submitted in plain text format to the server application, and graph data is returned in JSON format. The server application employs Neo4j’s Cypher query language to search for genes by ID. Genes can also be found by sequence search using a local BLAST [34] database. Gene nodes can be found by sequence search. Daisychain supports single nucleotide sequences as query as well as multi FASTA formatted sequences. Any special characters are removed from individual nucleotide sequences. The server application detects whether the sequence is nucleotide or protein based on the contained characters. Nucleotide or protein sequences are compared with the database using BLAST. This returns a list of gene or protein IDs, which are then displayed in graph format. Genes or protein nodes are retrieved from the homology database using Neo4Js Cypher query language.
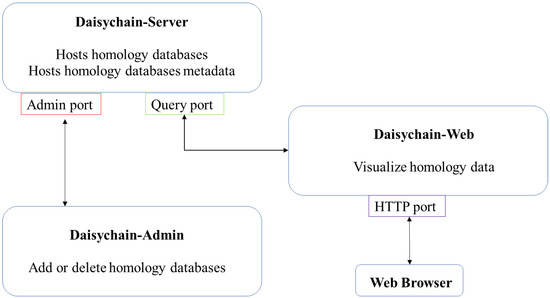
Figure 1.
Daisychain architecture. The three modules (Daisychain-Server, Daisychain-Web and Daisychain-Admin) communicate via distinct ports. Access to the Admin port can be restricted to local applications. Daisychain-Web hosts the web service.
Daisychain-Web’s underlying Cytoscape.js library accepts graph data in JSON format and produces a new graph visualisation or extends an existing data structure. Daisychain builds homology databases from genomic sequences in FASTA format and annotation data in GFF3 file format. Daisychain-Admin is used to import data, configure the GFF3 parser, and initiate the build of a new homology database. Homology databases are built from multiple assemblies. Each assembly needs to be represented by one genome FASTA file and one GFF3 annotation file. Genome data and annotation data are converted into CSV files and two FASTA files containing all coding genes or all translated sequences, respectively. The Python package gffutils is used in this step (https://pythonhosted.org/gffutils/ (accessed on 21 August 2021)). GFF3 parser configurations are set using Daisychain-Admin before initializing the homology database build. Homology clustering is flexible, using either BLAST+ or the MCL cluster algorithm [35]. For all BLAST searches, an E-value cutoff value of 1 × 10−5 is used. Query sequence length, hit sequence length, and the number of identical letters are used to calculate the total sequence identity. Clustering the BLAST results into gene and protein homolog clusters is performed as described before (https://micans.org/mcl/man/clmprotocols.html#blast (accessed on 21 August 2021)). Three different inflation values are used (1.4, 5.0, 10.0). These values are represented as “clustering sensitivity” in Daisychain. Changing the clustering sensitivity in Daisychain-Web changes the granularity of clusters. Higher inflation values typically create smaller clusters of homologs. It is possible to use other clustering tool results by manually inserting the output of these tools within Daisychain-Server.
3. Results
Daisychain’s three modules run autonomously and can be placed on different computers in the network (Figure 1). Network addresses, port numbers and paths to executables are defined in module-specific config files. Daisychain-Server and Daisychain-Web are run as a daemon service, while Daisychain-Server is accessed via the standalone tool Daisychain-Admin. The web service hosted by Daisychain-Web can be reached via any web browser that supports JavaScript. Daisychain-Admin is an interactive command-line tool used to manage homology databases hosted by Daisychain-server. It shows running and completed database management tasks and can be employed to shut down or delete homology databases.
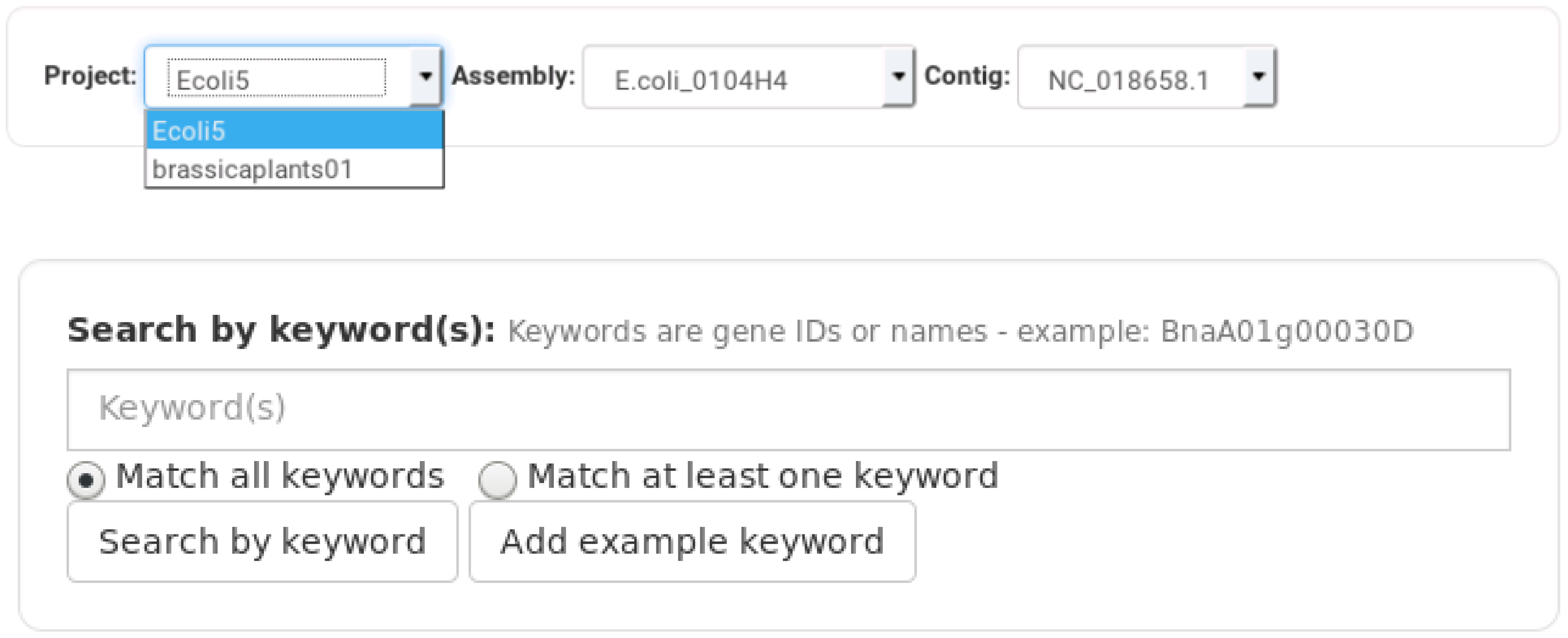
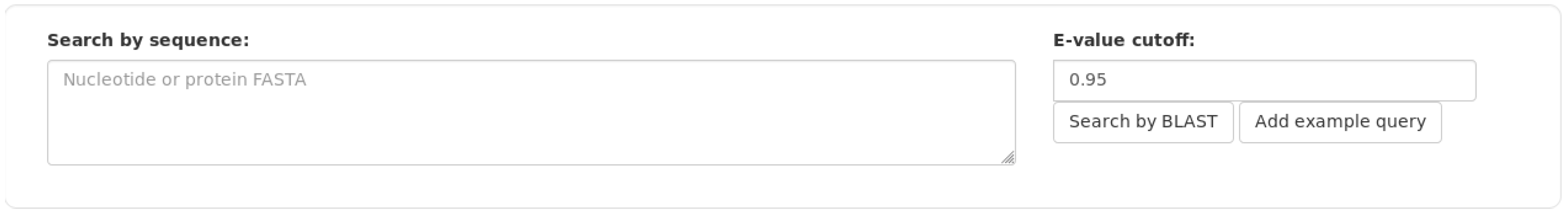
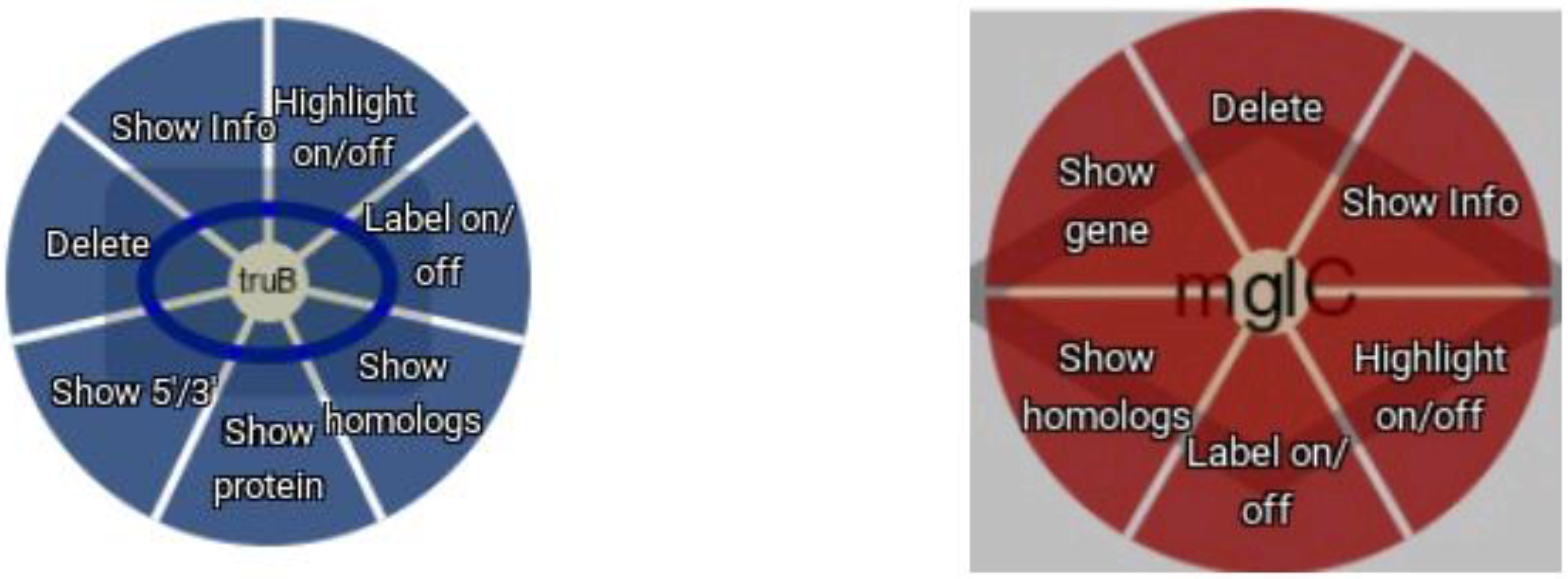
The web interface provided by Daisychain-Web consists of three areas: A search bar, the graph visualisation and a colour legend. The search bar lists all active homology databases hosted by Daisychain-Server. Search requests can be restricted to a particular assembly and a specified chromosome/contig. Gene annotations can be searched by entering one or multiple IDs or one or multiple nucleotide or protein sequences in FASTA format (Figure 2). The search results are visualised as graphs with genes shown as nodes (Figure 3). Edges represent either homology relationships or display the relative positioning of genes on the contig. The gene nodes are colour-coded, with each assembly having one specific colour assigned. Homology relations between genes are represented by coloured edges, representing the degree of sequence identity. Homology edges are coloured based on the percentage identity between the two genes or proteins connected by this edge. Identity values are converted to colors by this formula: rgb = [(((100 − perc_match) × 255)/100), ((perc_match/100) × 255), 0]. The colour of the edge provides a measure of the degree of identity between the homologs. Individual nodes can be highlighted and additional information about a gene or protein is available via the item info layover and tooltips. Gene neighbours, their homologs and proteins can be added to the visualisation via the node context menu (Figure 4). Protein nodes possess a diamond shape and are also coloured by assembly. As for genes, homolog edges represent sequence identity. Deletion of nodes and edges is possible via their context menus. Nodes can be rearranged freely with edges adjusting automatically to a new node position, while the graph outline can be set by one of the integrated graph layouts. For downstream analysis or publication, graph data can be exported as an image or tabular format, and the legend maps each assembly name to its assigned colour. Clicking one of the buttons toggles the visibility state of an assembly (Figure 4). This feature and the possibility to remove and re-add individual nodes facilitates the analysis of complex homology networks.
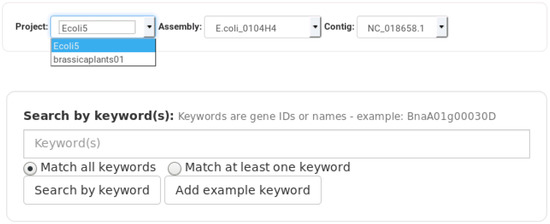

Figure 2.
Daisychain-Web search bar. Searches are performed for one specific homology database. The search is performed for all or one assembly and for all or one contig found in the selection of assemblies. Genes can be searched for by keywords. At least one keyword needs to be entered. Search results either match all or at least one keyword. Keywords are matched against gene names and gene descriptions. Genes can also be searched for by sequence(s).
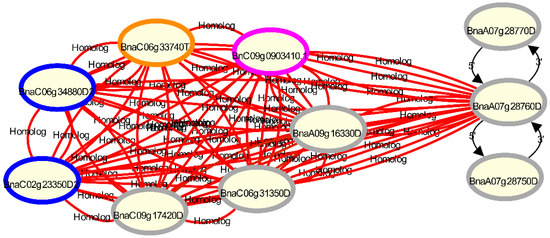
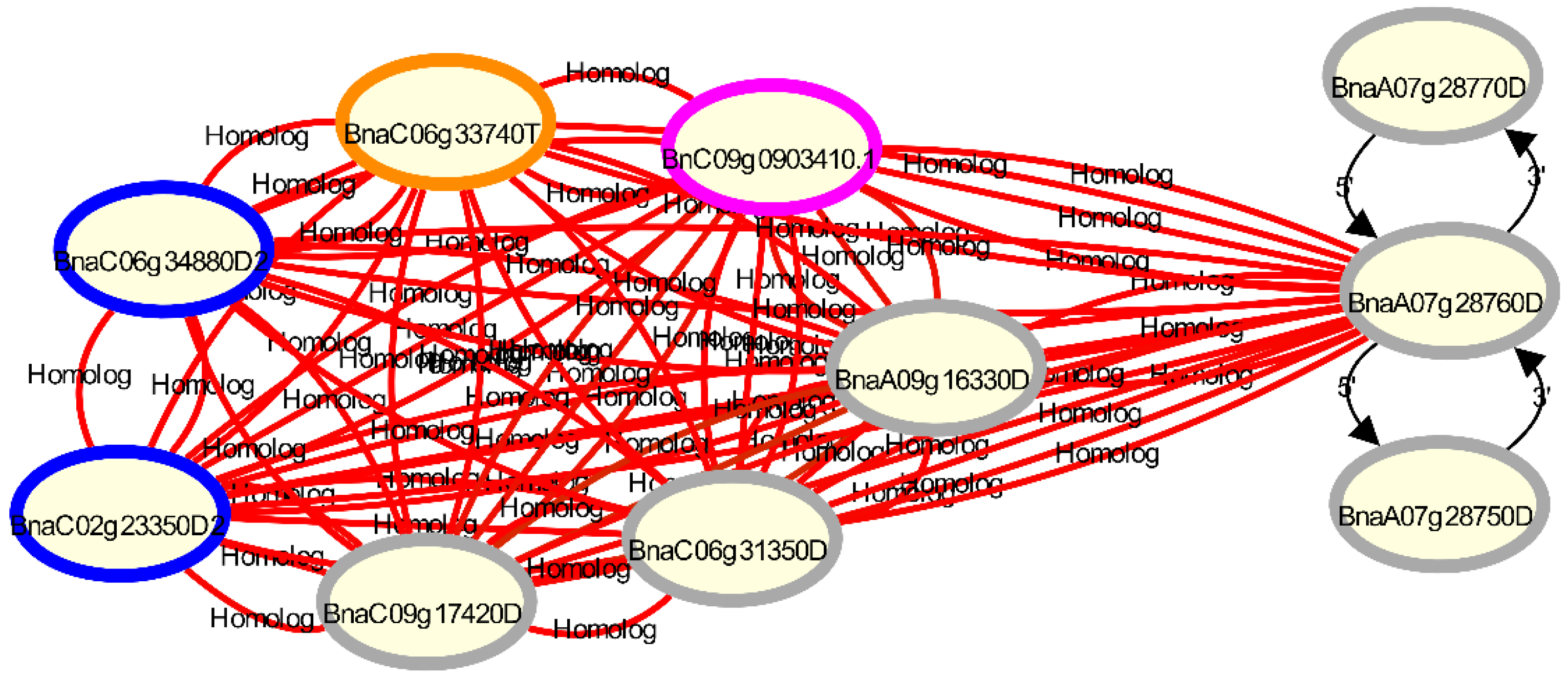
Figure 3.
Visualisation of gene homologies with Daisychain-Web for the disease resistance gene Rlm1 candidate gene BnaA07g28760D [36], its 3′ and 5′ neighbors and other homologs in the B. napus v4.1 annotation [12] (grey circles), compared with the B. napus ZS11 annotation [16] (pink circle), the B. napus Tapidor annotation (orange circles) and the Darmor-bzh v8.1 annotation (blue circles) [14]. The B. napus v4.1 annotation contains 3 homologs for the candidate gene, while the ZS11 and Tapidor annotations both contain only one homolog. Gene homologs are connected by undirected edges. The colour of the edge represents the sequence identity between the two connected genes. Gene neighbours are connected via differently styled edges, while gene nodes are coloured by their assembly. Clicking a node or an edge adds a tooltip to the visualisation with information about the assembly and contig the gene is located on. More information is shown in a layover menu when the “Show Info” option in the context menu is selected (not shown).
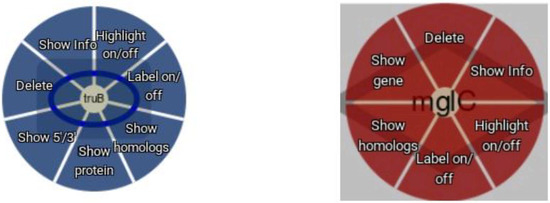
Figure 4.
Node context menu. Both gene (blue) and protein (red) nodes offer a context menu.
A publicly available instance of Daisychain is available at http://daisychain.appliedbioinformatics.com.au/ (accessed on 21 August 2021). This instance currently hosts comparisons for E. coli and B. napus assemblies. The B. napus instance currently compares four available genomes and one pangenome with a total of 367,159 genes. The E. coli instance compares four available genomes with a total of 20,399 compared genes. It is not possible for users to upload their own data but local instances of Daisychain with different datasets can be hosted using the code publicly available at https://github.com/AppliedBioinformatics/gene-daisychain (accessed on 21 August 2021) under GNU Lesser General Public License v3.
4. Discussion
Daisychain is a tool for analysing and visualising gene and protein homologies from custom collections of assemblies. The same genes within different assembly versions or assemblies of related individuals can be identified and visualised based on sequence similarity and syntenic relationship. Daisychain offers an intuitive and interactive visual representation otherwise only found in major public homology web resources. Its server/client structure permits homology data to be shared, for example, among members of a research group or a more widespread community. The publicly available Daisychain instance currently hosts assemblies for E. coli and B. napus and we plan to add more assemblies in future.
Author Contributions
Conceptualisation, O.S., C.-K.K.C., P.E.B., A.S., K.H.-P. and D.E.; methodology, O.S., J.P. and C.-K.K.C.; resources, A.S., K.H.-P., J.B. and D.E.; supervision, D.E. All authors have read and agreed to the published version of the manuscript.
Funding
This work is funded by the Australia Research Council (Projects DP1601004497, LP160100030 and DP200100762, DE210100398). AS and KHP are funded by the UK Biotechnology and Biological Sciences Research Council (Project: BB/P016855/1, DFW).
Acknowledgments
This work was supported by resources provided by the Pawsey Supercomputing Centre with funding from the Australian Government and the Government of Western Australia.
Conflicts of Interest
The authors declare no conflict of interest.
References
- NCBI, R.C. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016, 44, D7–D19. [Google Scholar] [CrossRef] [Green Version]
- Kriventseva, E.V.; Tegenfeldt, F.; Petty, T.J.; Waterhouse, R.M.; Simão, F.A.; Pozdnyakov, I.A.; Ioannidis, P.; Zdobnov, E.M. OrthoDB v8: Update of the hierarchical catalog of orthologs and the underlying free software. Nucleic Acids Res. 2015, 43, D250–D256. [Google Scholar] [CrossRef] [PubMed]
- Sonnhammer, E.L.L.; Östlund, G. InParanoid 8: Orthology analysis between 273 proteomes, mostly eukaryotic. Nucleic Acids Res. 2015, 43, D234–D239. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. SHOOT: Phylogenetic gene search and ortholog inference. bioRxiv 2021. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [Green Version]
- Fischer, S.; Brunk, B.P.; Chen, F.; Gao, X.; Harb, O.S.; Iodice, J.B.; Shanmugam, D.; Roos, D.S.; Stoeckert, C.J. Using OrthoMCL to assign proteins to OrthoMCL-DB groups or to cluster proteomes into new ortholog groups. Curr. Protoc. Bioinform. 2011, 35, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a Versatile Software Package for Scalable and Robust Microbial Pangenome Analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef] [Green Version]
- Contreras-Moreira, B.; Cantalapiedra, C.P.; Garcia-Pereira, M.J.; Gordon, S.P.; Vogel, J.P.; Igartua, E.; Casas, A.M.; Vinuesa, P. Analysis of Plant Pan-Genomes and Transcriptomes with GET_HOMOLOGUES-EST, a Clustering Solution for Sequences of the Same Species. Front. Plant Sci. 2017, 8, 184. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, F.; Fan, G.; Hu, Q.; Zhou, Y.; Guan, M.; Tong, C.; Li, J.; Du, D.; Qi, C.; Jiang, L.; et al. The high-quality genome of Brassica napus cultivar ‘ZS11’ reveals the introgression history in semi-winter morphotype. Plant J. 2017, 92, 452–468. [Google Scholar] [CrossRef] [Green Version]
- Bayer, P.E.; Hurgobin, B.; Golicz, A.A.; Chan, C.K.; Yuan, Y.; Lee, H.; Renton, M.; Meng, J.; Li, R.; Long, Y.; et al. Assembly and comparison of two closely related Brassica napus genomes. Plant Biotechnol. J. 2017, 15, 1602–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.-M.; Guan, Z.; Hu, J.; Guo, C.; Yang, Z.; Wang, S.; Liu, D.; Wang, B.; Lu, S.; Zhou, R. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 2020, 6, 34–45. [Google Scholar] [CrossRef]
- Chen, X.; Tong, C.; Zhang, X.; Song, A.; Hu, M.; Dong, W.; Chen, F.; Wang, Y.; Tu, J.; Liu, S.; et al. A high-quality Brassica napus genome reveals expansion of transposable elements, subgenome evolution and disease resistance. Plant Biotechnol. J. 2021, 19, 615–630. [Google Scholar] [CrossRef]
- Bayer, P.E.; Scheben, A.; Golicz, A.A.; Yuan, Y.; Faure, S.; Lee, H.; Chawla, H.S.; Anderson, R.; Bancroft, I.; Raman, H.; et al. Modelling of gene loss propensity in the pangenomes of three Brassica species suggests different mechanisms between polyploids and diploids. Plant Biotechnol. J. 2021, 19, 2488–2500. [Google Scholar] [CrossRef] [PubMed]
- Horesh, G.; Blackwell, G.A.; Tonkin-Hill, G.; Corander, J.; Heinz, E.; Thomson, N.R. A comprehensive and high-quality collection of Escherichia coli genomes and their genes. Microb. Genom. 2021, 7, 000499. [Google Scholar] [CrossRef]
- Golicz, A.A.; Bayer, P.E.; Bhalla, P.L.; Batley, J.; Edwards, D. Pangenomics comes of age: From bacteria to plant and animal applications. Trends Genet. 2020, 36, 132–145. [Google Scholar] [CrossRef]
- Bayer, P.E.; Golicz, A.A.; Scheben, A.; Batley, J.; Edwards, D. Plant pan-genomes are the new reference. Nat. Plants 2020, 6, 914–920. [Google Scholar] [CrossRef]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golicz, A.A.; Bayer, P.E.; Barker, G.C.; Edger, P.P.; Kim, H.; Martinez, P.A.; Chan, C.K.K.; Severn-Ellis, A.; McCombie, W.R.; Parkin, I.A.P.; et al. The pangenome of an agronomically important crop plant Brassica oleracea. Nat. Commun. 2016, 7, 13390. [Google Scholar] [CrossRef] [PubMed]
- Hurgobin, B.; Golicz, A.A.; Bayer, P.E.; Chan, C.K.K.; Tirnaz, S.; Dolatabadian, A.; Schiessl, S.V.; Samans, B.; Montenegro, J.D.; Parkin, I.A. Homoeologous exchange is a major cause of gene presence/absence variation in the amphidiploid Brassica napus. Plant Biotechnol. J. 2018, 16, 1265–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Du, H.; Li, P.; Shen, Y.; Peng, H.; Liu, S.; Zhou, G.A.; Zhang, H.; Liu, Z.; Shi, M.; et al. Pan-Genome of Wild and Cultivated Soybeans. Cell 2020, 182, 162–176.e13. [Google Scholar] [CrossRef] [PubMed]
- Bayer, P.E.; Valliyodan, B.; Hu, H.; Marsh, J.I.; Yuan, Y.; Vuong, T.D.; Patil, G.; Song, Q.; Batley, J.; Varshney, R.K. Sequencing the USDA core soybean collection reveals gene loss during domestication and breeding. Plant Genome 2021, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Bayer, P.; Ruperao, P.; Saxena, R.; Khan, A.; Golicz, A.; Nguyen, H.; Batley, J.; Edwards, D.; Varshney, R. Trait associations in the pangenome of pigeon pea (Cajanus cajan). Plant Biotechnol. J. 2020, 18, 1946–1954. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Golicz, A.A.; Lu, K.; Dossa, K.; Zhang, Y.; Chen, J.; Wang, L.; You, J.; Fan, D.; Edwards, D. Insight into the evolution and functional characteristics of the pan-genome assembly from sesame landraces and modern cultivars. Plant Biotechnol. J. 2019, 17, 881–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Feng, Q.; Lu, H.; Li, Y.; Wang, A.; Tian, Q.; Zhan, Q.; Lu, Y.; Zhang, L.; Huang, T.; et al. Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat. Genet. 2018, 50, 278–284. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Chebotarov, D.; Kudrna, D.; Llaca, V.; Lee, S.; Rajasekar, S.; Mohammed, N.; Al-Bader, N.; Sobel-Sorenson, C.; Parakkal, P. A platinum standard pan-genome resource that represents the population structure of Asian rice. Sci. Data 2020, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Rijzaani, H.; Bayer, P.E.; Rouard, M.; Doležel, J.; Batley, J.; Edwards, D. The pangenome of banana highlights differences between genera and genomes. Plant Genome 2021, e20100. [Google Scholar] [CrossRef]
- Montenegro, J.D.; Golicz, A.A.; Bayer, P.E.; Hurgobin, B.; Lee, H.; Chan, C.K.K.; Visendi, P.; Lai, K.; Doležel, J.; Batley, J. The pangenome of hexaploid bread wheat. Plant J. 2017, 90, 1007–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Rawlings, C.; Hassani-Pak, K. KnetMaps: A BioJS component to visualize biological knowledge networks [version 1; peer review: 3 approved, 1 approved with reservations]. F1000Research 2018, 7, 1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franz, M.; Lopes, C.T.; Huck, G.; Dong, Y.; Sumer, O.; Bader, G.D. Cytoscape.js: A graph theory library for visualisation and analysis. Bioinformatics 2016, 32, 309–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dongen, S.M. Graph Clustering by Flow Simulation. Ph.D. Thesis, Universiteit Utrecht, Utrecht, The Netherlands, 2000. [Google Scholar]
- Cantila, A.Y.; Saad, N.S.M.; Amas, J.C.; Edwards, D.; Batley, J. Recent Findings Unravel Genes and Genetic Factors Underlying Leptosphaeria maculans Resistance in Brassica napus and Its Relatives. Int. J. Mol. Sci. 2020, 22, 313. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).