
Integrative Transcriptome and Metabolome Analyses of the Interaction of Oat–Oat Stem Rust


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Material and Culture Conditions
2.2. Physiological Parameters
2.3. Metabolite Extraction
2.4. Data Preprocessing and Statistical Analysis
2.5. RNA Extraction, cDNA Library Construction, and Transcriptome Sequencing
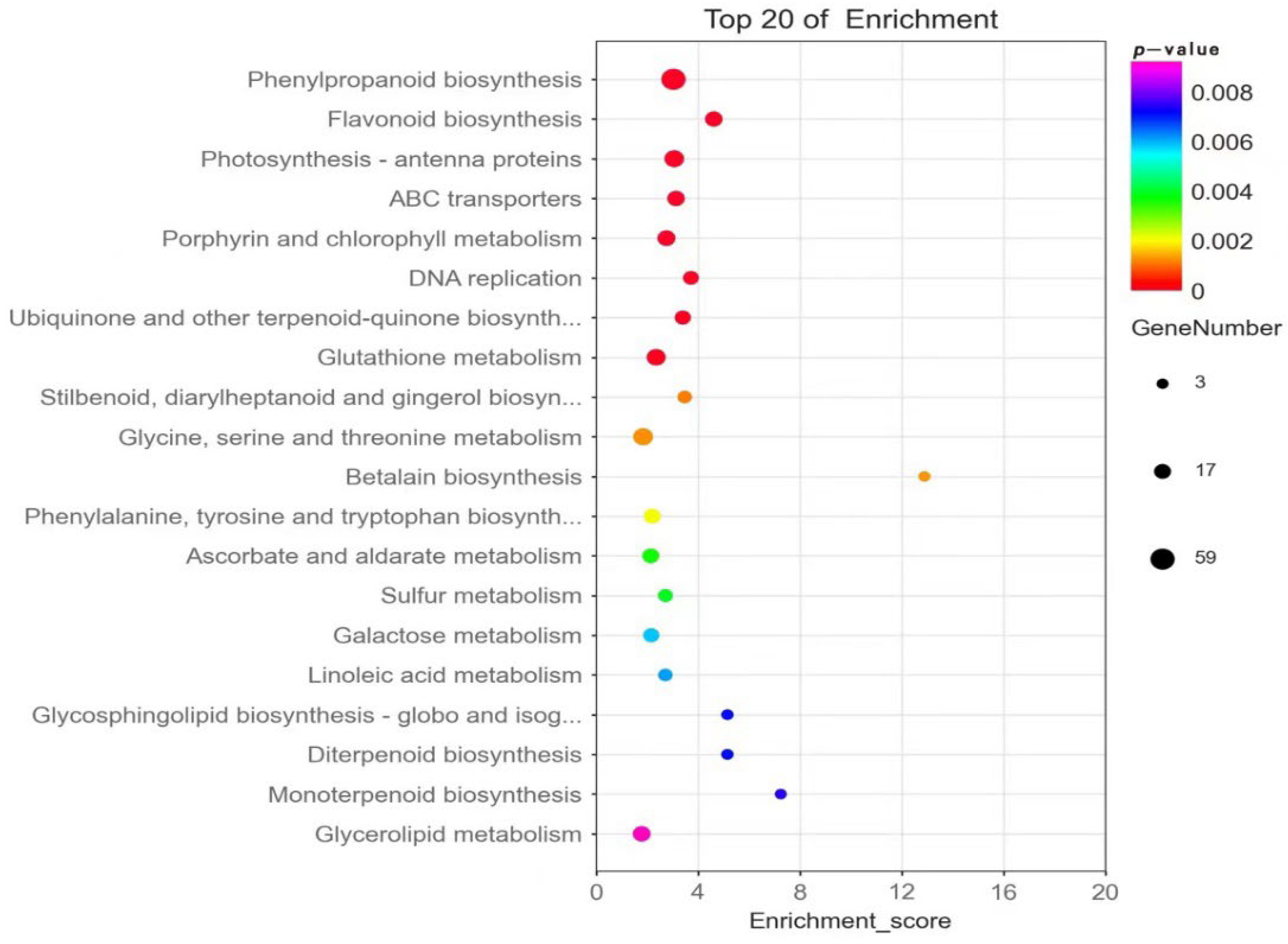
2.6. Gene Annotation, Differential Expression, and Enrichment Analyses
2.7. Pearson Correlation Analysis and Co-Expression Analysis
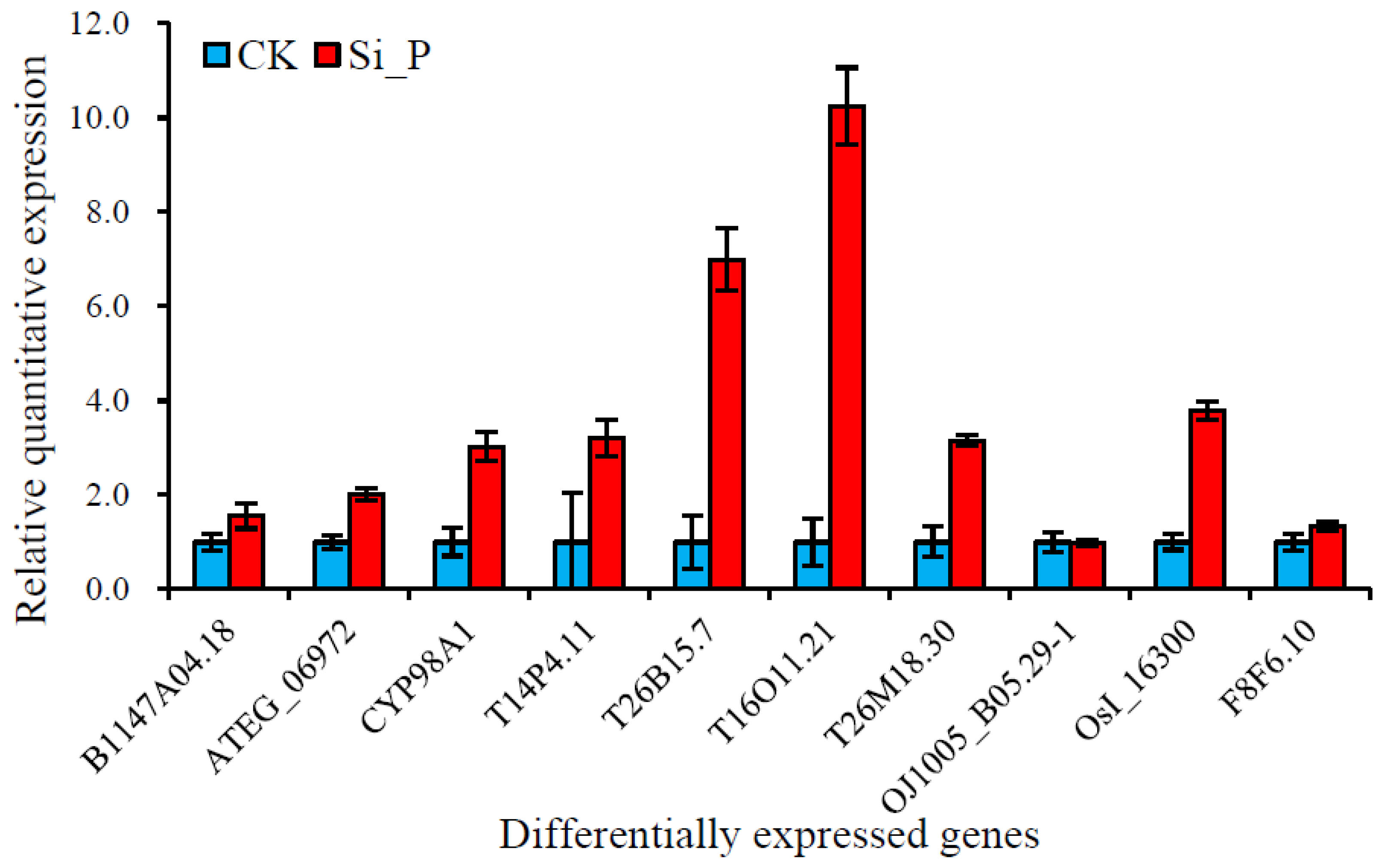
2.8. Real-Time Quantitative Reverse Transcription PCR Analysis
3. Results
3.1. Phenotypes
3.2. Physiological Changes
3.3. Metabolite Profile
3.4. RNA Sequencing and Identification of Differentially Expressed Genes
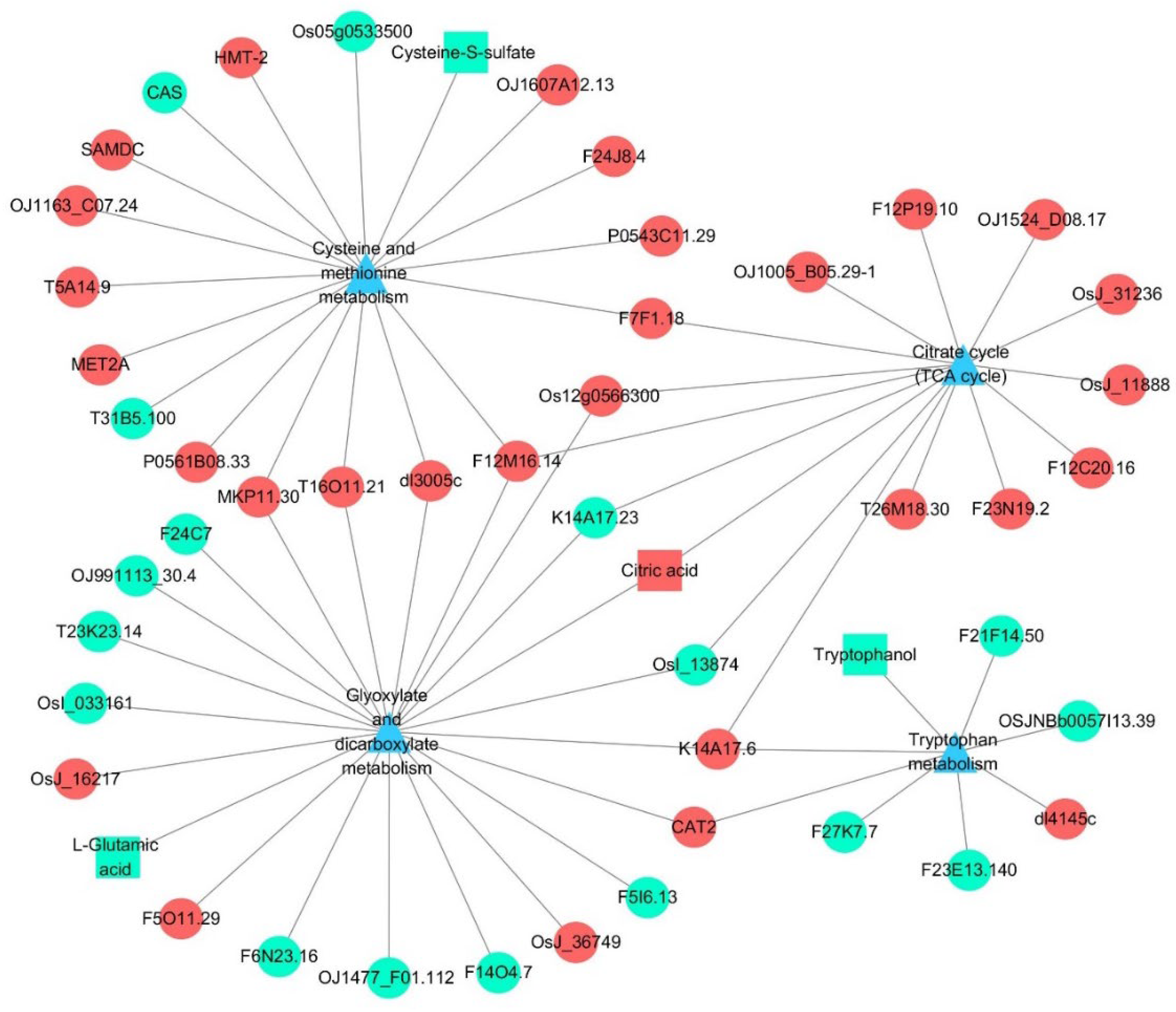
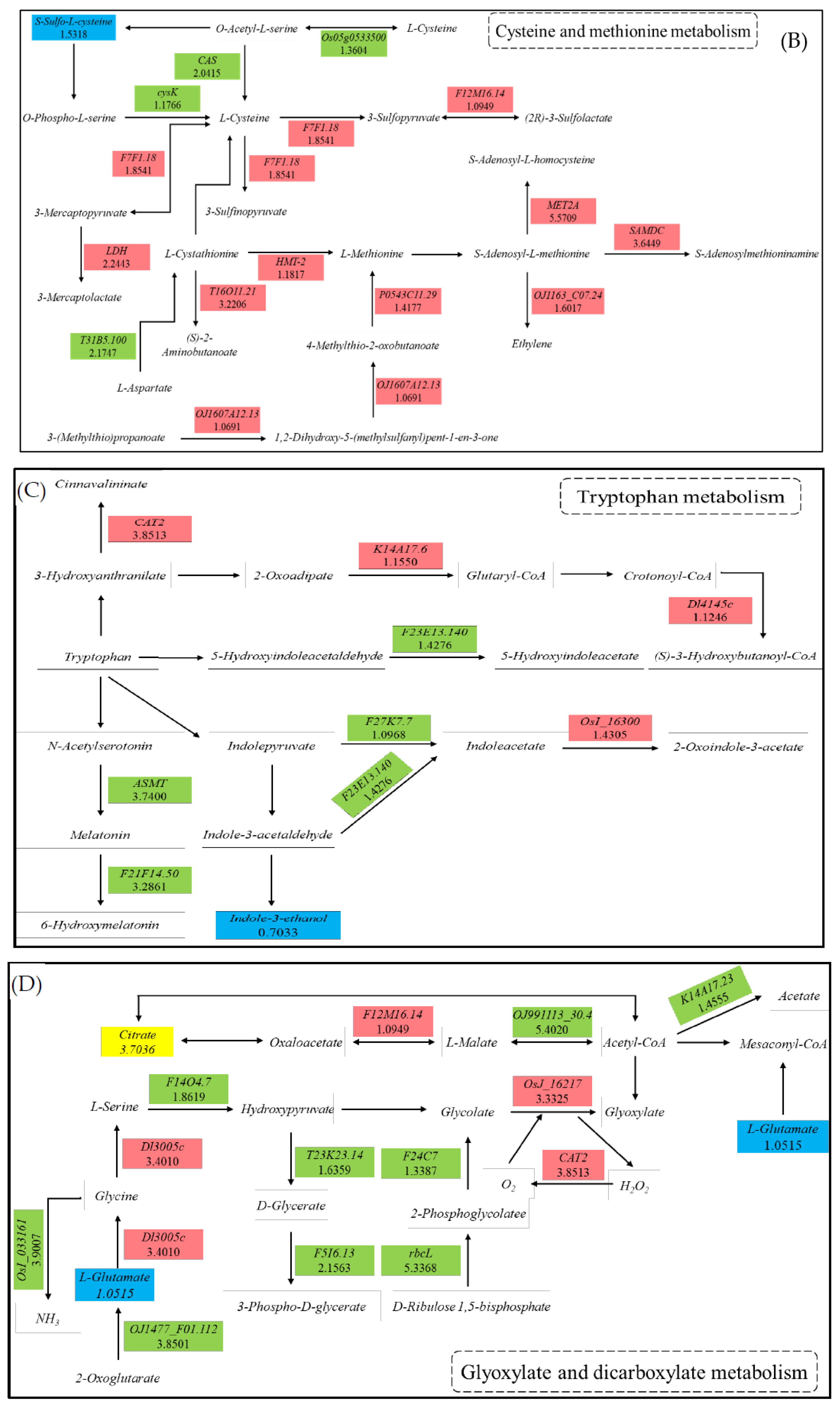
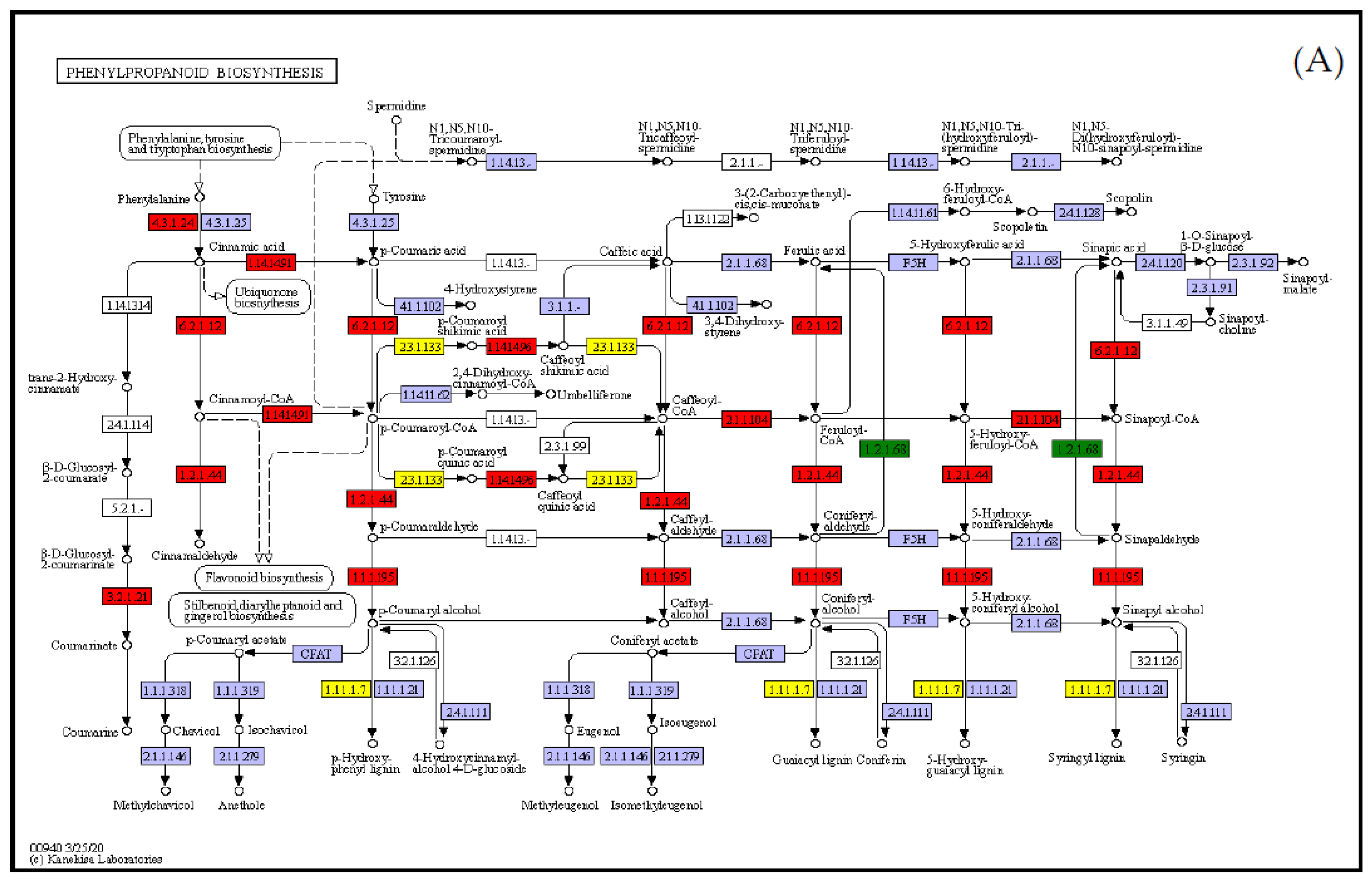
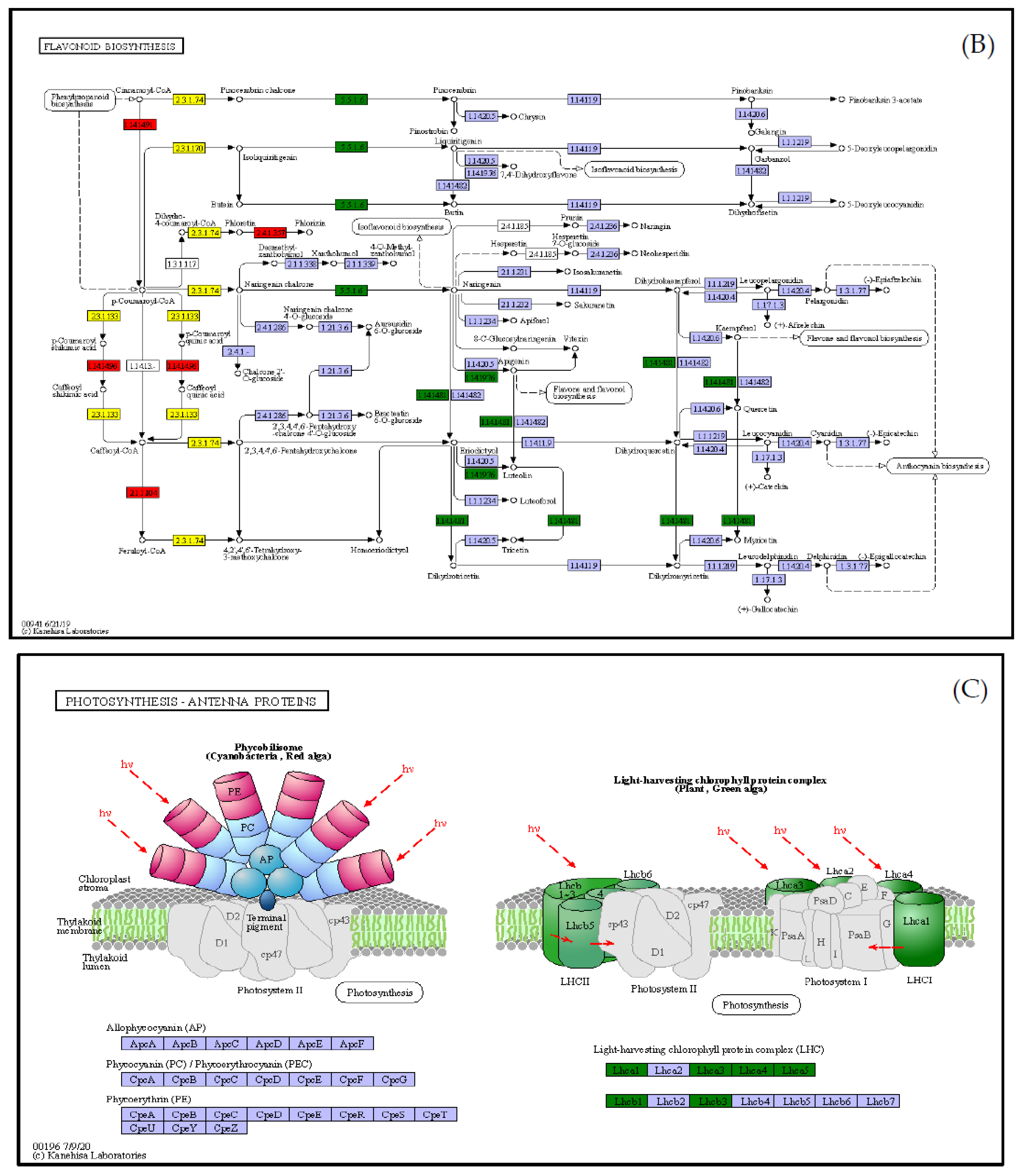
3.5. Integrated Analysis of the Transcriptome and Metabolome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bekele, W.A.; Wight, C.P.; Chao, S.; Howarth, C.J.; Tinker, N.A. Haplotype-based genotyping-by-sequencing in oat genome research. Plant Biotechnol. J. 2018, 16, 1452–1463. [Google Scholar] [CrossRef]
- Han, L.; Eneji, A.E.; Steinberger, Y.; Wang, W.; Yu, S.; Liu, H.; Liu, J. Comparative biomass production of six oat varieties in a saline soil ecology. Commun. Soil Sci. Plant Anal. 2014, 45, 2552–2564. [Google Scholar] [CrossRef]
- Gorash, A.; Armoniene, R.; Fetch, J.M.; Liatukas, Ž.; Danytė, V. Aspects in oat breeding: Nutrition quality, nakedness and disease resistance, challenges and perspectives. Ann. Appl. Biol. 2017, 171, 281–302. [Google Scholar] [CrossRef]
- Marshall, A.; Cowan, S.; Edwards, S.; Griffiths, I.; Howarth, C.; Langdon, T.; White, E. Crops that feed the world 9. Oats-a cereal crop for human and livestock feed with industrial applications. Food Secur. 2013, 5, 13–33. [Google Scholar] [CrossRef]
- Fetch, T.G., Jr. Physiologic specialization of Puccinia graminis on wheat, barley, and oat in Canada in 2000. Can. J. Plant Pathol. 2003, 25, 174–181. [Google Scholar] [CrossRef]
- Martinelli, F.; Uratsu, S.L.; Albrecht, U.; Reagan, R.L.; Phu, M.L.; Britton, M.; Buffalo, V.; Fass, J.; Leicht, E.; Zhao, W. Transcriptome Profiling of Citrus Fruit Response to Huanglongbing Disease. PLoS ONE 2012, 7, e38039. [Google Scholar] [CrossRef]
- Li, P.; Ruan, Z.; Fei, Z.; Yan, J.; Tnag, G. Integrated Transcriptome and Metabolome Analysis Revealed That Flavonoid Biosynthesis May Dominate the Resistance of Zanthoxylum bungeanum against Stem Canker. J. Agric. Food Chem. 2021, 69, 6360–6378. [Google Scholar] [CrossRef]
- Kushalappa, A.C.; Yogendra, K.N.; Karre, S. Plant innate immune response: Qualitative and quantitative resistance. Crit. Rev. Plant Sci. 2016, 35, 38–55. [Google Scholar] [CrossRef]
- Gnanesh, B.N.; Fetch, J.M.; Zegeye, T.; McCartney, C.A.; Fetch, T. Oat. In Alien Gene Transfer in Crop Plants; Pratap, A., Kumar, J., Eds.; Springer: New York, NY, USA, 2014; Volume 2, pp. 51–73. [Google Scholar]
- Gnocato, F.S.; Dracatos, P.M.; Karaoglu, H.; Zhang, P.; Berlin, A.; Park, R.F. Development, characterization and application of genomic SSR markers for the oat stem rust pathogen Puccinia graminis f. sp. avenae. Plant Pathol. 2018, 67, 457–466. [Google Scholar] [CrossRef]
- Li, T.Y.; Wu, L.X.; Wang, H.; Chen, S.; Cao, Y.Y. The race and virulence patterns of Puccinia graminis f. sp. avenae in China. Acta Triticeae Sin. 2014, 34, 552–556. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Sade, D.; Shriki, O.; Cuadros-Inostroza, A.; Tohge, T.; Semel, Y.; Haviv, Y.; Willmitzer, L.; Fernie, A.R.; Czosnek, H.; Brotman, Y. Comparative metabolomics and transcriptomics of plant response to Tomato yellow leaf curl virus infection in resistant and susceptible tomato cultivars. Metabolomics 2015, 11, 81–97. [Google Scholar] [CrossRef]
- Vogt, T. Phenylpropanoid biosynthesis. Mol. Plant 2015, 3, 2–20. [Google Scholar] [CrossRef]
- La Camera, S.; Gouzerh, G.; Dhondt, S.; Hoffmann, L.; Fritig, B.; Legrand, M.; Heitz, T. Metabolic reprogramming in plant innate immunity: The contributions of phenylpropanoid and oxylipin pathways. Immunol. Rev. 2004, 198, 267–284. [Google Scholar] [CrossRef]
- Bellincampi, D.; Cervone, F.; Lionetti, V. Plant cell wall dynamics and wall-related susceptibility in plant-pathogen interactions. Front. Plant Sci. 2014, 5, 228. [Google Scholar] [CrossRef]
- Jaeck, E.; Dumas, B.; Geoffroy, P.; Favet, N.; Inze, D.; Van Montagu, M.; Fritig, B.; Legrand, M. Regulation of enzymes involved in lignin biosynthesis: Induction of O-methyltransferase mRNAs during the hypersensitive reaction of tobacco to tobacco mosaic virus. Mol. Plant Microbe Interact. 1992, 5, 294–300. [Google Scholar] [CrossRef]
- Starr, J.L.; Yang, W.; Yan, Y.; Crutcher, F.; Kolomiets, M. Expression of phenylalanine ammonia lyase genes in maize lines differing in susceptibility to Meloidogyne incognita. J. Nematol. 2014, 46, 360–364. [Google Scholar]
- Kim, D.S.; Hwang, B.K. An important role of the pepper phenylalanine ammonia-lyase gene (PAL1) in salicylic acid-dependent signalling of the defence response to microbial pathogens. J. Exp. Bot. 2014, 65, 2295–2306. [Google Scholar] [CrossRef]
- Yadav, V.; Wang, Z.; Wei, C.; Amo, A.; Ahmed, B.; Yang, X.; Zhang, X. Phenylpropanoid pathway engineering: An emerging approach towards plant defense. Pathogens 2020, 9, 312. [Google Scholar] [CrossRef]
- Gill, U.S.; Uppalapati, S.R.; Gallego-Giraldo, L.; Ishiga, Y.; Dixon, R.A.; Mysore, K.S. Metabolic flux towards the (iso) flavonoid pathway in lignin modified alfalfa lines induces resistance against Fusarium oxysporum f. sp. medicaginis. Plant Cell Environ. 2018, 41, 1997–2007. [Google Scholar]
- Hua, J. Modulation of plant immunity by light, circadian rhythm, and temperature. Curr. Opin. Plant Biol. 2013, 16, 406–413. [Google Scholar] [CrossRef]
- Horton, P.; Ruban, A.; Walters, R. Regulation of light harvesting in green plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1996, 47, 655–684. [Google Scholar] [CrossRef]
- Kumudini, B.S.; Jayamohan, N.; Patil, S.; Govardhana, M. Chapter 11—Primary Plant Metabolism During Plant-Pathogen Interactions and Its Role in Defense. In Plant Metabolites and Regulation Under Environmental Stress; Ahmad, P., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 215–229. [Google Scholar]
- Melvin, D.B. Primary metabolism and plant defense-fuel for the fire. Mol. Plant Microbe Interact. 2009, 22, 487–497. [Google Scholar]
- Dong, N.Q.; Lin, H.X. Contribution of phenylpropanoid metabolism to plant development and plant-environment interactions. J. Integr. Plant Biol. 2020, 63, 180–209. [Google Scholar] [CrossRef]
- Li, L.; Li, T.; Jiang, Y.; Yang, Y.; Zhang, L.; Jiang, Z.; Wei, C.; Wan, X.; Yang, H. Alteration of local and systemic amino acids metabolism for the inducible defense in tea plant (Camellia sinensis) in response to leaf herbivory by Ectropis oblique. Arch. Biochem. Biophys. 2020, 683, 108301. [Google Scholar] [CrossRef]
- Wang, M.; Gu, Z.C.; Wang, R.R.; Guo, J.J.; Ling, N.; Firbank, L.G.; Guo, S.W. Plant Primary Metabolism Regulated by Nitrogen Contributes to Plant-Pathogen Interactions. Plant Cell Physiol. 2019, 60, 329–342. [Google Scholar] [CrossRef]
- Cebeci, O.; Kokturk, B.; Ergen, N.; Ozturk, L.; Cakmak, I.; Budak, H. Differential expression of wheat transcriptomes in response to varying cadmium concentrations. Biol. Plant. 2008, 52, 703–708. [Google Scholar] [CrossRef]
- Gai, Z.S.; Wang, Y.; Ding, Y.Q.; Qian, W.J.; Qiu, C.; Xie, H.; Sun, L.T.; Jiang, Z.W.; Ma, Q.P.; Wang, L.J.; et al. Exogenous abscisic acid induces the lipid and flavonoid metabolism of tea plants under drought stress. Sci. Rep. 2020, 10, 12275. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A Guide to Immunometabolism for Immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef]
- Williams, N.C.; O’Neill, L.A.J. A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front. Immunol. 2018, 9, 141. [Google Scholar] [CrossRef]
- Barth, C.; Jander, G. Arabidopsis myrosinases TGG1 and TGG2 have redundant function in glucosinolate breakdown and insect defense. Plant J. 2006, 46, 549–562. [Google Scholar] [CrossRef]
- Bednarek, P.; Osbourn, A. Plant-microbe interactions: Chemical diversity in plant defense. Science 2009, 324, 746–748. [Google Scholar] [CrossRef]
- Bednarek, P. Chemical warfare or modulators of defence responses-the function of secondary metabolites in plant immunity. Curr. Opin. Plant Biol. 2012, 15, 407–414. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Lv, P.; Mi, J.; Zhao, B.; Liu, J. Integrative Transcriptome and Metabolome Analyses of the Interaction of Oat–Oat Stem Rust. Agronomy 2022, 12, 2353. https://doi.org/10.3390/agronomy12102353
Li Y, Lv P, Mi J, Zhao B, Liu J. Integrative Transcriptome and Metabolome Analyses of the Interaction of Oat–Oat Stem Rust. Agronomy. 2022; 12(10):2353. https://doi.org/10.3390/agronomy12102353
Chicago/Turabian StyleLi, Yinghao, Pin Lv, Junzhen Mi, Baoping Zhao, and Jinghui Liu. 2022. "Integrative Transcriptome and Metabolome Analyses of the Interaction of Oat–Oat Stem Rust" Agronomy 12, no. 10: 2353. https://doi.org/10.3390/agronomy12102353
APA StyleLi, Y., Lv, P., Mi, J., Zhao, B., & Liu, J. (2022). Integrative Transcriptome and Metabolome Analyses of the Interaction of Oat–Oat Stem Rust. Agronomy, 12(10), 2353. https://doi.org/10.3390/agronomy12102353