
Deterministic Process Dominated Belowground Community Assembly When Suffering Tomato Bacterial Wilt Disease

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Plant and Soil Samples
2.2. Soil Physicochemical Analysis
2.3. DNA Extraction, Gene Amplification and Sequencing
2.4. Fluorescence Quantitative PCR (qPCR) Quantification of R. Solanacearum Density
2.5. Bioinformatic Analysis
2.6. Predicted Bacterial Gene Function Changes Analysis
2.7. The Analysis of Co-Occurrence Network
2.8. The Calculation of Community Assembly Process
2.9. Statistical Analysis
3. Results
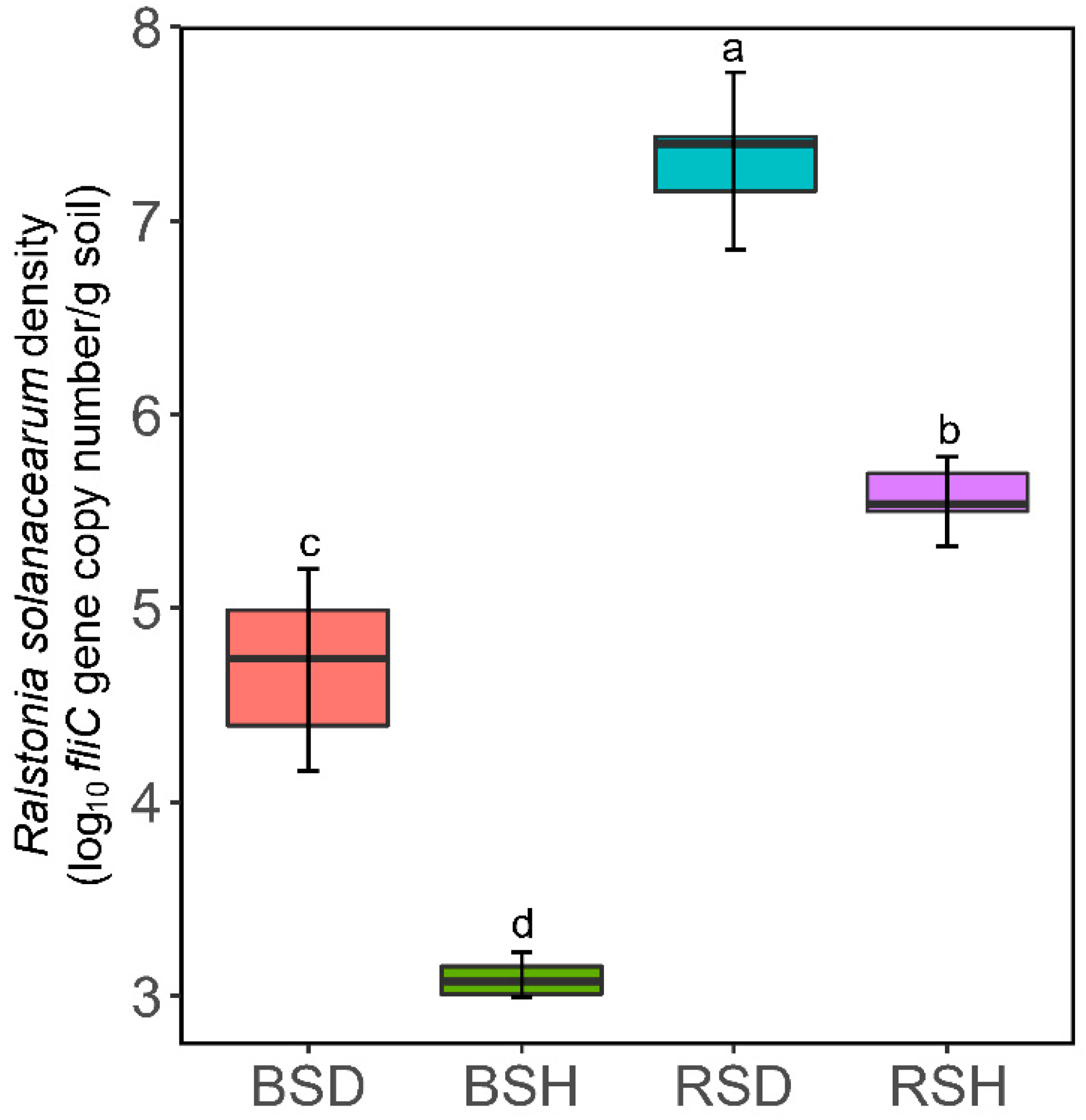
3.1. Plant Growth, Pathogen Density and Soil Properties
3.2. Microbial Composition, Diversity and Community Structure in Response to BWD
3.3. Influences of BWD on the Microbial Function
3.4. Influences of BWD on the Microbial Co-Occurrence Network
3.5. Bacterial Community Assembly Processes in Response to the BWD
4. Discussion
4.1. The Effect of Increased Pathogen Density on the Bacterial Community Composition
4.2. Increased Pathogen Density Altered the Relative Contribution of Stochastic and Deterministic Processes
4.3. The Effect of Homogeneous Selection on Bacterial Community
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bailey, K.; Lazarovits, G. Suppressing soil-borne diseases with residue management and organic amendments. Soil Till. Res. 2003, 72, 169–180. [Google Scholar] [CrossRef]
- Wu, H.; Gao, Z.; Zhou, X.; Shi, X.; Wang, M.; Shang, X.; Liu, Y.; Gu, D.; Wang, W. Microbial dynamics and natural remediation patterns of Fusarium-infested watermelon soil under 3-yr of continuous fallow condition. Soil Use Manag. 2013, 29, 220–229. [Google Scholar] [CrossRef]
- Hayward, A.C. Biology and Epidemiology of Bacterial Wilt Caused by Pseudomonas solanacearum. Annu. Rev. Phytopathol. 1991, 29, 65–87. [Google Scholar] [CrossRef]
- Raaijmakers, J.M.; Paulitz, T.C.; Steinberg, C.; Alabouvette, C.; Moenne-Loccoz, Y. The rhizosphere: A playground and battlefield for soilborne pathogens and beneficial microorganisms. Plant Soil 2009, 321, 341–361. [Google Scholar] [CrossRef] [Green Version]
- Garbeva, P.; Veen Jorge, J.A.; van Elsas, J. Microbial diversity in soil: Selection of the microbial populations by plant and soil type and implementations for disease suppressiveness. Annu. Rev. Phytopathol. 2004, 42, 243–270. [Google Scholar] [CrossRef] [PubMed]
- Mendes, L.; Raaijmakers, J.; Hollander, M.; Mendes, R.; Tsai, S. Influence of resistance breeding in common bean on rhizosphere microbiome composition and function. ISME J. 2017, 12, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Bulgarelli, D.; Schlaeppi, K.; Spaepen, S.; van Themaat, E.V.L.; Schulze-Lefert, P. Structure and Functions of the Bacterial Microbiota of Plants. Annu. Rev. Plant Biol. 2013, 64, 807–838. [Google Scholar] [CrossRef] [Green Version]
- Castrillo, G.; Teixeira, P.J.; Herrera Paredes, S.; Law, F.; Lorenzo, L.; Feltcher, M.; Finkel, O.; Breakfield, N.; Mieczkowski, P.; Jones, C.; et al. Root microbiota drive direct integration of phosphorus stress and immunity. Nature 2017, 543, 513–518. [Google Scholar] [CrossRef]
- Mendes, R.; Garbeva, P.; Raaijmakers, J.M. The rhizosphere microbiome: Significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol. Rev. 2013, 37, 634–663. [Google Scholar] [CrossRef]
- Carrión, V.; Perez-Jaramillo, J.E.; Cordovez, V.; Tracanna, V.; de Hollander, M.; Ruiz-Buck, D.; Mendes, L.W.; van Ijcken, W.F.J.; Gomez-Exposito, R.; Elsayed, S.S.; et al. Pathogen-induced activation of disease-suppressive functions in the endophytic root microbiome. Science 2019, 366, 606–612. [Google Scholar] [CrossRef]
- Mendes, R.; Kruijt, M.; de Bruijn, I.; Dekkers, E.; van der Voort, M.; Schneider, J.H.; Piceno, Y.M.; DeSantis, T.Z.; Andersen, G.L.; Bakker, P.A.; et al. Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 2011, 332, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Philippot, L.; Raaijmakers, J.M.; Lemanceau, P.; Van, D.P.; Wim, H. Going back to the roots: The microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 2013, 11, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Van Elsas, J.D.; Kastelein, P.; De Vries, P.M.; van Overbeek, L. Effects of ecological factors on the survival and physiology of Ralstonia solanacearum bv. 2 in irrigation water. Can. J. Microbiol. 2001, 47, 842–854. [Google Scholar] [CrossRef]
- Van Elsas, J.D.; Chiurazzi, M.; Mallon, C.A.; Elhottovā, D.; Kristůfek, V.; Salles, J.F. Microbial diversity determines the invasion of soil by a bacterial pathogen. Proc. Natl. Acad. Sci. USA 2012, 109, 1159–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Yang, T.; Friman, V.P.; Xu, Y.; Shen, Q.; Jousset, A. Trophic network architecture of root-associated bacterial communities determines pathogen invasion and plant health. Nat. Commun. 2015, 6, 8413. [Google Scholar] [CrossRef] [Green Version]
- Tilman, D. Niche tradeoffs, neutrality, and community structure: A stochastic theory of resource competition, invasion, and community assembly. Proc. Natl. Acad. Sci. USA 2004, 101, 10854–10861. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, R.; Pieterse, C.; Bakker, P. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef]
- Wei, Z.; Huang, J.; Yang, T.; Jousset, A.; Xu, Y.; Shen, Q.; Friman, V.P. Seasonal variation in the biocontrol efficiency of bacterial wilt is driven by temperature-mediated changes in bacterial competitive interactions. J. Appl. Ecol. 2017, 54, 1440–1448. [Google Scholar] [CrossRef] [Green Version]
- Mallon, C.; Poly, F.; Roux, X.; Marring, I.; Elsas, J.A.N.; Salles, J. Resource pulses can alleviate the biodiversity–invasion relationship in soil microbial communities. Ecology 2015, 96, 915–926. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Chen, R.; Stegen, J.; Guo, Z.; Zhang, J.; Li, Z.; Lin, X. Two key features influencing community assembly processes at regional scale: Initial state and degree of change in environmental conditions. Mol. Ecol. 2018, 27, 5238–5251. [Google Scholar] [CrossRef]
- Liu, W.B.; Graham, E.; Zhong, L.; Zhang, J.; Li, W.; Li, Z.; Lin, X.; Feng, Y. Dynamic microbial assembly processes correspond to soil fertility in sustainable paddy agroecosystems. Funct. Ecol. 2020, 34, 1244–1256. [Google Scholar] [CrossRef]
- Mendes, L.W.; Kuramae, E.E.; Navarrete, A.A.; van Veen, J.A.; Tsai, S.M. Taxonomical and functional microbial community selection in soybean rhizosphere. ISME J. 2014, 8, 1577–1587. [Google Scholar] [CrossRef] [PubMed]
- Dumbrell, A.; Nelson, M.; Helgason, T.; Dytham, C.; Fitter, A. Relative roles of niche and neutral processes in structuring a soil microbial community. ISME J. 2010, 4, 337–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langenheder, S.; Székely, A. Species sorting and neutral processes are both important during the initial assembly of bacterial communities. ISME J. 2011, 5, 1086–1094. [Google Scholar] [CrossRef] [Green Version]
- Graham, E.B.; Crump, A.R.; Resch, C.T.; Fansler, S.J.; Arntzen, E.V.; Kennedy, D.W.; Fredrickson, J.K.; Stegen, J.C. Deterministic influences exceed dispersal effects on hydrologically-connected microbiomes. Environ. Microbiol. 2017, 19, 1552–1567. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, W.; Ye, D.; Jiang, Y.H.; Wang, A. Stochastic Assembly Leads to Alternative Communities with Distinct Functions in a Bioreactor Microbial Community. MBio 2013, 4, e00584-12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dini-Andreote, F.; Stegen, J.; Elsas, J.; Salles, J. Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession. Proc. Natl. Acad. Sci. USA 2015, 112, E1326–E1332. [Google Scholar] [CrossRef] [Green Version]
- Stegen, J.; Lin, X.; Konopka, A.; Fredrickson, J. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J. 2012, 6, 1653–1664. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Graham, E.; Dong, Y.; Zhong, L.; Zhang, J.; Qiu, C.; Chen, R.; Lin, X.; Feng, Y. Balanced stochastic versus deterministic assembly processes benefit diverse yet uneven ecosystem functions in representative agroecosystems. Environ. Microbiol. 2020, 23, 391–404. [Google Scholar] [CrossRef]
- Tripathi, B.; Stegen, J.; Kim, M.; Dong, K.; Adams, J.; Lee, Y.K. Soil pH mediates the balance between stochastic and deterministic assembly of bacteria. ISME J. 2018, 12, 1072–1083. [Google Scholar] [CrossRef]
- Zhang, K.; Shi, Y.; Cui, X.; Yue, P.; Li, K.; Liu, X.; Tripathi, B.; Chu, H. Salinity Is a Key Determinant for Soil Microbial Communities in a Desert Ecosystem. mSystems 2019, 4, e00225-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stegen, J.; Lin, X.; Fredrickson, J.; Chen, X.; Kennedy, D.; Murray, C.; Rockhold, M.; Konopka, A. Quantifying community assembly processes and identifying features that impose them. ISME J. 2013, 7, 2069–2079. [Google Scholar] [CrossRef] [PubMed]
- Ning, D.; Deng, Y.; Tiedje, J.; Zhou, J. A general framework for quantitatively assessing ecological stochasticity. Proc. Natl. Acad. Sci. USA 2019, 116, 16892–16898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F. Ralstonia solanacearum Species Complex and Bacterial Wilt Disease. J. Bacteriol. Parasitol. 2013, 4, e119. [Google Scholar] [CrossRef]
- Peng, J.; Liu, H.; Shen, M.; Chen, R.; Li, J.; Dong, Y. The inhibitory effects of different types of Brassica seed meals on the virulence of Ralstonia solanacearum. Pest Manag. Sci. 2021, 77, 5129–5138. [Google Scholar] [CrossRef]
- Liu, H.; Dong, Y.H.; Shen, M.; Sun, F.; Wang, X.; Liu, J.; Li, J. Characteristics of Rhizosphere Microbial Communities in a Disease-suppressive Soil of Tomato Bacterial Wilt and its Disease-suppressive Transmission Mechanism. Acta Pedol. Sin. 2021. [Google Scholar] [CrossRef]
- Lu, R.K. Analytical Methods of Soil and Agricultural Chemistry; China Agricultural Science and Technology Press: Beijing, China, 1999. [Google Scholar]
- Zhang, Z.; Liu, H.; Liu, X.; Chen, Y.; Lu, Y.; Shen, M.; Dang, K.; Zhao, Y.; Dong, Y.; Li, Q.; et al. Organic fertilizer enhances rice growth in severe saline-alkali soil by increasing soil bacterial diversity. Soil Use Manag. 2021, 38, 964–977. [Google Scholar] [CrossRef]
- Schonfeld, J.; Heuer, H.; van Elsas, J.D.; Smalla, K. Specific and sensitive detection of Ralstonia solanacearum in soil on the basis of PCR amplification of fliC fragments. Appl. Environ. Microb. 2003, 69, 7248–7256. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.; Lauber, C.; Walters, W.; Berg-Lyons, D.; Lozupone, C.; Turnbaugh, P.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Deng, Y.; Shen, L.; Wen, C.; Yan, Q.; Ning, D.; Qin, Y.; Xue, K.; Wu, L.; He, Z.; et al. Temperature mediates continental-scale diversity of microbes in forest soils. Nat. Commun. 2016, 7, 12083. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Jiang, Y.-H.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Deng, Y.; Luo, F.; He, Z.; Tu, Q.; Zhi, X.Y. Functional Molecular Ecological Networks. MBio 2010, 1, e00169-10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.; Wang, J.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Dalsing, B.; Allen, C. Nitrate Assimilation Contributes to Ralstonia solanacearum Root Attachment, Stem Colonization, and Virulence. J. Bacteriol. 2013, 196, 949–960. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Wei, Z.; Friman, V.P.; Xu, Y.; Shen, Q.; Kowalchuk, G.; Jousset, A. Resource availability modulates biodiversity-invasion relationships by altering competitive interactions. Environ. Microbiol. 2017, 19, 2984–2991. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Ding, W.; Li, S.; Liu, Y.; Wang, J.; Yang, L.; Zhang, S. Soil Acidification Aggravates the Occurrence of Bacterial Wilt in South China. Front. Microbiol. 2017, 8, 703. [Google Scholar] [CrossRef] [Green Version]
- Dalsing, B.; Truchon, A.; Gonzalez-Orta, E.; Milling, A.; Allen, C. Ralstonia solanacearum Uses Inorganic Nitrogen Metabolism for Virulence, ATP Production, and Detoxification in the Oxygen-Limited Host Xylem Environment. MBio 2015, 6, e02471-14. [Google Scholar] [CrossRef] [Green Version]
- Edwards, J.; Johnson, C.; Santos-Medellín, C.; Lurie, E.; Natarajkumar, P.; Bhatnagar, S.; Eisen, J.; Sundaresan, V. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA 2015, 112, E911–E920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhang, N.; Qiu, M.; Feng, H.; Vivanco, J.; Shen, Q.; Zhang, R. Enhanced rhizosphere colonization of beneficial Bacillus amyloliquefaciens SQR9 by pathogen infection. FEMS Microbio. Lett. 2014, 353, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stringlis, I.; Yu, K.; Feussner, K.; de Jonge, R.; van Bentum, S.; Verk, M.; Berendsen, R.; Bakker, P.; Feussner, I.; Pieterse, C. MYB72-dependent coumarin exudation shapes root microbiome assembly to promote plant health. Proc. Natl. Acad. Sci. USA 2018, 115, E5213–E5222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Procópio, R.; Silva, I.; Martins, M.; Azevedo, J.; Araújo, J. Antibiotics produced by Streptomyces. Braz. J. Infect. Dis. Off. Publ. Braz. Soc. Infect. Dis. 2012, 16, 466–471. [Google Scholar] [CrossRef]
- Nion, Y.A.; Toyota, K. Recent Trends in Control Methods for Bacterial Wilt Diseases Caused by Ralstonia solanacearum. Microbes Environ. 2015, 30, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Yang, X.; Yin, S.; Shen, Q.; Ran, W.; Xu, Y. Efficacy of Bacillus-fortified organic fertiliser in controlling bacterial wilt of tomato in the field. Appl. Soil Ecol. 2011, 48, 152–159. [Google Scholar] [CrossRef]
- Laksmanan, V.; Selvaraj, G.; Bais, H. Functional Soil Microbiome: Belowground Solutions to an Aboveground Problem. Plant Physiol. 2014, 166, 689–700. [Google Scholar] [CrossRef] [Green Version]
- Kéfi, S.; Berlow, E.L.; Wieters, E.A.; Navarrete, S.A.; Petchey, O.L.; Wood, S.A.; Boit, A.; Joppa, L.N.; Lafferty, K.D.; Williams, R.J. More than a meal… integrating non-feeding interactions into food webs. Ecol. Lett. 2012, 15, 291–300. [Google Scholar] [CrossRef]
- Thébault, E.; Fontaine, C. Stability of Ecological Communities and the Architecture of Mutualistic and Trophic Networks. Science 2010, 329, 853–856. [Google Scholar] [CrossRef]
- Hu, J.; Wei, Z.; Friman, V.P.; Gu, S.H.; Wang, X.F.; Eisenhauer, N.; Yang, T.J.; Ma, J.; Shen, Q.R.; Xu, Y.C.; et al. Probiotic diversity enhances rhizosphere microbiome function and plant disease suppression. MBio 2016, 7, 8. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Barahona, M.; Tan, Y.J.; Deng, H.Z. Natural Connectivity of Complex Networks. Chin. Phys. Lett. 2010, 7, 295–298. [Google Scholar] [CrossRef]
- Xiong, W.; Song, Y.; Yang, K.; Gu, Y.; Wei, Z.; Kowalchuk, G.; Xu, Y.; Jousset, A.; Shen, Q.; Geisen, S. Rhizosphere protists are key determinants of plant health. Microbiome 2020, 8, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Xiong, C.; Gao, C.; Tsui, C.; Wang, M.; Zhou, X.; Zhang, A.; Cai, L. Disease-induced changes in plant microbiome assembly and functional adaptation. Microbiome 2021, 9, 187. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Gu, Y.; Hu, J.; Yin, S.; Xu, Y.; Jousset, A.; Shen, Q.; Friman, V.P. Ralstonia solanacearum pathogen disrupts bacterial rhizosphere microbiome during an invasion. Soil Biol. Biochem. 2018, 118, 8–17. [Google Scholar] [CrossRef]
- Nemergut, D.; Schmidt, S.; Fukami, T.; O’Neill, S.; Bilinski, T.; Stanish, L.; Knelman, J.; Darcy, J.; Lynch, R.; Wickey, P.; et al. Patterns and Processes of Microbial Community Assembly. Microbiol. Mol. Biol. R. 2013, 77, 342–356. [Google Scholar] [CrossRef] [Green Version]
- Bahram, M.; Kohout, P.; Anslan, S.; Harend, H.; Abarenkov, K.; Tedersoo, L. Stochastic distribution of small soil eukaryotes resulting from high dispersal and drift in a local environment. ISME J. 2015, 10, 885–896. [Google Scholar] [CrossRef] [Green Version]
- Jiao, S.; Lu, Y. Abundant fungi adapt to broader environmental gradients than rare fungi in agricultural fields. Glob. Chang. Biol. 2020, 26, 4506–4520. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Properties | Diseased Samples | Healthy Samples |
|---|---|---|
| Plant | ||
| Plant height (cm) | 68.63 ± 4.93 a | 78.00 ± 6.57 a |
| Disease index (%) | 83.33 ± 0.06 a | 12.50 ± 0.05 b |
| Soil | ||
| moisture | 0.14 ± 0.01 a | 0.08 ± 0.01 b |
| pH | 6.20 ± 0.09 b | 6.61 ± 0.03 a |
| Ec (mS cm−1) | 0.30 ± 0.03 a | 0.19 ± 0.01 b |
| NO3−-N (mg kg−1) | 45.68 ± 4.11 a | 19.13 ± 3.41 b |
| NH4+-N (mg kg−1) | 14.23 ± 0.28 a | 10.92 ± 0.77 b |
| SOC (g kg−1) | 7.67 ± 0.18 b | 8.97 ± 0.14 a |
| TN (g kg−1) | 1.38 ± 0.04 a | 1.33 ± 0.06 a |
| TP (g kg−1) | 1.34 ± 0.03 a | 1.30 ± 0.07 a |
| TK (g kg−1) | 15.54 ± 0.34 a | 15.08 ± 0.25 a |
| AP (mg kg−1) | 116.47 ± 5.91 a | 97.78 ± 7.83 a |
| AK (mg kg−1) | 270.33 ± 5.11 b | 316.20 ± 18.51 a |
| Sample | Richness | Shannon | Chao 1 | Faith’s PD |
|---|---|---|---|---|
| BSD | 2151 ± 75 a | 9.15 ± 0.07 a | 2796 ± 88 a | 114 ± 4.37 a |
| BSH | 2119 ± 108 a | 9.10 ± 0.16 a | 2702 ± 135 a | 150 ± 6.14 a |
| RSD | 912 ± 71 c | 2.79 ± 0.29 c | 1481 ± 101 c | 78 ± 4.45 c |
| RSH | 1515 ± 70 b | 7.02 ± 0.26 b | 2117 ± 144 b | 116 ± 4.67 b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Sun, F.; Peng, J.; Shen, M.; Li, J.; Dong, Y. Deterministic Process Dominated Belowground Community Assembly When Suffering Tomato Bacterial Wilt Disease. Agronomy 2022, 12, 1024. https://doi.org/10.3390/agronomy12051024
Liu H, Sun F, Peng J, Shen M, Li J, Dong Y. Deterministic Process Dominated Belowground Community Assembly When Suffering Tomato Bacterial Wilt Disease. Agronomy. 2022; 12(5):1024. https://doi.org/10.3390/agronomy12051024
Chicago/Turabian StyleLiu, Hong, Feifei Sun, Junwei Peng, Minchong Shen, Jiangang Li, and Yuanhua Dong. 2022. "Deterministic Process Dominated Belowground Community Assembly When Suffering Tomato Bacterial Wilt Disease" Agronomy 12, no. 5: 1024. https://doi.org/10.3390/agronomy12051024
APA StyleLiu, H., Sun, F., Peng, J., Shen, M., Li, J., & Dong, Y. (2022). Deterministic Process Dominated Belowground Community Assembly When Suffering Tomato Bacterial Wilt Disease. Agronomy, 12(5), 1024. https://doi.org/10.3390/agronomy12051024