
Integrated Transcriptome and Proteome Analyses of Maize Inbred lines in Response to Salt Stress

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Proteome Sequencing and Analysis of the Differentially Expressed Proteins
2.3. Transcriptome Sequencing and Analysis of Differentially Expressed Genes
2.4. Association Analysis
2.5. Bioinformatics Analysis
2.6. RNA Extraction and qRT-PCR
3. Results
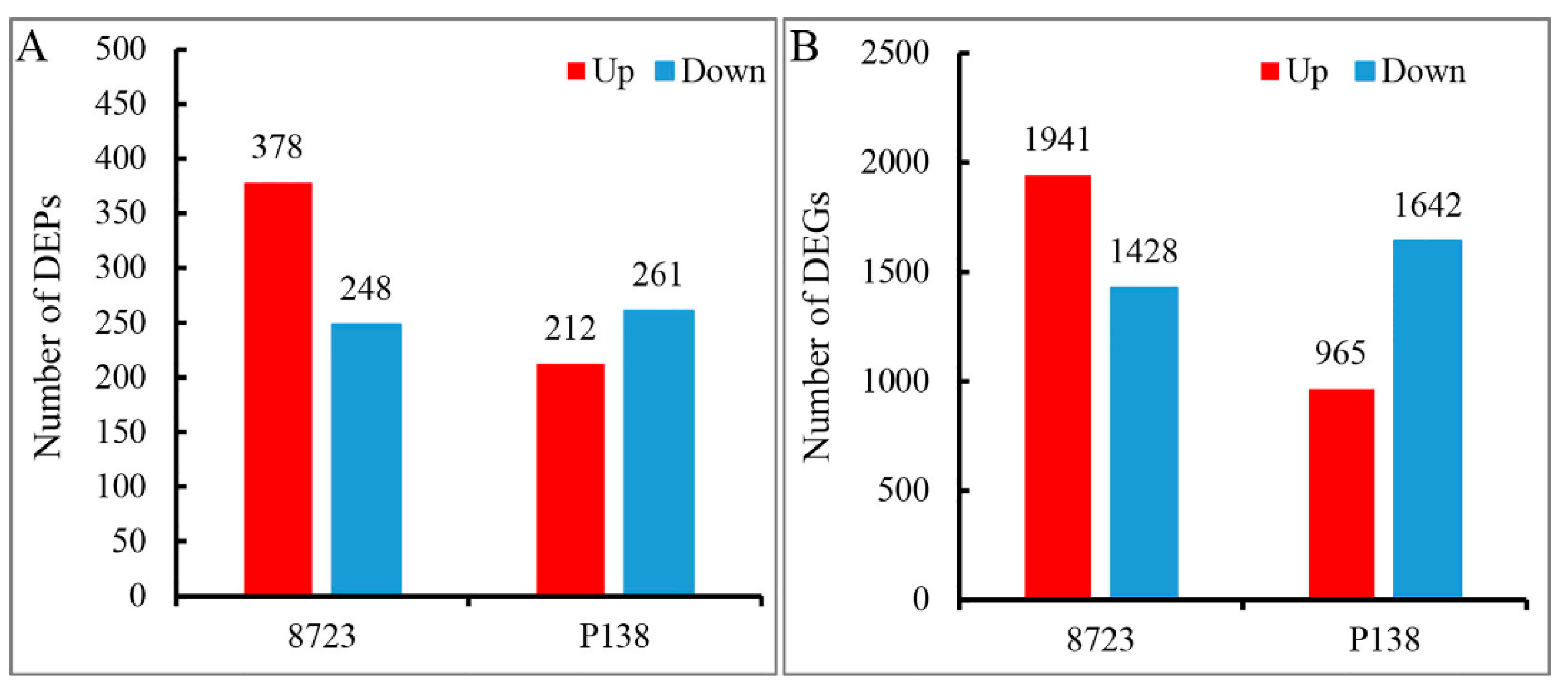
3.1. Proteome and Transcriptome Data Analysis
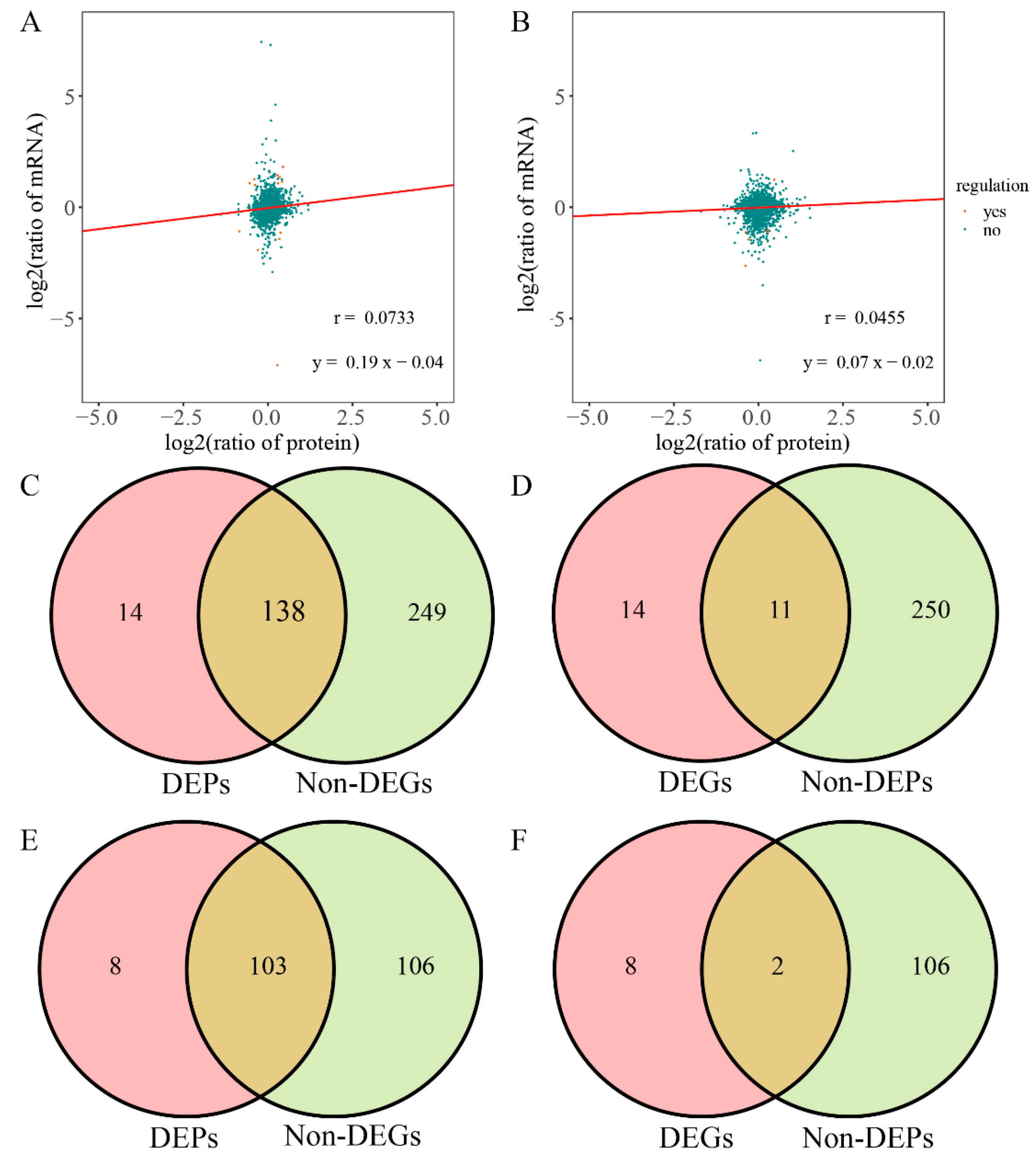
3.2. Correlation Analysis between Transcriptome and Protein
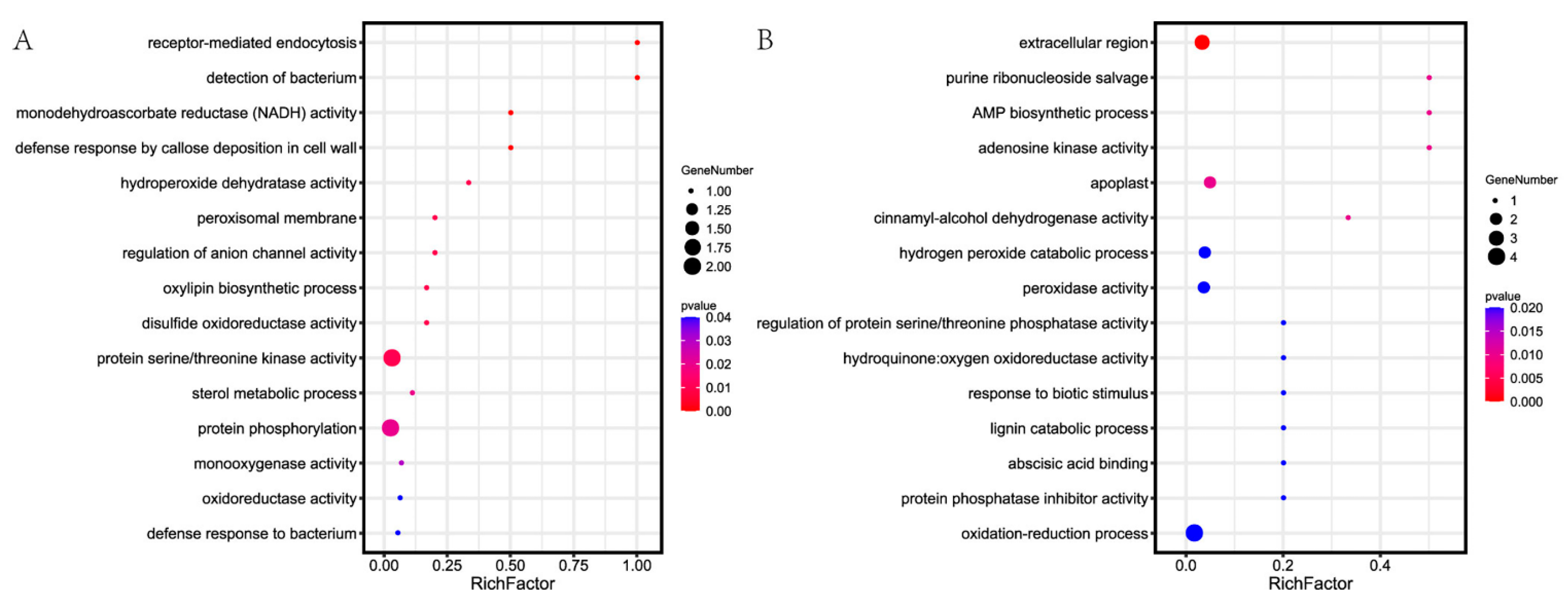
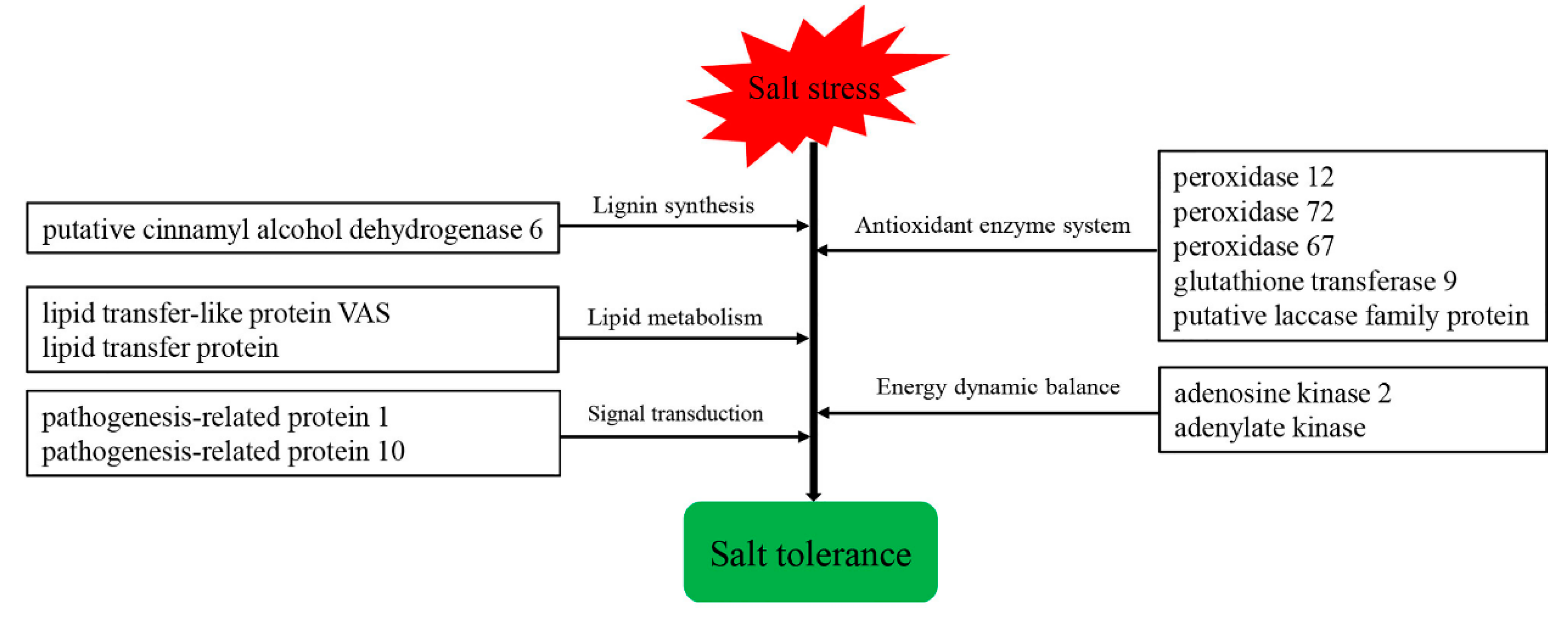
3.3. Bioinformatics Analysis of Related DEPs/DEGs
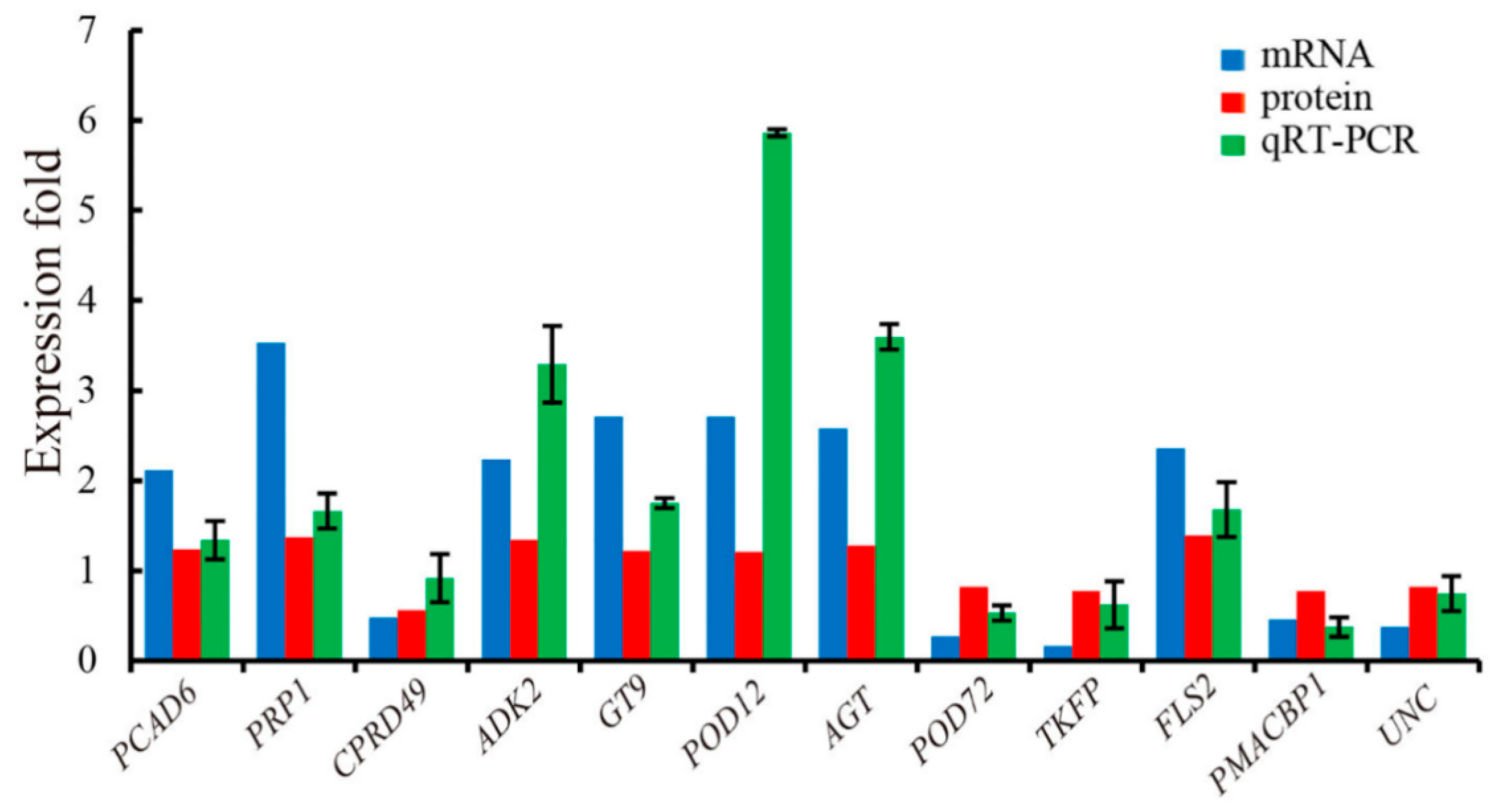
3.4. qRT-PCR Verification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, J.; Xing, S.; Li, J.; Makeschin, F.; Song, Y. Agroforestry and its application in amelioration of saline soils in eastern China coastal region. For. Stud. China 2004, 6, 27–33. [Google Scholar] [CrossRef]
- Abbasi, G.; Javaid, A.; Anwar-ul-Haq, M.; Ahmad, N. Screening of maize hybrids for salt tolerance at seedling stage under hydroponic condition. Soil Environ. 2012, 31, 83–90. [Google Scholar]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabala, S. Learning from halophytes: Physiological basis and strategies to improve abiotic stress tolerance in crops. Ann. Bot. 2013, 112, 1209–1221. [Google Scholar] [CrossRef] [PubMed]
- Li, G.K.; Gao, J.; Peng, H.; Shen, Y.O.; Ding, H.P.; Zhang, Z.M.; Pan, G.T.; Lin, H.J. Proteomic changes in maize as a response to heavy metal (lead) stress revealed by iTRAQ quantitative proteomics. Genet. Mol. Res. 2016, 15, 7254. [Google Scholar] [CrossRef]
- Campos, H.; Cooper, M.; Habben, J.; Edmeades, G.; Schussler, J. Improving drought tolerance in maize: A view from industry. Field Crops Res. 2004, 90, 19–34. [Google Scholar] [CrossRef]
- Min, H.; Chen, C.; Wei, S.; Shang, X.; Sun, M.; Xia, R.; Liu, X.; Hao, D.; Chen, H.; Xie, Q. Identification of drought tolerant mechanisms in maize seedlings based on transcriptome analysis of recombination inbred lines. Front. Plant Sci. 2016, 7, 1080. [Google Scholar] [CrossRef]
- Cui, D.; Wu, D.; Liu, J.; Li, D.; Xu, C.; Li, S.; Li, P.; Zhang, H.; Liu, X.; Jiang, C.; et al. Proteomic analysis of seedling roots of two maize inbred lines that differ significantly in the salt stress response. PLoS ONE 2015, 10, e0116697. [Google Scholar] [CrossRef]
- Nader, K.; Hoorn, J.W.; Hamdy, A.; Mastrorilli, M. Salinity effect on crop development and yield, analysis of salt tolerance according to several classification methods. Agric. Water Manag. 2003, 62, 37–66. [Google Scholar] [CrossRef]
- Gu, L.; Liu, Y.; Zong, X.; Liu, L.; Li, D.P.; Li, D.Q. Overexpression of maize mitogen-activated protein kinase gene, ZmSIMK1 in Arabidopsis increases tolerance to salt stress. Mol. Biol. Rep. 2010, 37, 4067–4073. [Google Scholar] [CrossRef]
- Wu, J.; Jiang, Y.; Liang, Y.; Chen, L.; Chen, W.; Cheng, B. Expression of the maize MYB transcription factor ZmMYB3R enhances drought and salt stress tolerance in transgenic plants. Plant Physiol. Biochem. 2019, 137, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cai, H.; Lu, M.; Wei, Q.; Xu, L.; Bo, C.; Ma, Q.; Zhao, Y.; Cheng, B. A maize stress-responsive Di19 transcription factor, ZmDi19-1, confers enhanced tolerance to salt in transgenic Arabidopsis. Plant Cell Rep. 2019, 38, 1563–1578. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.D.; Holzinger, E.R.; Li, R.; Pendergrass, S.A.; Kim, D. Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef]
- Maier, T.; Güell, M.; Serrano, L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009, 583, 3966–3973. [Google Scholar] [CrossRef] [Green Version]
- Muers, M. Gene expression: Transcriptome to proteome and back to genome. Nat. Rev. Genet. 2011, 12, 518. [Google Scholar] [CrossRef]
- Liu, J.-F.; Xing, J.; Fang, J.; Ai, P.; Cheng, Y. New insight into ovary abortion during ovary development of hazelnut through a combined proteomic and transcriptomic analysis. Sci. Hortic. 2018, 234, 36–48. [Google Scholar] [CrossRef]
- Jiang, C.; Bi, Y.; Mo, J.; Zhang, R.; Qu, M.; Feng, S.; Essemine, J. Proteome and transcriptome reveal the involvement of heat shock proteins and antioxidant system in thermotolerance of Clematis florida. Sci. Rep. 2020, 10, 8883. [Google Scholar] [CrossRef]
- Ye, X.; Wang, H.; Chen, P.; Fu, B.; Zhang, M.; Li, J.; Zheng, X.; Tan, B.; Feng, J. Combination of iTRAQ proteomics and RNA-seq transcriptomics reveals multiple levels of regulation in phytoplasma-infected Ziziphus jujuba Mill. Hortic. Res. 2017, 4, 17080. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Zhang, H.; Wang, T.; Chen, S.; Dai, S. Proteomics-based investigation of salt-responsive mechanisms in plant roots. J. Proteom. 2013, 82, 230–253. [Google Scholar] [CrossRef]
- Peng, Y.; Li, W.; Wang, K.; Wang, H. Effects of salt stress on seed germination and seedlings growth of salt-tolerant line and salt-sensitive line of maize. Acta Prataculturae Sin. 2012, 21, 62–71. [Google Scholar]
- Yun, S.H.; Park, G.W.; Kim, J.Y.; Kwon, S.O.; Choi, C.W.; Leem, S.H.; Kwon, K.H.; Yoo, J.S.; Lee, C.; Kim, S.; et al. Proteomic characterization of the Pseudomonas putida KT2440 global response to a monocyclic aromatic compound by iTRAQ analysis and 1DE-MudPIT. J. Proteom. 2011, 74, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.G.; Cheng, W.H.; Tian, W.G.; Li, Y.J.; Liu, F.; Xue, F.; Zhu, Q.H.; Sun, Y.Q.; Sun, J. iTRAQ-based comparative proteomic analysis provides insights into somatic embryogenesis in Gossypium hirsutum L. Plant Mol. Biol. 2018, 96, 89–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Chen, F.; Fang, P.; Peng, Y.; Zeng, W.; Zhao, X.; Ding, Y.; Zhuang, Z.; Gao, Q.; Ren, B. Comparative proteomics of salt-tolerant and salt-sensitive maize inbred lines to reveal the molecular mechanism of salt tolerance. Int. J. Mol. Sci. 2019, 20, 4725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Fang, P.; Zeng, W.; Ding, Y.; Zhuang, Z.; Peng, Y. Comparing transcriptome expression profiles to reveal the mechanisms of salt tolerance and exogenous glycine betaine mitigation in maize seedlings. PLoS ONE 2020, 15, e0233616. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Lan, P.; Li, W.; Schmidt, W. Complementary proteome and transcriptome profiling in phosphate-deficient Arabidopsis roots reveals multiple levels of gene regulation. Mol. Cell. Proteom. 2012, 11, 1156–1166. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Kitano, H. Systems biology: A brief overview. Science 2002, 295, 1662–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Xu, Z.; Zhang, Y.; Chai, L.; Yi, H.; Deng, X. An integrative analysis of the transcriptome and proteome of the pulp of a spontaneous late-ripening sweet orange mutant and its wild type improves our understanding of fruit ripening in citrus. J. Exp. Bot. 2014, 65, 1651–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Zheng, G.; Pei, C.; Zhang, Z. Correlation Analysis on proteomic and transcriptomic of salt-response in leaves of southern type alfalfa. J. Nucl. Agric. Sci. 2016, 30, 1706–1715. [Google Scholar]
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Karas, M.; Hillenkamp, F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal. Chem. 1988, 60, 2299–2301. [Google Scholar] [CrossRef]
- Betz, G.A.; Knappe, C.; Lapierre, C.; Olbrich, M.; Welzl, G.; Langebartels, C.; Heller, W.; Sandermann, H.; Ernst, D. Ozone affects shikimate pathway transcripts and monomeric lignin composition in European beech (Fagus sylvatica L.). Eur. J. For. Res. 2009, 128, 109–116. [Google Scholar] [CrossRef]
- Tronchet, M.; Balagué, C.; Kroj, T.; Jouanin, L.; Roby, D. Cinnamyl alcohol dehydrogenases-C and D, key enzymes in lignin biosynthesis, play an essential role in disease resistance in Arabidopsis. Mol. Plant Pathol. 2010, 11, 83–92. [Google Scholar] [CrossRef]
- Zhao, Q.; Dixon, R.A. Transcriptional networks for lignin biosynthesis: More complex than we thought? Trends Plant Sci. 2011, 16, 227–233. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Aguayo, I.; Rodríguez-Galán, J.M.; García, R.; Torreblanca, J.; Pardo, J.M. Salt stress enhances xylem development and expression of S-adenosyl-L-methionine synthase in lignifying tissues of tomato plants. Planta 2004, 220, 278–285. [Google Scholar] [CrossRef]
- Bi, C.; Chen, F.; Jackson, L.; Gill, B.; Li, W. Expression of lignin biosynthetic genes in wheat during development and upon infection by fungal pathogens. Plant Mol. Biol. Rep. 2011, 29, 149–161. [Google Scholar] [CrossRef]
- Kaur, H.; Shaker, K.; Heinzel, N.; Ralph, J.; Gális, I.; Baldwin, I.T. Environmental stresses of field growth allow cinnamyl alcohol dehydrogenase-deficient Nicotiana attenuata plants to compensate for their structural deficiencies. Plant Physiol. 2012, 159, 1545–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sibout, R.; Eudes, A.; Pollet, B.; Goujon, T.; Mila, I.; Granier, F.; Séguin, A.; Lapierre, C.; Jouanin, L. Expression pattern of two paralogs encoding cinnamyl alcohol dehydrogenases in Arabidopsis. Isolation and characterization of the corresponding mutants. Plant Physiol. 2003, 132, 848–860. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.P.; Zhong, N.Q.; Qu, Z.L.; Wang, F.; Liu, N.; Xia, G.X. Root and vascular tissue-specific expression of glycine-rich protein AtGRP9 and its interaction with AtCAD5, a cinnamyl alcohol dehydrogenase, in Arabidopsis thaliana. J. Plant Res. 2007, 120, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.C.; Farese, R.V., Jr. Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 2012, 81, 687–714. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Lee, S.B.; Kim, H.J.; Min, M.K.; Hwang, I.; Suh, M.C. Characterization of glycosylphosphatidylinositol-anchored lipid transfer protein 2 (LTPG2) and overlapping function between LTPG/LTPG1 and LTPG2 in cuticular wax export or accumulation in Arabidopsis thaliana. Plant Cell Physiol. 2012, 53, 1391–1403. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Ge, X.; Ma, H. The rice OsDIL gene plays a role in drought tolerance at vegetative and reproductive stages. Plant Mol. Biol. 2013, 82, 239–253. [Google Scholar] [CrossRef]
- Yu, K.; Soares, J.M.; Mandal, M.K.; Wang, C.; Chanda, B.; Gifford, A.N.; Fowler, J.S.; Navarre, D.; Kachroo, A.; Kachroo, P. A feedback regulatory loop between G3P and lipid transfer proteins DIR1 and AZI1 mediates azelaic-acid-induced systemic immunity. Cell Rep. 2013, 3, 1266–1278. [Google Scholar] [CrossRef] [Green Version]
- Pitzschke, A.; Datta, S.; Persak, H. Salt stress in Arabidopsis: Lipid transfer protein AZI1 and its control by mitogen-activated protein kinase MPK3. Mol. Plant 2014, 7, 722–738. [Google Scholar] [CrossRef] [Green Version]
- Torres-Schumann, S.; Godoy, J.A.; Pintor-Toro, J.A. A probable lipid transfer protein gene is induced by NaCl in stems of tomato plants. Plant Mol. Biol. 1992, 18, 749–757. [Google Scholar] [CrossRef]
- Pan, Y.; Li, J.; Jiao, L.; Li, C.; Zhu, D.; Yu, J. A non-specific setaria italica lipid transfer protein gene plays a critical role under abiotic stress. Front. Plant Sci. 2016, 7, 1752. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; He, X.; Wenmin, Q.; Lu, Z.; Zhang, Y.; Chen, S.; Liu, M.; Qiao, G.; Renying, Z. Pathogenesis-related protein PR10 from Salix matsudana Koidz exhibits resistance to salt stress in transgenic Arabidopsis thaliana. Environ. Exp. Bot. 2017, 141, 74–82. [Google Scholar] [CrossRef]
- Jain, S.; Kumar, D.; Jain, M.; Chaudhary, P.; Deswal, R.; Sarin, N.B. Ectopic overexpression of a salt stress-induced pathogenesis-related class 10protein (PR10) gene from peanut (Arachis hypogaea L.) affords broad spectrum abiotic stress tolerance in transgenic tobacco. Plant Cell Tissue Organ Cult. 2012, 109, 19–31. [Google Scholar] [CrossRef]
- Seo, P.J.; Lee, A.K.; Xiang, F.; Park, C.M. Molecular and functional profiling of Arabidopsis pathogenesis-related genes: Insights into their roles in salt response of seed germination. Plant Cell Physiol. 2008, 49, 334–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weretilnyk, E.A.; Alexander, K.J.; Drebenstedt, M.; Snider, J.D.; Summers, P.S.; Moffatt, B.A. Maintaining methylation activities during salt stress. The involvement of adenosine kinase. Plant Physiol. 2001, 125, 856–865. [Google Scholar] [CrossRef] [Green Version]
- Lange, P.R.; Geserick, C.; Tischendorf, G.; Zrenner, R. Functions of chloroplastic adenylate kinases in Arabidopsis. Plant Physiol. 2008, 146, 492–504. [Google Scholar] [CrossRef] [Green Version]
- Dunwell, J.; Gibbings, G.; Mahmood, T.; Naqvi, S.M.S. Germin and germin-like proteins: Evolution, structure, and function. Crit. Rev. Plant Sci. 2008, 27, 342–375. [Google Scholar] [CrossRef]
- Barman, A.R.; Banerjee, J. Versatility of germin-like proteins in their sequences, expressions, and functions. Funct. Integr. Genom. 2015, 15, 533–548. [Google Scholar] [CrossRef]
- Banerjee, J.; Maiti, M.K. Functional role of rice germin-like protein1 in regulation of plant height and disease resistance. Biochem. Biophys. Res. Commun. 2010, 394, 178–183. [Google Scholar] [CrossRef]
- Banerjee, J.; Das, N.; Dey, P.; Maiti, M.K. Transgenically expressed rice germin-like protein1 in tobacco causes hyper-accumulation of H2O2 and reinforcement of the cell wall components. Biochem. Biophys. Res. Commun. 2010, 402, 637–643. [Google Scholar] [CrossRef]
- Banerjee, J.; Gantait, S.; Maiti, M.K. Physiological role of rice germin-like protein 1 (OsGLP1) at early stages of growth and development in indica rice cultivar under salt stress condition. Plant Cell Tissue Organ Cult. 2017, 131, 127–137. [Google Scholar] [CrossRef]
- Sandalio, L.M.; Rodríguez-Serrano, M.; Romero-Puertas, M.C.; del Río, L.A. Role of peroxisomes as a source of reactive oxygen species (ROS) signaling molecules. Sub-Cell. Biochem. 2013, 69, 231–255. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, H.; Sun, L.; Jiao, Y.; Zhang, G.; Miao, C.; Hao, F. NADPH oxidase AtrbohD and AtrbohF function in ROS-dependent regulation of Na⁺/K⁺ homeostasis in Arabidopsis under salt stress. J. Exp. Bot. 2012, 63, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Zenda, T.; Liu, S.; Wang, X.; Jin, H.; Liu, G.; Duan, H. Comparative proteomic and physiological analyses of two divergent maize inbred lines provide more insights into drought-stress tolerance mechanisms. Int. J. Mol. Sci. 2018, 19, 3225. [Google Scholar] [CrossRef] [Green Version]
- Azevedo Neto, A.; Prisco, J.T.; Enéas-Filho, J.; Abreu, C.; Gomes-Filho, E. Effect of salt stress on antioxidative enzymes and lipid peroxidation in leaves and roots of salt-tolerant and salt-sensitive maize genotypes. Environ. Exp. Bot. 2006, 56, 87–94. [Google Scholar] [CrossRef]
- Qi, Y.C.; Liu, W.Q.; Qiu, L.Y.; Zhang, S.M.; Ma, L.; Zhang, H. Overexpression of glutathione S-transferase gene increases salt tolerance of Arabidopsis. Russ. J. Plant Physiol. 2010, 57, 233–240. [Google Scholar] [CrossRef]
- Fernández-Pérez, F.; Pomar, F.; Pedreño, M.A.; Novo-Uzal, E. Suppression of Arabidopsis peroxidase 72 alters cell wall and phenylpropanoid metabolism. Plant Sci. Int. J. Exp. Plant Biol. 2015, 239, 192–199. [Google Scholar] [CrossRef]
- Berthet, S.; Demont-Caulet, N.; Pollet, B.; Bidzinski, P.; Cézard, L.; Le Bris, P.; Borrega, N.; Hervé, J.; Blondet, E.; Balzergue, S.; et al. Disruption of LACCASE4 and 17 results in tissue-specific alterations to lignification of Arabidopsis thaliana stems. Plant Cell 2011, 23, 1124–1137. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Nakashima, J.; Chen, F.; Yin, Y.; Fu, C.; Yun, J.; Shao, H.; Wang, X.; Wang, Z.Y.; Dixon, R.A. Laccase is necessary and nonredundant with peroxidase for lignin polymerization during vascular development in Arabidopsis. Plant Cell 2013, 25, 3976–3987. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparable Group | Gene ID | Protein ID | DEGs-FC | DEPs-FC | Gene Description |
|---|---|---|---|---|---|
| Line 8723S vs. line 8723CK | 100272868 | B4FR97 | 2.11 | 1.24 | Putative cinnamyl alcohol dehydrogenase 6 |
| 103653695 | A0A1D6Q3Y3 | 0.01 | 1.22 | BAF250_C domain-containing protein | |
| 103626441 | A0A1D6GW54 | 0.37 | 1.27 | Lipid transfer-like protein VAS | |
| 103634525 | A0A1D6JZU3 | 3.53 | 1.37 | Pathogenesis-related protein 1 | |
| 100502492 | A0A1D6G901 | 2.02 | 0.77 | Putative laccase family protein | |
| 103642203 | A0A1D6JI74 | 0.48 | 0.56 | GDSL esterase/lipase CPRD49 | |
| 103653801 | K7UH13 | 2.24 | 1.34 | Adenosine kinase 2 | |
| 542629 | Q9FQD0 | 2.71 | 1.22 | Glutathione transferase 9 | |
| 100194034 | B4FH68 | 2.71 | 1.21 | Peroxidase 12 | |
| 100286113 | B6UEL1 | 2.11 | 0.69 | Germin-like protein subfamily 1 member 11 | |
| 103629444 | A0A1D6LKL1 | 0.45 | 1.30 | Zinc finger family protein | |
| 100281608 | B6SWX3 | 2.58 | 1.28 | Anthocyanidin 3-O-glucosyltransferase | |
| 100216875 | B4FK56 | 0.27 | 0.82 | Peroxidase 72 | |
| 103627158 | A0A1D6H878 | 2.40 | 0.76 | DUF679 domain membrane protein 7 | |
| P138S vs. P138CK | 103648046 | A0A1D6F6D8 | 0.45 | 1.21 | UPF0481 protein At3g47200 |
| 100193724 | A0A1D6E1G8 | 0.16 | 0.77 | Tyrosine kinase family protein | |
| 103645931 | A0A1D6DZ88 | 2.36 | 1.39 | LRR receptor-like serine/threonine-protein kinase FLS2 | |
| 103654805 | K7U6R9 | 0.34 | 1.21 | Allene oxide synthase 3 | |
| 100193942 | B4FGU5 | 0.47 | 1.22 | Putidaredoxin reductase homolog1 | |
| 100273295 | B4FUC4 | 0.46 | 0.77 | Plasma membrane-associated cation-binding protein 1 | |
| 100283521 | B6TJ40 | 0.49 | 1.21 | Lipid transfer protein | |
| 100283518 | B6TJ32 | 0.37 | 0.82 | Uncharacterized LOC100283518 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, F.; Ji, X.; Zhuang, Z.; Peng, Y. Integrated Transcriptome and Proteome Analyses of Maize Inbred lines in Response to Salt Stress. Agronomy 2022, 12, 1053. https://doi.org/10.3390/agronomy12051053
Chen F, Ji X, Zhuang Z, Peng Y. Integrated Transcriptome and Proteome Analyses of Maize Inbred lines in Response to Salt Stress. Agronomy. 2022; 12(5):1053. https://doi.org/10.3390/agronomy12051053
Chicago/Turabian StyleChen, Fenqi, Xiangzhuo Ji, Zelong Zhuang, and Yunling Peng. 2022. "Integrated Transcriptome and Proteome Analyses of Maize Inbred lines in Response to Salt Stress" Agronomy 12, no. 5: 1053. https://doi.org/10.3390/agronomy12051053
APA StyleChen, F., Ji, X., Zhuang, Z., & Peng, Y. (2022). Integrated Transcriptome and Proteome Analyses of Maize Inbred lines in Response to Salt Stress. Agronomy, 12(5), 1053. https://doi.org/10.3390/agronomy12051053