
Diversity Chromosome Evolution of Ty1-copia Retrotransposons in Pennisetum purpureum Revealed by FISH


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and DNA Isolation
2.2. PCR Products and Characterization of Ty1-copia RT Sequences
2.3. Sequence Analysis, Comparisons, and Phylogenetic Trees
2.4. Dot-Blot Hybridization
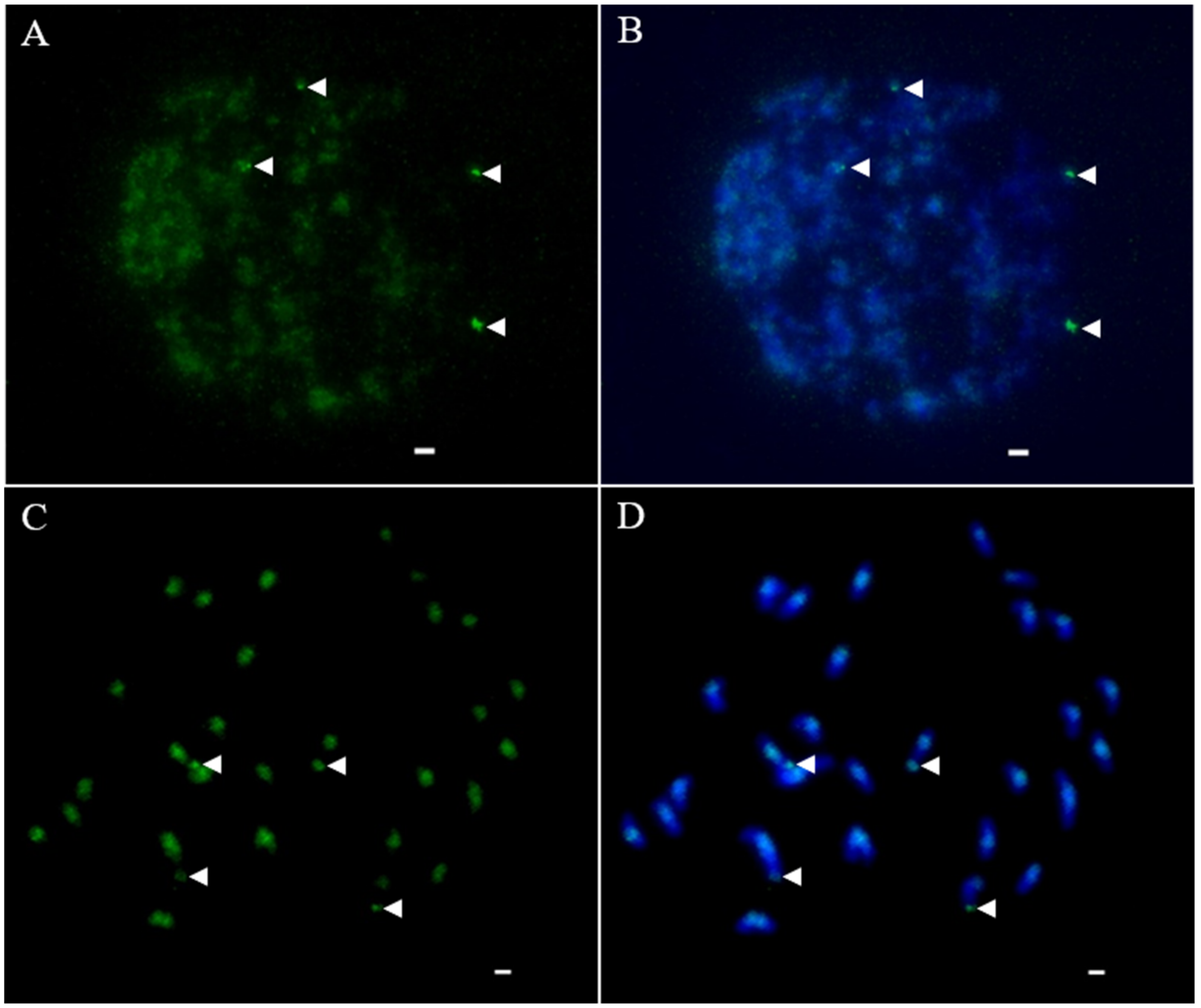
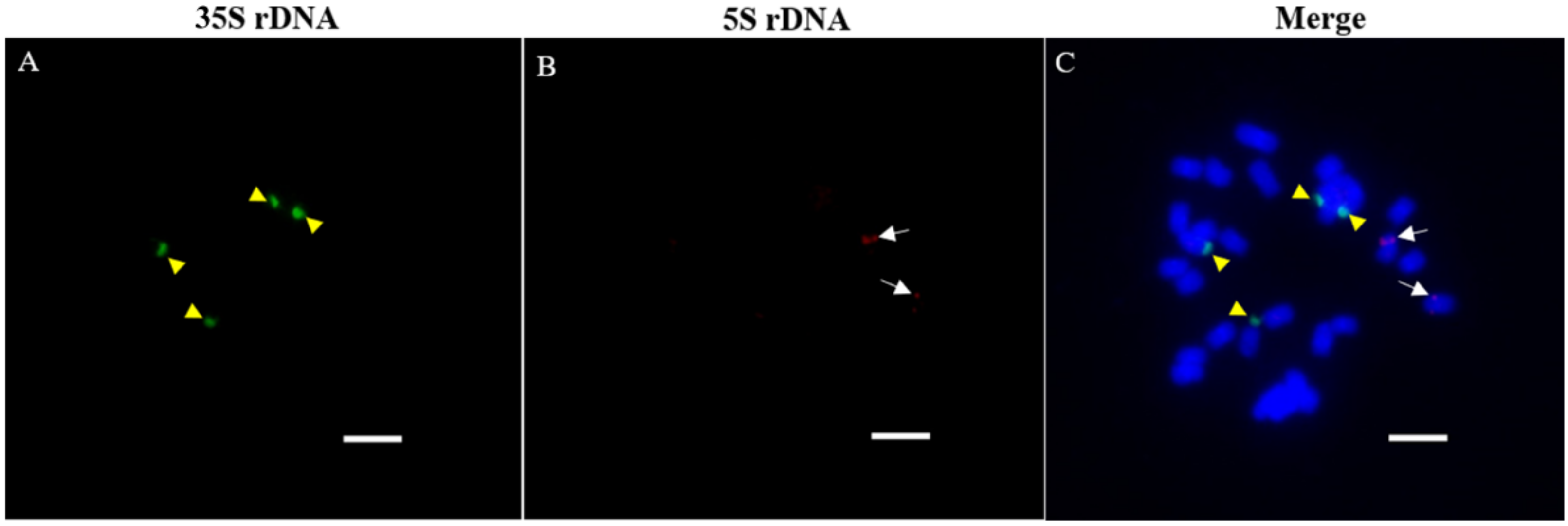
2.5. FISH
3. Results
3.1. PCR Products and Characterization of Ty1-copia RT Sequences
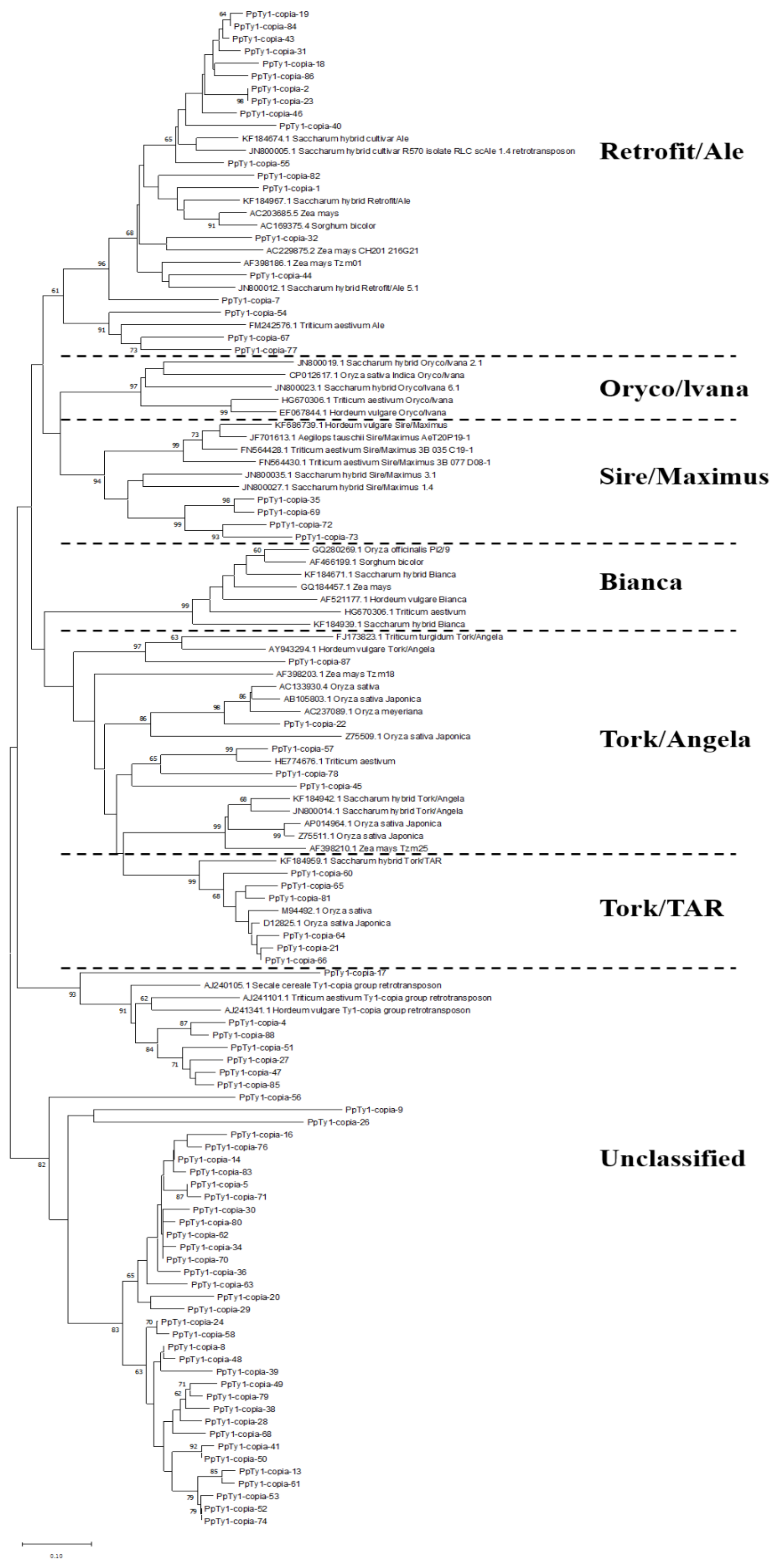
3.2. Classification and Phylogenetic Analysis of Identified Ty1-copia RT Sequences from P. purpureum
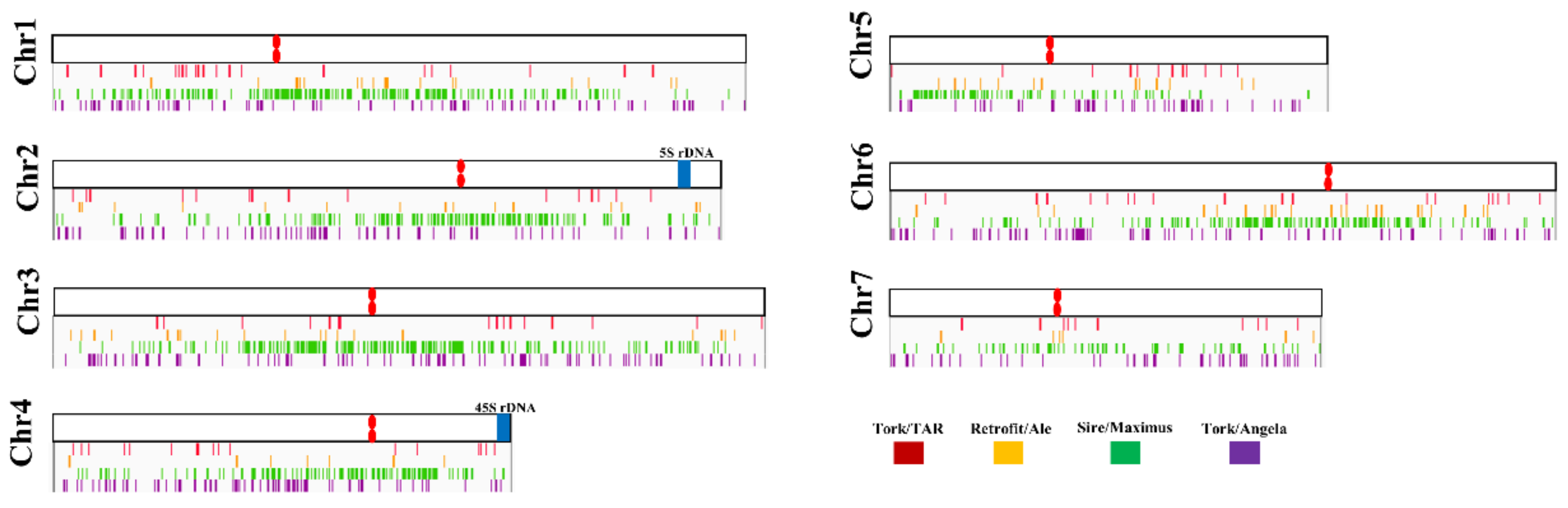
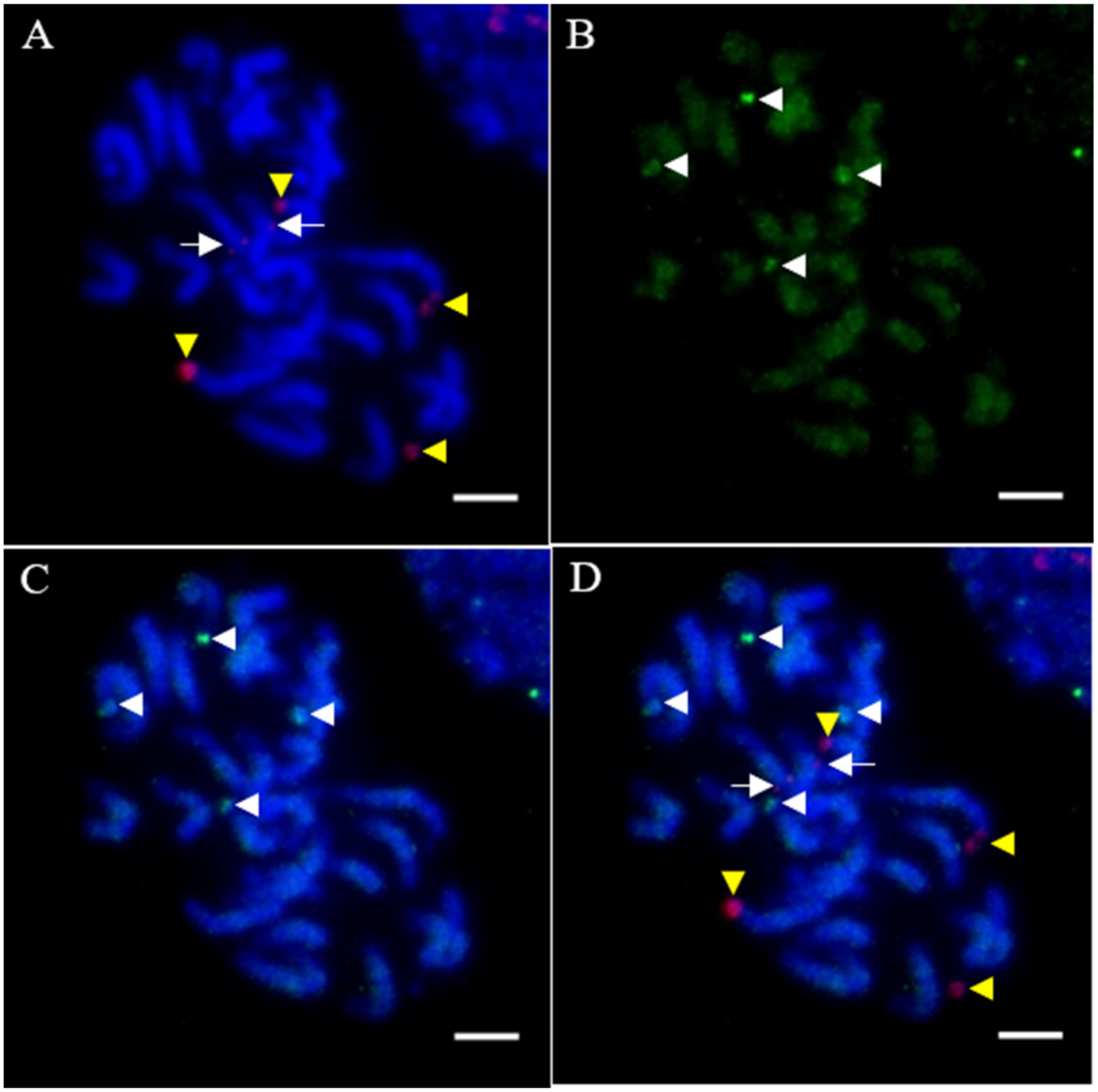
3.3. Relative Abundance in P. purpureum Genome and Chromosomal Patterns of RTs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Menegol, D.; Scholl, A.L.; Fontana, R.C.; Dillon, A.J.P.; Camassola, M. Potential of a Penicillium echinulatum enzymatic complex produced in either submerged or solid-state cultures for enzymatic hydrolysis of elephant grass. Fuel 2014, 133, 232–240. [Google Scholar] [CrossRef]
- Han, Y.; Zhang, T.; Thammapichai, P.; Weng, Y.; Jiang, J. Chromosome-specific painting in Cucumis species using bulked oligonucleotides. Genetics 2015, 200, 771–779. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.-M.; Zhang, X.-Q.; Dong, Z.-X.; Guo, H.-R. Dynamic changes of lignin contents of MT-1 elephant grass and its closely related cultivars. Biomass Bioenergy 2011, 35, 1732–1738. [Google Scholar] [CrossRef]
- Li, H.-Q.; Li, C.-L.; Sang, T.; Xu, J. Pretreatment on Miscanthus lutarioriparious by liquid hot water for efficient ethanol production. Biotechnol. Biofuels 2013, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.-G.; Long, S.P.; Ort, D.R. What is the maximum efficiency with which photosynthesis can convert solar energy into biomass? Curr. Opin. Biotechnol. 2008, 19, 153–159. [Google Scholar] [CrossRef]
- Somerville, C.; Youngs, H.; Taylor, C.; Davis, S.C.; Long, S.P. Feedstocks for lignocellulosic biofuels. Science 2010, 329, 790–792. [Google Scholar] [CrossRef] [Green Version]
- Woodard, K.; Prine, G. Dry matter accumulation of elephantgrass, energycane, and elephantmillet in a subtropical climate. Crop Sci. 1993, 33, 818–824. [Google Scholar] [CrossRef]
- Martel, E.; De Nay, D.; Siljak-Yakoviev, S.; Brown, S.; Sarr, A. Genome size variation and basic chromosome number in pearl millet and fourteen related Pennisetum species. J. Hered. 1997, 88, 139–143. [Google Scholar] [CrossRef]
- Vicient, C.M.; Jaaskelainen, M.J.; Kalendar, R.; Schulman, A.H. Active retrotransposons are a common feature of grass genomes. Plant Physiol. 2001, 125, 1283–1292. [Google Scholar] [CrossRef] [Green Version]
- Hanson, R.E.; Islam-Faridi, M.N.; Crane, C.F.; Zwick, M.S.; Czeschin, D.G.; Wendel, J.F.; McKnight, T.D.; Price, H.J.; Stelly, D.M. Ty1-copia-retrotransposon behavior in a polyploid cotton. Chromosome Res. 2000, 8, 73–76. [Google Scholar] [CrossRef]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Kawahara, Y.; de la Bastide, M.; Hamilton, J.P.; Kanamori, H.; McCombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 2013, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef] [Green Version]
- Paterson, A.H.; Bowers, J.E.; Bruggmann, R.; Dubchak, I.; Grimwood, J.; Gundlach, H.; Haberer, G.; Hellsten, U.; Mitros, T.; Poliakov, A. The Sorghum bicolor genome and the diversification of grasses. Nature 2009, 457, 551–556. [Google Scholar] [CrossRef] [Green Version]
- Ammiraju, J.S.; Zuccolo, A.; Yu, Y.; Song, X.; Piegu, B.; Chevalier, F.; Walling, J.G.; Ma, J.; Talag, J.; Brar, D.S. Evolutionary dynamics of an ancient retrotransposon family provides insights into evolution of genome size in the genus Oryza. Plant J. 2007, 52, 342–351. [Google Scholar] [CrossRef]
- Cheng, Z.; Dong, F.; Langdon, T.; Ouyang, S.; Buell, C.R.; Gu, M.; Blattner, F.R.; Jiang, J. Functional rice centromeres are marked by a satellite repeat and a centromere-specific retrotransposon. Plant Cell 2002, 14, 1691–1704. [Google Scholar] [CrossRef] [Green Version]
- Devos, K.M.; Brown, J.K.; Bennetzen, J.L. Genome size reduction through illegitimate recombination counteracts genome expansion in Arabidopsis. Genome Res. 2002, 12, 1075–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, D.; Gill, N.; Kim, H.R.; Walling, J.G.; Zhang, W.; Fan, C.; Yu, Y.; Ma, J.; SanMiguel, P.; Jiang, N. A lineage-specific centromere retrotransposon in Oryza brachyantha. Plant J. 2009, 60, 820–831. [Google Scholar] [CrossRef]
- Jiang, N.; Bao, Z.; Temnykh, S.; Cheng, Z.; Jiang, J.; Wing, R.A.; McCouch, S.R.; Wessler, S.R. Dasheng: A recently amplified nonautonomous long terminal repeat element that is a major component of pericentromeric regions in rice. Genetics 2002, 161, 1293–1305. [Google Scholar] [CrossRef]
- Xu, Y.; Du, J. Young but not relatively old retrotransposons are preferentially located in gene-rich euchromatic regions in tomato (Solanum lycopersicum) plants. Plant J. 2014, 80, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Bennetzen, J.L. Retrotransposons: Central players in the structure, evolution and function of plant genomes. Trends Plant Sci. 2000, 5, 509–510. [Google Scholar] [CrossRef]
- De la Chaux, N.; Wagner, A. BEL/Pao retrotransposons in metazoan genomes. BMC Evol. Biol. 2011, 11, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flavell, A. Ty1-copia group retrotransposons and the evolution of retroelements in the eukaryotes. In Transposable Elements and Evolution; Springer: Dordrecht, The Netherlands, 1993; pp. 258–274. [Google Scholar]
- Goodwin, T.; Poulter, R. A group of deuterostome Ty3/gypsy-like retrotransposons with Ty1/copia-like pol-domain orders. Mol. Genet. Genom. 2002, 267, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Kolano, B.; Bednara, E.; Weiss-Schneeweiss, H. Isolation and characterization of reverse transcriptase fragments of LTR retrotransposons from the genome of Chenopodium quinoa (Amaranthaceae). Plant Cell Rep. 2013, 32, 1575–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Sun, H.; Zhao, G.; Dai, H.; Gao, X.; Li, H.; Zhang, Z. Isolation and characterization of genomic retrotransposon sequences from octoploid strawberry (Fragaria × ananassa Duch.). Plant Cell Rep. 2008, 27, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Pereira, V. Insertion bias and purifying selection of retrotransposons in the Arabidopsis thaliana genome. Genome Biol. 2004, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Santini, S.; Cavallini, A.; Natali, L.; Minelli, S.; Maggini, F.; Cionini, P. Ty1/copia-and Ty3/gypsy-like DNA sequences in Helianthus species. Chromosoma 2002, 111, 192–200. [Google Scholar] [CrossRef]
- Abd El-Twab, M.H.; Kondo, K. Physical mapping of 45S rDNA loci by fluorescent in situ hybridization and Evolution among polyploid Dendranthema species. Chromosome Sci. 2003, 7, 71–76. [Google Scholar]
- Wu, J.; Huang, Y.; Lin, Y.; Fu, C.; Liu, S.; Deng, Z.; Li, Q.; Huang, Z.; Chen, R.; Zhang, M. Unexpected inheritance pattern of Erianthus arundinaceus chromosomes in the intergeneric progeny between Saccharum spp. and Erianthus arundinaceus. PLoS ONE 2014, 9, e110390. [Google Scholar] [CrossRef] [Green Version]
- D’Hont, A.; Ison, D.; Alix, K.; Roux, C.; Glaszmann, J.C. Determination of basic chromosome numbers in the genus Saccharum by physical mapping of ribosomal RNA genes. Genome 1998, 41, 221–225. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, X.; Tian, L.; Chen, L.; Yu, W. Identification of peanut (Arachis hypogaea) chromosomes using a fluorescence in situ hybridization system reveals multiple hybridization events during tetraploid peanut formation. New Phytol. 2016, 211, 1424–1439. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.N., Jr.; Via, L.E. A rapid CTAB DNA isolation technique useful for RAPD fingerprinting and other PCR applications. BioTechniques 1993, 14, 748–750. [Google Scholar] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547. [Google Scholar] [CrossRef] [PubMed]
- Ranwez, V.; Harispe, S.; Delsuc, F.; Douzery, E.J. MACSE: Multiple Alignment of Coding SEquences accounting for frameshifts and stop codons. PLoS ONE 2011, 6, e22594. [Google Scholar] [CrossRef]
- Santos, G.; Maia, I.M.; Araújo, F.F.; Pacheco, A.; Medeiros, S.; Gadelha, C.R.; Silva, S.C.; Araujo-Filho, R.; Oliveira, D. HiperMUSCLE: An Intuitive Graphical User Interface for the Multiple Sequence Alignment Program MUSCLE (Edgar, 2004); 2008; pp. 818–824. [Google Scholar]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Luo, L.; Hu, X.; Yu, F.; Yang, Y.; Deng, Z.; Wu, J.; Chen, R.; Zhang, M. Characterization, genomic organization, abundance, and chromosomal distribution of Ty1-copia retrotransposons in Erianthus arundinaceus. Front. Plant Sci. 2017, 8, 924. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Dos Reis, G.B.; Mesquita, A.T.; Torres, G.A.; Andrade-Vieira, L.F.; Vander Pereira, A.; Davide, L.C. Genomic homeology between Pennisetum purpureum and Pennisetum glaucum (Poaceae). Comp. Cytogenet. 2014, 8, 199. [Google Scholar]
- Hawkins, J.S.; Kim, H.; Nason, J.D.; Wing, R.A.; Wendel, J.F. Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Res. 2006, 16, 1252–1261. [Google Scholar] [CrossRef] [Green Version]
- Kidwell, M.G.; Lisch, D. Transposable elements as sources of variation in animals and plants. Proc. Natl. Acad. Sci. USA 1997, 94, 7704–7711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisch, D. How important are transposons for plant evolution? Nat. Rev. Genet. 2013, 14, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; SanMiguel, P.; Lai, J.; Messing, J.; Bennetzen, J.L. DNA rearrangement in orthologous orp regions of the maize, rice and sorghum genomes. Genetics 2005, 170, 1209–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zedek, F.; Šmerda, J.; Šmarda, P.; Bureš, P. Correlated evolution of LTR retrotransposons and genome size in the genus Eleocharis. BMC Plant Biol. 2010, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Alipour, A.; Tsuchimoto, S.; Sakai, H.; Ohmido, N.; Fukui, K. Structural characterization of copia-type retrotransposons leads to insights into the marker development in a biofuel crop, Jatropha curcas L. Biotechnol. Biofuels 2013, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Pearce, S.R.; Pich, U.; Harrison, G.; Flavell, A.J.; Heslop-Harrison, J.; Schubert, I.; Kumar, A. TheTy1-copia group retrotransposons ofAllium cepa are distributed throughout the chromosomes but are enriched in the terminal heterochromatin. Chromosome Res. 1996, 4, 357–364. [Google Scholar] [CrossRef]
- McCarthy, E.M.; Liu, J.; Lizhi, G.; McDonald, J.F. Long terminal repeat retrotransposons of Oryza sativa. Genome Biol. 2002, 3, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kato, A.; Lamb, J.C.; Birchler, J.A. Chromosome painting using repetitive DNA sequences as probes for somatic chromosome identification in maize. Proc. Natl. Acad. Sci. USA 2004, 101, 13554–13559. [Google Scholar] [CrossRef] [Green Version]
- Malik, H.S.; Henikoff, S. Major evolutionary transitions in centromere complexity. Cell 2009, 138, 1067–1082. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Liu, N.; Peng, K.; Zhang, Q. The distribution and copy number of copia-like retrotransposons in rice (Oryza sativa L.) and their implications in the organization and evolution of the rice genome. Proc. Natl. Acad. Sci. USA 1999, 96, 6824–6828. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clones Number | Amino Acid Similarity | Stop Coding or Frameshift Mutations | Clones Number | Amino Acid Similarity | Stop Coding or Frameshift Mutations |
|---|---|---|---|---|---|
| 1 | N/A | - | 43 | N/A | - |
| 2 | N/A | - | 44 | N/A | - |
| 3, 64 | 100% | - | 45 | N/A | Frameshift mutations |
| 4 | N/A | Stop coding | 46 | N/A | - |
| 5 | N/A | - | 49 | N/A | - |
| 6, 24 | 100% | Stop coding | 51 | N/A | - |
| 7 | N/A | Stop coding | 52 | N/A | - |
| 8 | N/A | 53 | N/A | - | |
| 9 | N/A | Frameshift mutations | 54 | N/A | - |
| 10, 30 | 100% | - | 55 | N/A | Frameshift mutations |
| 11, 33, 59, 62 | 100% | - | 56 | N/A | Stop coding |
| 12, 14 | 100% | - | 57 | N/A | - |
| 13 | N/A | - | 58 | N/A | - |
| 15,47 | 100% | - | 60 | N/A | Frameshift mutations |
| 16 | N/A | Stop coding | 61 | N/A | - |
| 17 | N/A | Stop coding | 63 | N/A | - |
| 18 | N/A | - | 65 | N/A | Stop coding |
| 19 | N/A | - | 66 | N/A | Stop coding |
| 20 | N/A | Frameshift mutations | 67 | N/A | Frameshift mutations |
| 21 | N/A | - | 68 | N/A | - |
| 22 | N/A | - | 69 | N/A | Frameshift mutations |
| 23 | N/A | - | 71 | N/A | - |
| 24 | N/A | Stop coding | 72 | N/A | Frameshift mutations |
| 25, 48 | 100% | - | 73 | N/A | Frameshift mutations |
| 26 | N/A | Stop coding | 74 | N/A | - |
| 27 | N/A | - | 75, 85 | N/A | - |
| 28 | N/A | - | 76 | N/A | - |
| 29 | N/A | Frameshift mutations | 77 | N/A | - |
| 31 | N/A | Stop coding | 78 | N/A | - |
| 32 | N/A | - | 79 | N/A | - |
| 34 | N/A | - | 80 | N/A | - |
| 35 | N/A | Frameshift mutations | 81 | N/A | - |
| 36 | N/A | - | 82 | N/A | - |
| 37,50 | 100% | - | 83 | N/A | - |
| 38 | N/A | Frameshift mutations | 84 | N/A | - |
| 39 | N/A | - | 86 | N/A | - |
| 40 | N/A | Frameshift mutations | 87 | N/A | - |
| 41 | N/A | - | 88 | N/A | - |
| 42, 70 | N/A | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Z.; Huang, Y.; Wu, J.; Zhang, M.; Deng, Z. Diversity Chromosome Evolution of Ty1-copia Retrotransposons in Pennisetum purpureum Revealed by FISH. Agronomy 2022, 12, 1312. https://doi.org/10.3390/agronomy12061312
Yu Z, Huang Y, Wu J, Zhang M, Deng Z. Diversity Chromosome Evolution of Ty1-copia Retrotransposons in Pennisetum purpureum Revealed by FISH. Agronomy. 2022; 12(6):1312. https://doi.org/10.3390/agronomy12061312
Chicago/Turabian StyleYu, Zehuai, Yongji Huang, Jiayun Wu, Muqing Zhang, and Zuhu Deng. 2022. "Diversity Chromosome Evolution of Ty1-copia Retrotransposons in Pennisetum purpureum Revealed by FISH" Agronomy 12, no. 6: 1312. https://doi.org/10.3390/agronomy12061312
APA StyleYu, Z., Huang, Y., Wu, J., Zhang, M., & Deng, Z. (2022). Diversity Chromosome Evolution of Ty1-copia Retrotransposons in Pennisetum purpureum Revealed by FISH. Agronomy, 12(6), 1312. https://doi.org/10.3390/agronomy12061312