
DNA Methylation and RNA-Sequencing Analysis to Identify Genes Related to Spontaneous Leaf Spots in a Wheat Variety ‘Zhongkenuomai No.1’

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Transcriptome Sequencing and Data Analysis
2.3. Differentially Expressed Gene Analysis
2.4. Gene Functional Annotation
2.5. Gene Set Enrichment Analysis
2.6. Protein–Protein Interaction (PPI) Network Construction and Modules Selection
2.7. MethylRAD Sequencing
2.8. MethylRAD Data Analysis
2.9. GO, KEGG Enrichment, and Integrated Analysis
2.10. qPCR Analysis
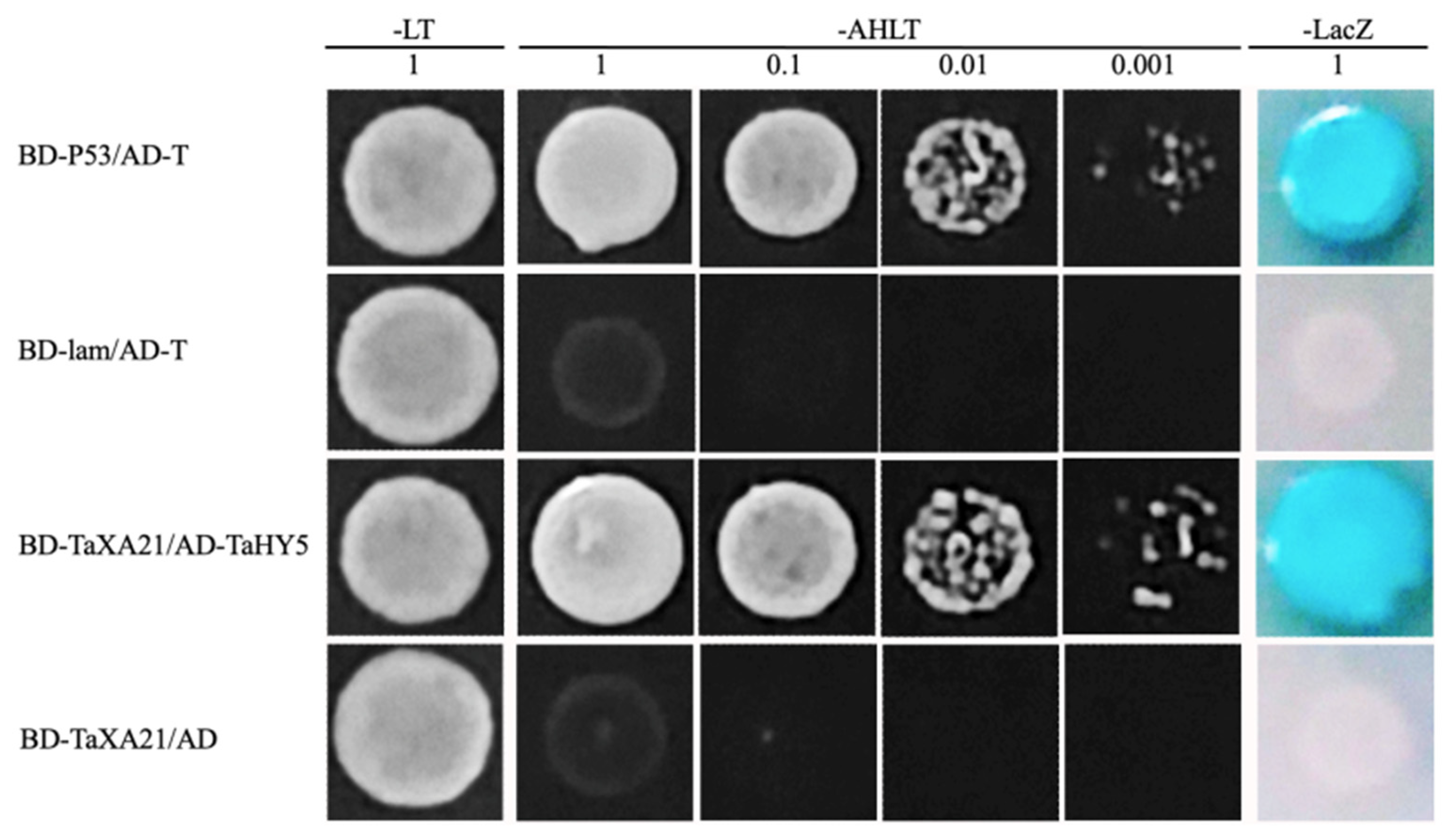
2.11. Yeast Two-Hybrid (Y2H)
3. Results
3.1. Transcriptome Assembly and Functional Annotation
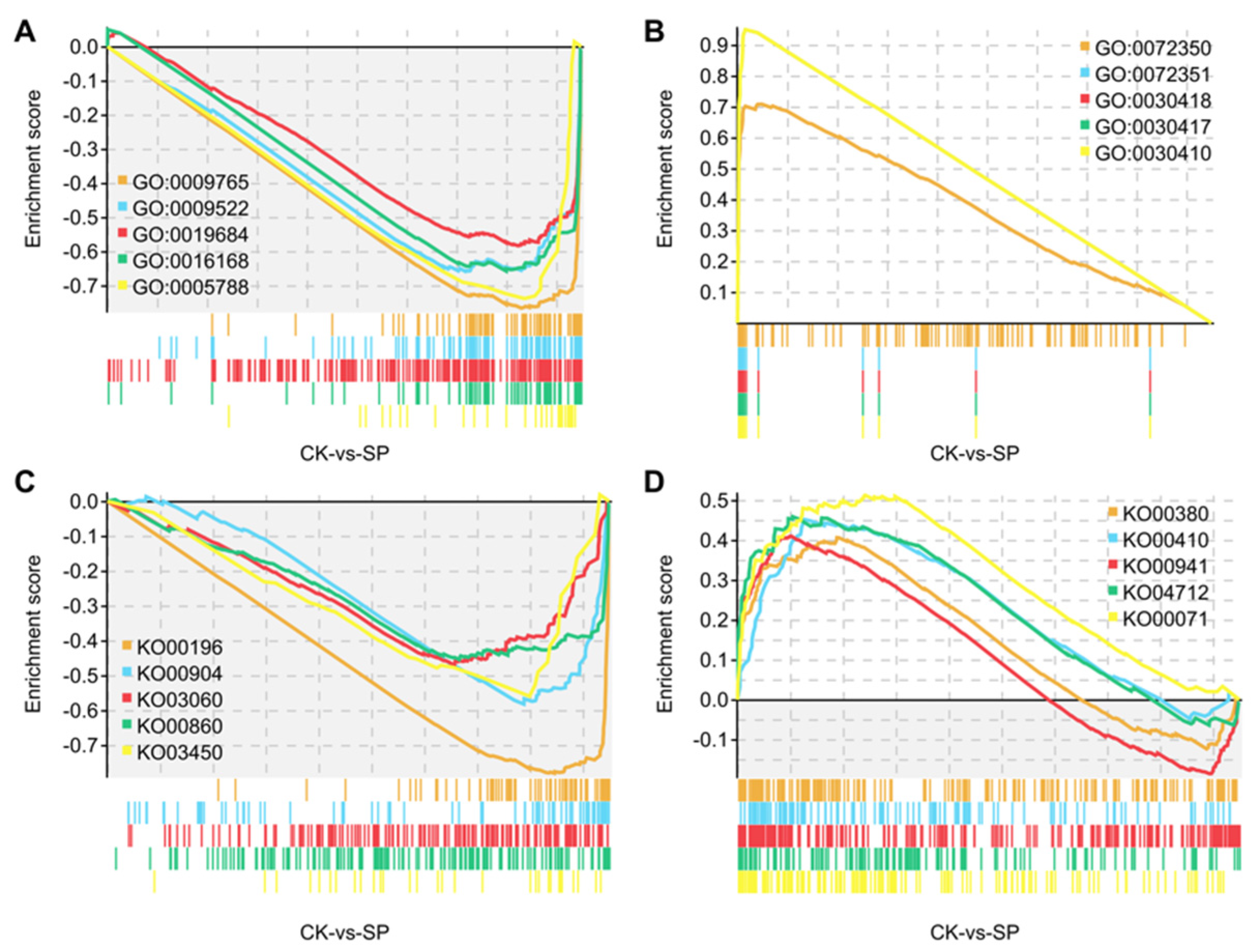
3.2. GSEA
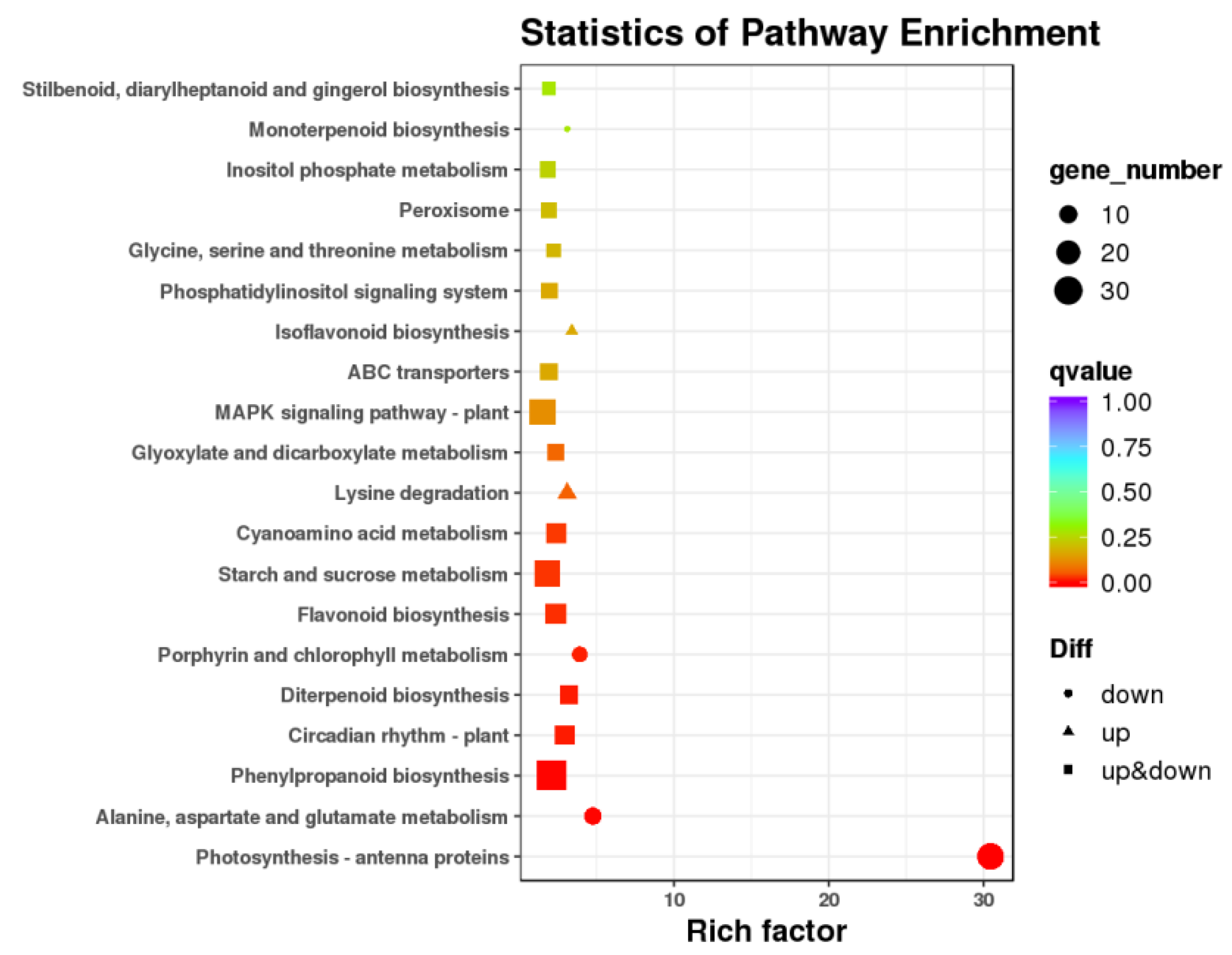
3.3. Differentially Expressed Genes and Functional Analysis
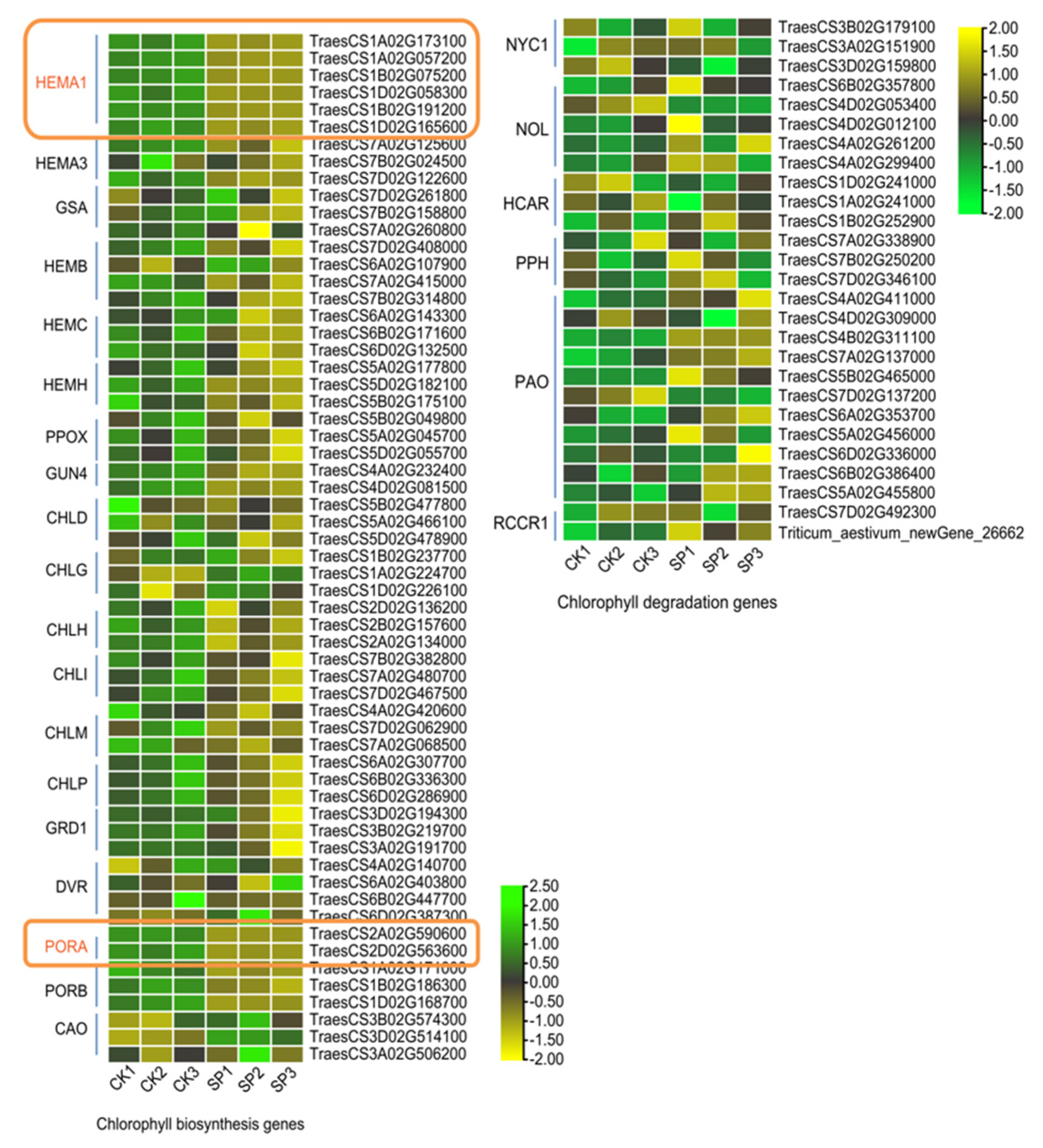
3.4. Pigmentation Pathways
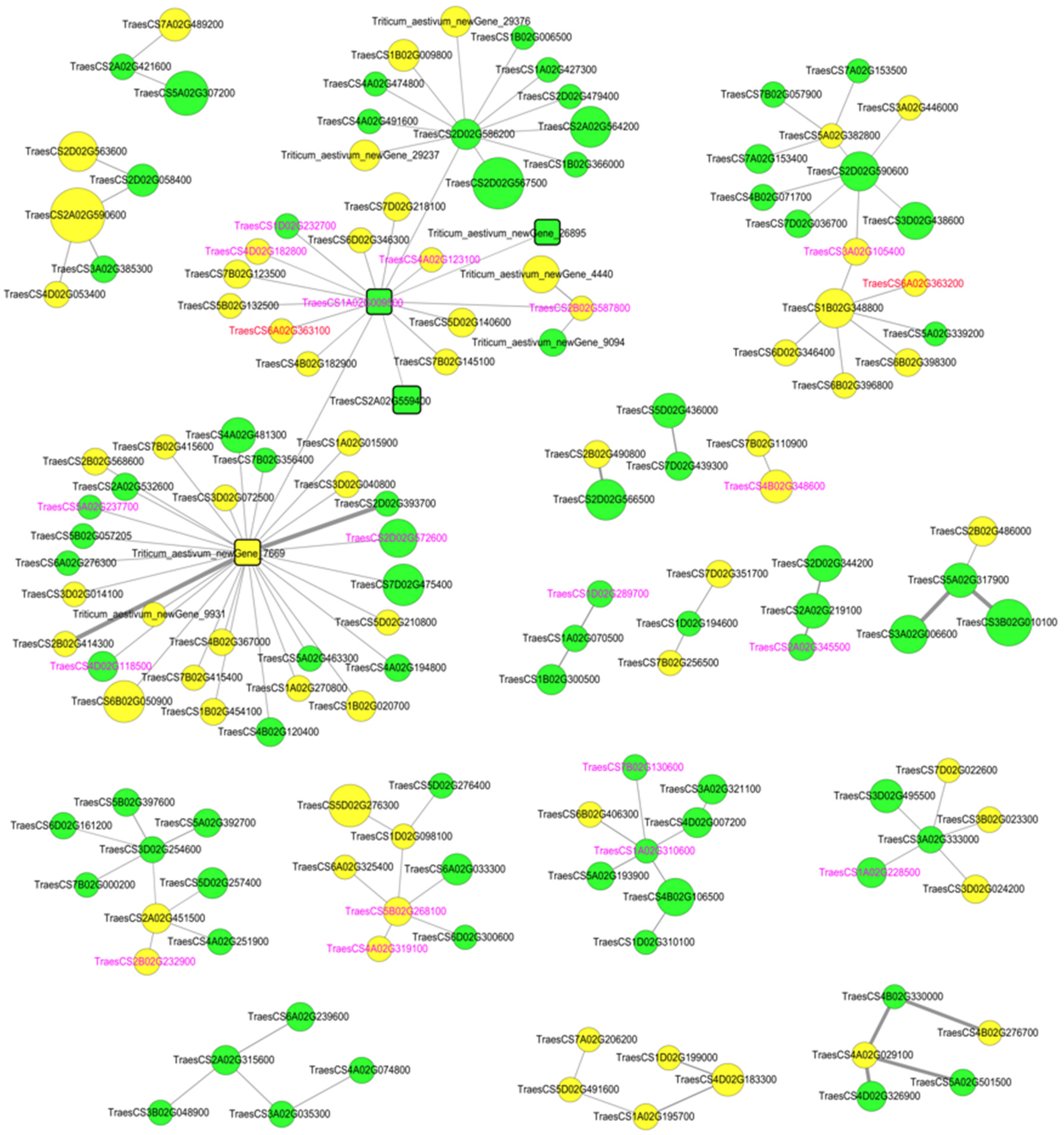
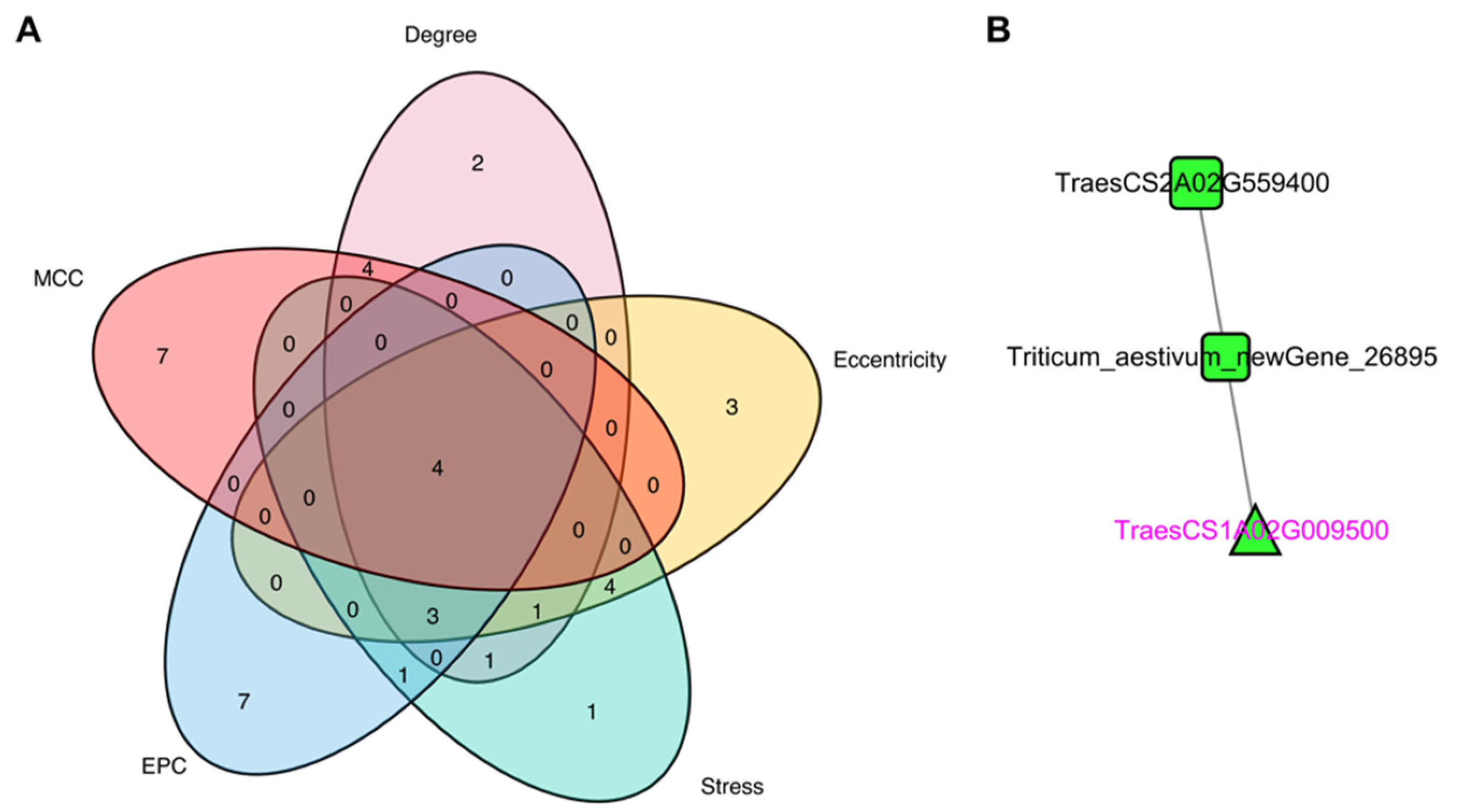
3.5. Identification of Hub Genes through PPI Network Analysis of DEGs
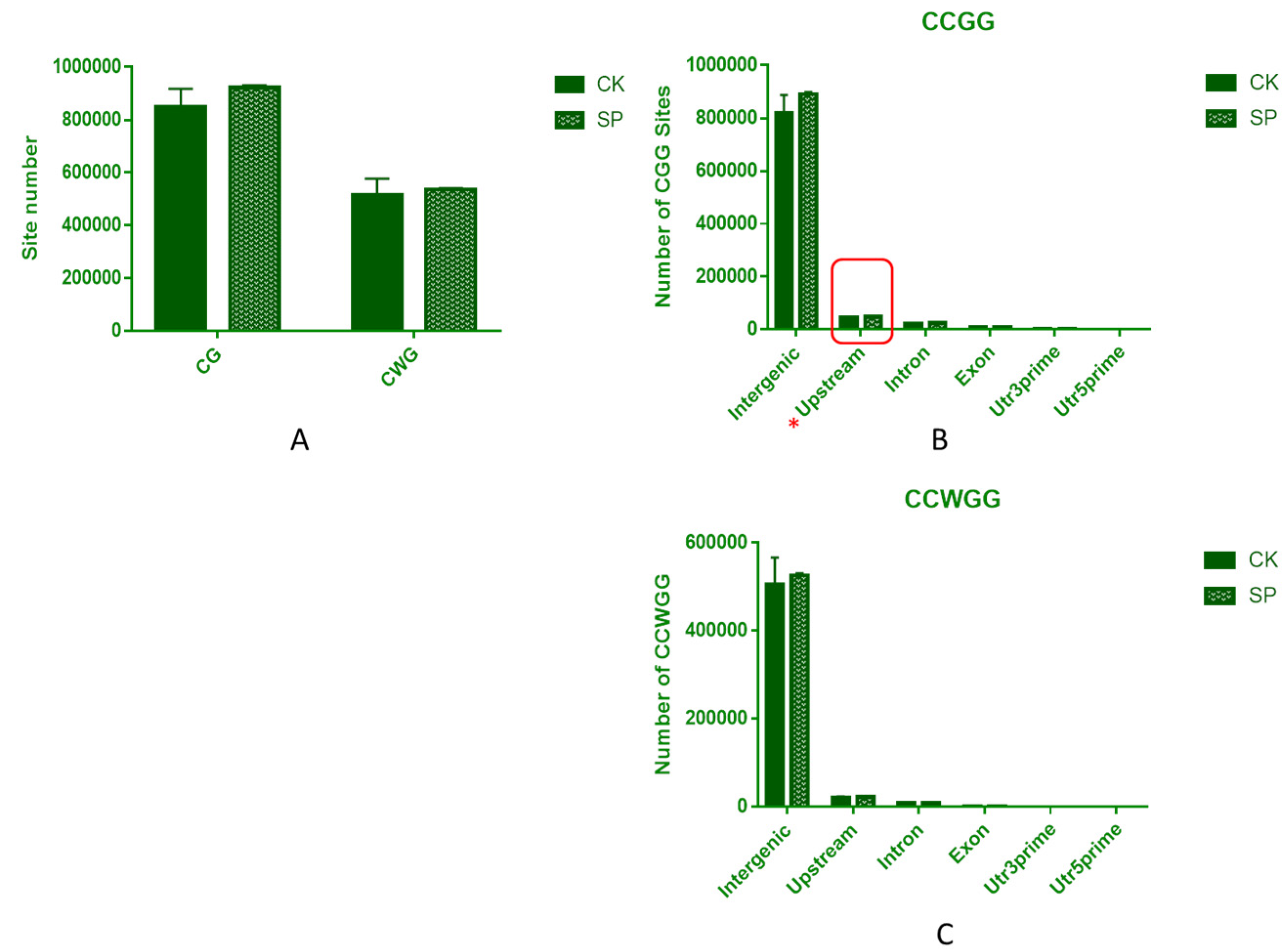
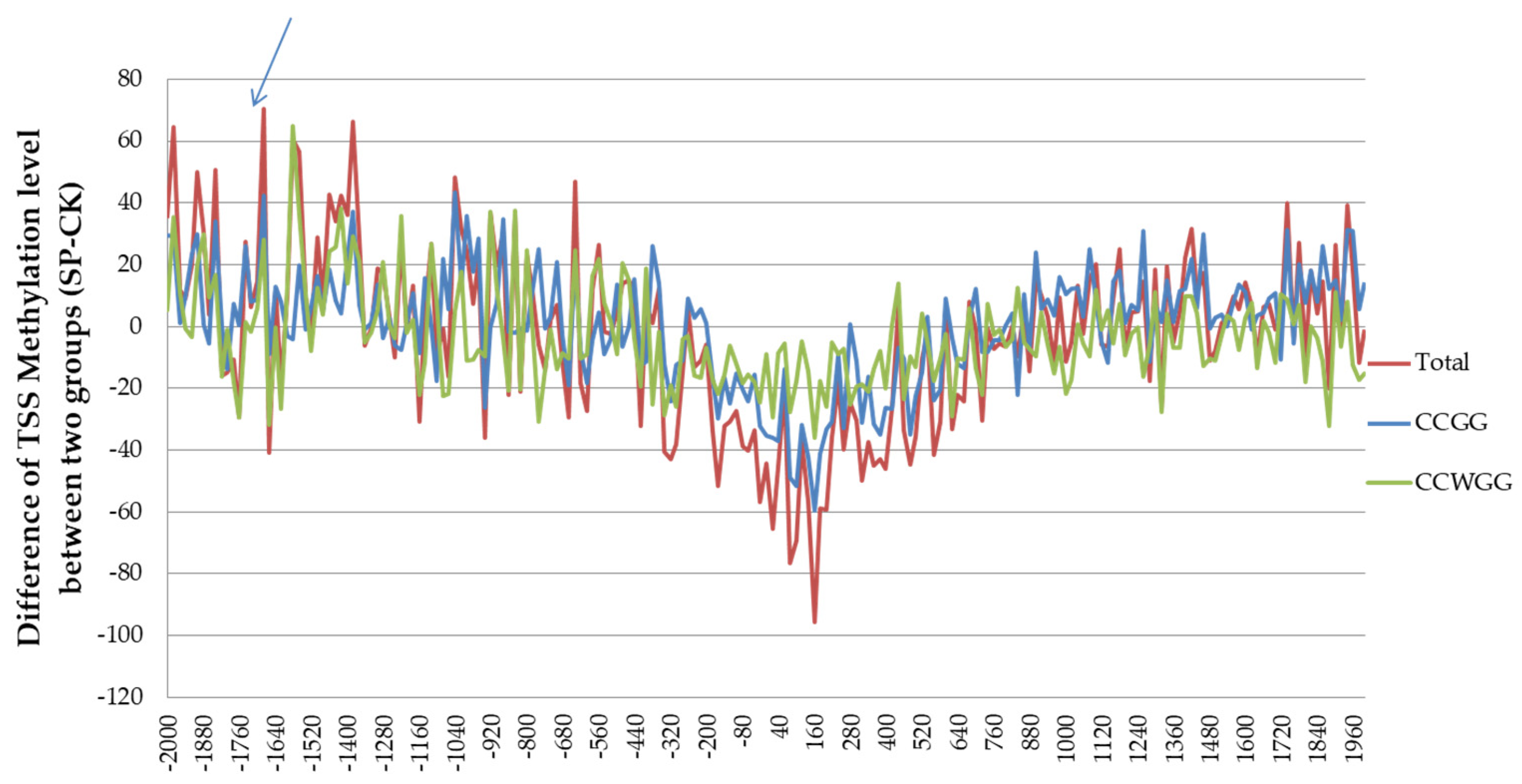
3.6. DNA MethylRAD Sequencing and DNA Methylation Levels
3.7. Distribution of Methylation Sites within Different Functional Elements and Gene Regions
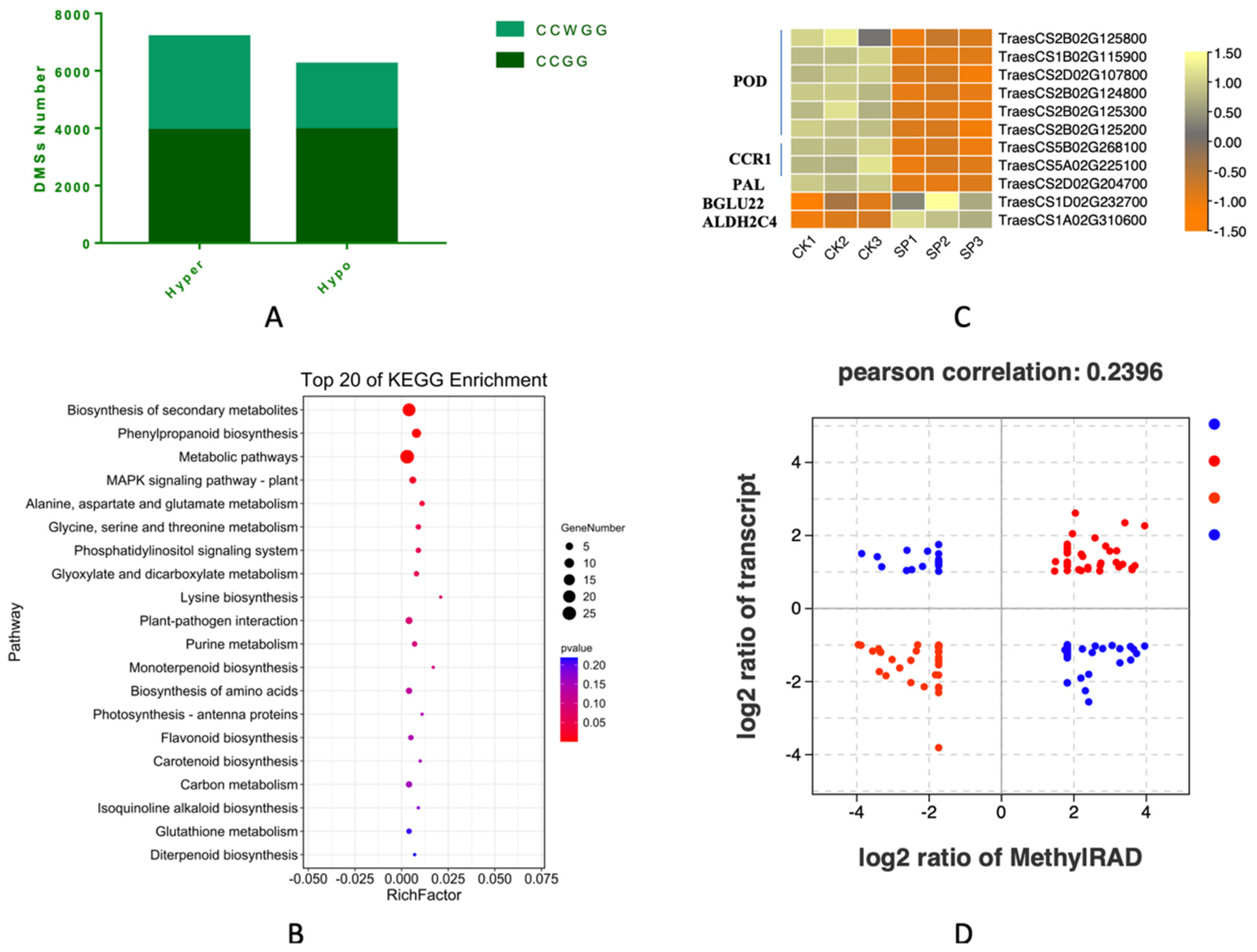
3.8. The Differentially Methylated Sites and Functional Analysis
3.9. Integrated Analysis of DMSs and DEGs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Godfray Brenchley, R.; Spannagl, M.; Pfeifer, M.; Barker, G.L.A.; D’Amore, R.; Allen, A.M.; McKenzie, N.; Kramer, M.; Kerhornou, A.; Bolser, D.; et al. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 2012, 491, 705–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godfray, H.C.; Beddington, J.R.; Crute, I.R.; Haddad, L.; Lawrence, D.; Muir, J.F.; Pretty, J.; Robinson, S.; Thomas, S.M.; Toulmin, C. Food security: The challenge of feeding 9 billion people. Science 2010, 327, 812–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilman, D.; Balzer, C.; Hill, J.; Befort, B.L. Global food demand and the sustainable intensification of agriculture. Proc. Natl. Acad. Sci. USA 2011, 108, 20260–20264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Shi, X.; Zheng, F.; Wu, D.; Li, A.-A.; Sun, F.-Y.; Li, C.-C.; Wu, J.-C.; Li, T. Transcriptome analysis of Dlm mutants reveals the potential formation mechanism of lesion mimic in wheat. Eur. J. Plant Pathol. 2016, 146, 987–997. [Google Scholar] [CrossRef]
- Wang, F.; Wu, W.; Wang, D.; Yang, W.; Sun, J.; Liu, D.; Zhang, A. Characterization and Genetic Analysis of a Novel Light-Dependent Lesion Mimic Mutant, lm3, Showing Adult-Plant Resistance to Powdery Mildew in Common Wheat. PLoS ONE 2016, 11, e0155358. [Google Scholar] [CrossRef]
- Wu, H.; Shi, N.; An, X.; Liu, C.; Fu, H.; Cao, L.; Feng, Y.; Sun, D.; Zhang, L. Candidate Genes for Yellow Leaf Color in Common Wheat (Triticum aestivum L.) and Major Related Metabolic Pathways according to Transcriptome Profiling. Int. J. Mol. Sci. 2018, 19, 1594. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Zhang, L.; Li, Y.; Ali Buttar, Z.; Wang, N.; Xie, Y.; Wang, C. Single Nucleotide Mutagenesis of the TaCHLI Gene Suppressed Chlorophyll and Fatty Acid Biosynthesis in Common Wheat Seedlings. Front. Plant Sci. 2020, 11, 97. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.-H.; Li, X.; Zhang, X.-X.; Zhang, H.; Zhao, X.-Y. Mutation Mechanism of Leaf Color in Plants: A Review. Forests 2020, 11, 851. [Google Scholar] [CrossRef]
- Tanaka, A.; Tanaka, R. Chlorophyll metabolism. Curr. Opin. Plant Biol. 2006, 9, 248–255. [Google Scholar] [CrossRef]
- Kumar, A.M.; Söll, D. Antisense HEMA1 RNA Expression Inhibits Heme and Chlorophyll Biosynthesis in Arabidopsis1. Plant Physiol. 2000, 122, 49–56. [Google Scholar] [CrossRef] [Green Version]
- McCormac, A.C.; Fischer, A.; Kumar, A.M.; Söll, D.; Terry, M.J. Regulation of HEMA1 expression by phytochrome and a plastid signal during de-etiolation in Arabidopsis thaliana. Plant J. 2001, 25, 549–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilag, L.L.; Kumar, A.M.; Söll, D. Light regulation of chlorophyll biosynthesis at the level of 5-aminolevulinate formation in Arabidopsis. Plant Cell 1994, 6, 265–275. [Google Scholar] [PubMed] [Green Version]
- Kumar, A.M.; Csankovszki, G.; Söll, D. A second and differentially expressed glutamyl-tRNA reductase gene from Arabidopsis thaliana. Plant Mol. Biol. 1996, 30, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Reinbothe, S.; Reinbothe, C.; Lebedev, N.; Apel, K. PORA and PORB, Two Light-Dependent Protochlorophyllide-Reducing Enzymes of Angiosperm Chlorophyll Biosynthesis. Plant Cell 1996, 8, 763–769. [Google Scholar] [CrossRef]
- Kang, S.-J.; Fang, Y.-X.; Zou, G.-X.; Ruan, B.-P.; Zhao, J.; Dong, G.-J.; Yan, M.-X.; Gao, Z.-Y.; Zhu, L. White-Green Leaf Gene Encoding Protochlorophyllide Oxidoreductase B Is Involved in Chlorophyll Synthesis of Rice. Crop Sci. 2015, 55, 284–293. [Google Scholar] [CrossRef]
- Holtorf, H.; Reinbothe, S.; Reinbothe, C.; Bereza, B.; Apel, K. Two routes of chlorophyllide synthesis that are differentially regulated by light in barley (Hordeum vulgare L.). Proc. Natl. Acad. Sci. USA 1995, 92, 3254–3258. [Google Scholar] [CrossRef] [Green Version]
- Peremarti, A.; Marè, C.; Aprile, A.; Roncaglia, E.; Cattivelli, L.; Villegas, D.; Royo, C. Transcriptomic and proteomic analyses of a pale-green durum wheat mutant shows variations in photosystem components and metabolic deficiencies under drought stress. BMC Genomics 2014, 15, 125. [Google Scholar] [CrossRef]
- Wang, T.; Lu, Q.; Song, H.; Hu, N.; Wei, Y.; Li, P.; Liu, Y.; Zhao, Z.; Liu, J.; Zhang, B.; et al. DNA Methylation and RNA-Sequencing Analysis Show Epigenetic Function during Grain Filling in Foxtail Millet (Setaria italica L.). Front. Plant Sci. 2021, 12, 741415. [Google Scholar] [CrossRef]
- N’Diaye, A.; Byrns, B.; Cory, A.T.; Nilsen, K.T.; Walkowiak, S.; Sharpe, A.; Robinson, S.J.; Pozniak, C.J. Machine learning analyses of methylation profiles uncovers tissue-specific gene expression patterns in wheat. Plant Genome 2020, 13, e20027. [Google Scholar] [CrossRef]
- Candaele, J.; Demuynck, K.; Mosoti, D.; Beemster, G.T.S.; Inzé, D.; Nelissen, H. Differential methylation during maize leaf growth targets developmentally regulated genes. Plant Physiol. 2014, 164, 1350–1364. [Google Scholar] [CrossRef] [Green Version]
- Ogneva, Z.; Dubrovina, A.; Kiselev, K. Age-associated alterations in DNA methylation and expression of methyltransferase and demethylase genes in Arabidopsis thaliana. Biol. Plant. 2016, 60, 628–634. [Google Scholar] [CrossRef]
- Herrera, C.M.; Bazaga, P. Epigenetic correlates of plant phenotypic plasticity: DNA methylation differs between prickly and nonprickly leaves in heterophyllous Ilex aquifolium (Aquifoliaceae) trees. Bot. J. Linn. Soc. 2013, 171, 441–452. [Google Scholar] [CrossRef] [Green Version]
- Cocciolone, S.M.; Cone, K.C. Pl-Bh, an anthocyanin regulatory gene of maize that leads to variegated pigmentation. Genetics 1993, 135, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.-M.; Wang, L.; Zhou, Y.; Cui, J.; Wang, Y.; Zhao, C. Leaf patterning of Clivia miniata var. variegata is associated with differential DNA methylation. Plant Cell Rep. 2016, 35, 167–184. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [Green Version]
- Natale, D.A.; Galperin, M.Y.; Tatusov, R.L.; Koonin, E.V. Using the COG Database to Improve Gene Recognition in Complete Genomes. Genetica 2000, 108, 9–17. [Google Scholar] [CrossRef]
- Muller, J.; Szklarczyk, D.; Julien, P.; Letunic, I.; Roth, A.; Kuhn, M.; Powell, S.; Von, M.C.; Doerks, T.; Jensen, L.J. eggNOG v2.0: Extending the evolutionary genealogy of genes with enhanced non-supervised orthologous groups, species and functional annotations. Nucleic Acids Res. 2010, 38, D190. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [Green Version]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [Green Version]
- Bader, G.D.; Hogue, C.W.V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Lv, J.; Zhang, L.; Dou, J.; Sun, Y.; Li, X.; Fu, X.; Dou, H.; Mao, J.; Hu, X.; et al. MethylRAD: A simple and scalable method for genome-wide DNA methylation profiling using methylation-dependent restriction enzymes. Open Biol. 2015, 5, 150130. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-J.; Li, D.; Gong, J.; Wang, Y.-B.; Chen, Z.-B.; Pang, B.-S.; Chen, X.-C.; Gao, J.-G.; Yang, W.-B.; Zhang, F.-T.; et al. Comparative transcriptome and DNA methylation analysis in temperature-sensitive genic male sterile wheat BS366. BMC Genomics 2021, 22, 911. [Google Scholar] [CrossRef] [PubMed]
- Feldman, M.; Levy, A.A. Genome Evolution Due to Allopolyploidization in Wheat. Genetics 2012, 192, 763–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Bian, Y.; Gou, X.; Zhu, B.; Xu, C.; Qi, B.; Li, N.; Rustgi, S.; Zhou, H.; Han, F.; et al. Persistent whole-chromosome aneuploidy is generally associated with nascent allohexaploid wheat. Proc. Natl. Acad. Sci. USA 2013, 110, 3447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormac, A.C.; Terry, M.J. Light-signalling pathways leading to the co-ordinated expression of HEMA1 and Lhcb during chloroplast development in Arabidopsis thaliana. Plant J. 2002, 32, 549–559. [Google Scholar] [CrossRef]
- Song, W.-Y.; Wang, G.-L.; Chen, L.-L.; Kim, H.-S.; Pi, L.-Y.; Holsten, T.; Gardner, J.; Wang, B.; Zhai, W.-X.; Zhu, L.-H.; et al. A Receptor Kinase-Like Protein Encoded by the Rice Disease Resistance Gene, Xa21. Science 1995, 270, 1804–1806. [Google Scholar] [CrossRef] [Green Version]
- Von Wettstein, D.; Gough, S.; Kannangara, C.G. Chlorophyll Biosynthesis. Plant Cell 1995, 7, 1039–1057. [Google Scholar] [CrossRef]
- Yao, Q.; Zhou, R.; Fu, T.; Wu, W.; Zhu, Z.; Li, A.; Jia, J. Characterization and mapping of complementary lesion-mimic genes lm1 and lm2 in common wheat. Theor. Appl. Genet. 2009, 119, 1005–1012. [Google Scholar] [CrossRef]
- Bhatia, H.; Khemka, N.; Jain, M.; Garg, R. Genome-wide bisulphite-sequencing reveals organ-specific methylation patterns in chickpea. Sci. Rep. 2018, 8, 9704. [Google Scholar] [CrossRef]
- Wibowo, A.; Becker, C.; Durr, J.; Price, J.; Spaepen, S.; Hilton, S.; Putra, H.; Papareddy, R.; Saintain, Q.; Harvey, S. Partial maintenance of organ-specific epigenetic marks during plant asexual reproduction leads to heritable phenotypic variation. Proc. Natl. Acad. Sci. USA 2018, 115, E9145–E9152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.; Li, K.-t.; Zhang, S.-y.; Zhang, J.; Song, T.-t; Zhu, Y.-j.; Yao, Y.-c. The Structure and Methylation Level of the McMYB10 Promoter Determine the Leaf Color of Malus Crabapple. HortScience Horts 2017, 52, 520–526. [Google Scholar] [CrossRef] [Green Version]
- Nie, W.-F. DNA methylation: From model plants to vegetable crops. Biochem. Soc. Trans. 2021, 49, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Soto-Cardinault, C.G.; Duarte-Aké, F.; De-la-Peña, C.; Góngora-Castillo, E. DNA Methylation and Transcriptomic Next-Generation Technologies in Cereal Genomics. In Cereal Genomics: Methods and Protocols; Vaschetto, L.M., Ed.; Springer: New York, NY, USA, 2020; pp. 65–84. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| #ID | CK vs. SP | Swiss_Prot_Annotation |
|---|---|---|
| Triticum_aestivum_newGene_7669 | down | Transcription factor HY5 OS = Solanum lycopersicum OX = 4081 GN = HY5 PE = 2 SV = 1 |
| Triticum_aestivum_newGene_26895 | up | -- |
| TraesCS1A02G009500 | up | Receptor kinase-like protein Xa21 OS = Oryza sativa subsp. japonica OX = 39947 GN = XA21 PE = 1 SV = 1 |
| TraesCS2A02G559400 | up | Probable LRR receptor-like serine/threonine-protein kinase At3g47570 OS = Arabidopsis thaliana OX = 3702 GN = At3g47570 PE = 2 SV = 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Z.; Wang, F.; Fan, X.; Feng, B.; Zhou, Q.; Yang, Q.; Wang, T. DNA Methylation and RNA-Sequencing Analysis to Identify Genes Related to Spontaneous Leaf Spots in a Wheat Variety ‘Zhongkenuomai No.1’. Agronomy 2022, 12, 1519. https://doi.org/10.3390/agronomy12071519
Xu Z, Wang F, Fan X, Feng B, Zhou Q, Yang Q, Wang T. DNA Methylation and RNA-Sequencing Analysis to Identify Genes Related to Spontaneous Leaf Spots in a Wheat Variety ‘Zhongkenuomai No.1’. Agronomy. 2022; 12(7):1519. https://doi.org/10.3390/agronomy12071519
Chicago/Turabian StyleXu, Zhibin, Fang Wang, Xiaoli Fan, Bo Feng, Qiang Zhou, Qichang Yang, and Tao Wang. 2022. "DNA Methylation and RNA-Sequencing Analysis to Identify Genes Related to Spontaneous Leaf Spots in a Wheat Variety ‘Zhongkenuomai No.1’" Agronomy 12, no. 7: 1519. https://doi.org/10.3390/agronomy12071519
APA StyleXu, Z., Wang, F., Fan, X., Feng, B., Zhou, Q., Yang, Q., & Wang, T. (2022). DNA Methylation and RNA-Sequencing Analysis to Identify Genes Related to Spontaneous Leaf Spots in a Wheat Variety ‘Zhongkenuomai No.1’. Agronomy, 12(7), 1519. https://doi.org/10.3390/agronomy12071519