

Identification of Allele-Specific Expression Genes Associated with Maize Heterosis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Phenotypic and Genotypic Data
2.2. Variance Components Analysis
2.3. Mapping of MPH
2.4. Retrieving Yield and Biomass Genes
2.5. Colocalization of MPH Significant Loci and ASEGs
2.6. Construction of Weighted Gene Co-Expression Networks
3. Results
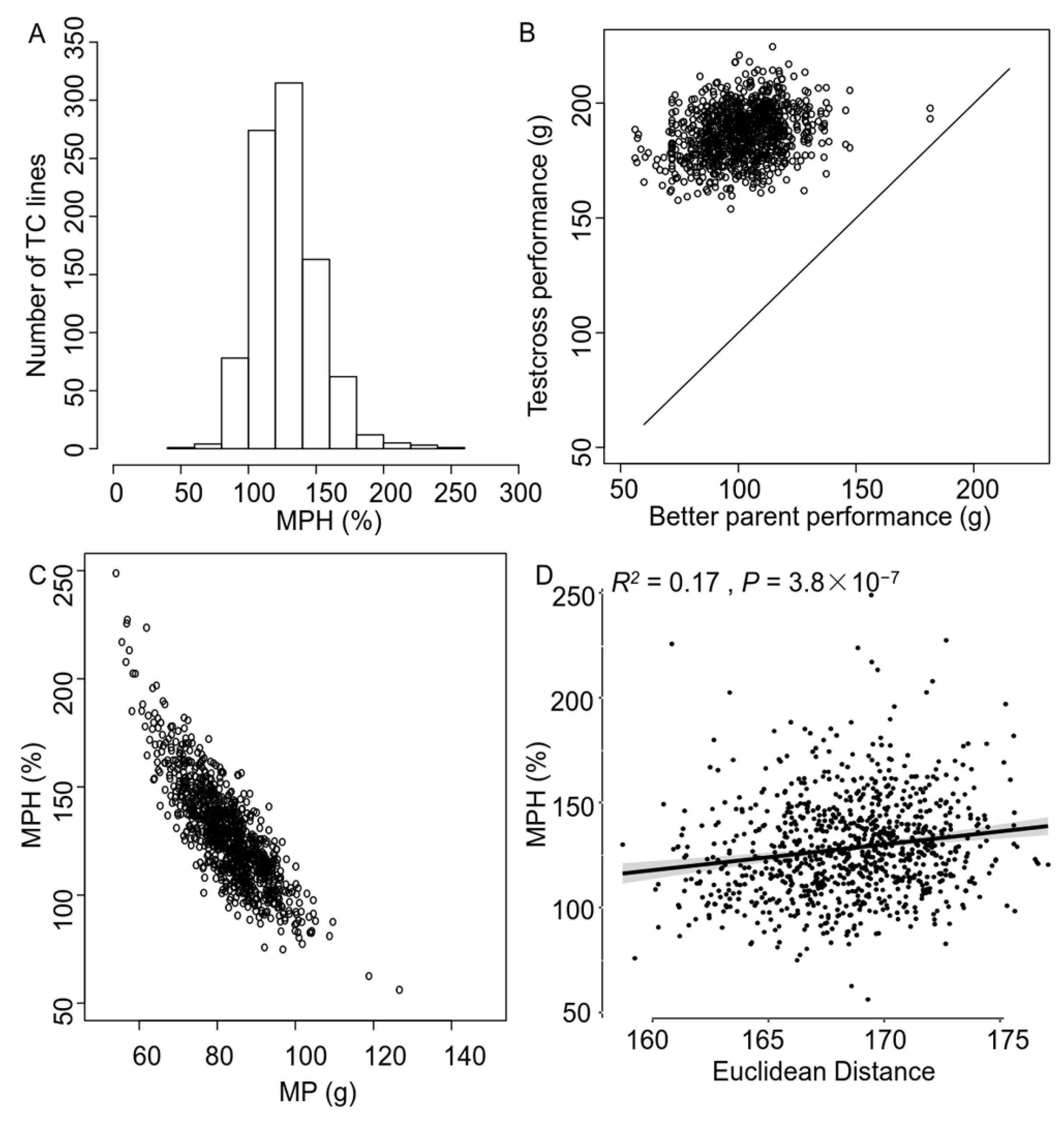
3.1. Observation of Strong Heterosis in the Testcross Population
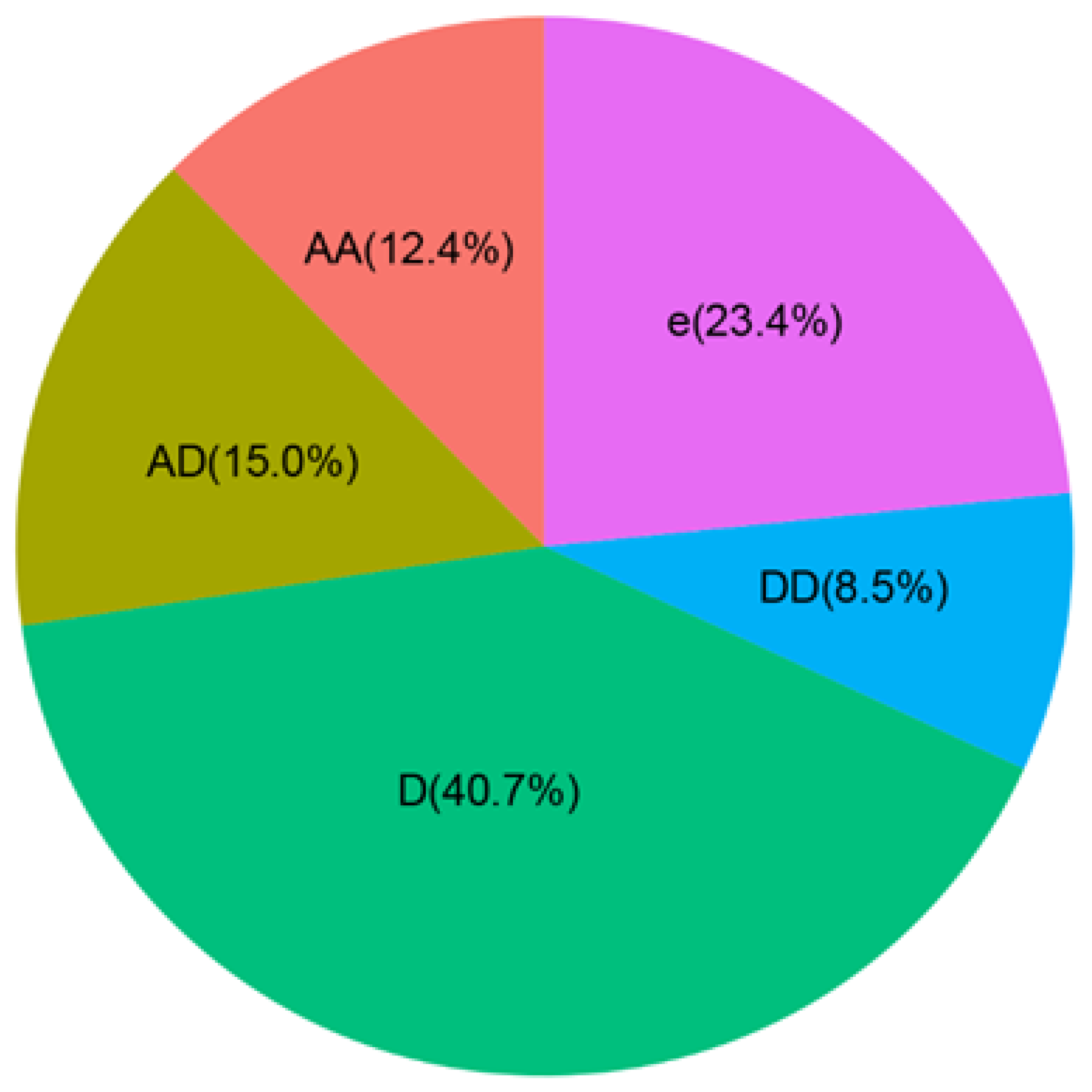
3.2. Genetic Dissection of MPH and Candidate Gene Identification
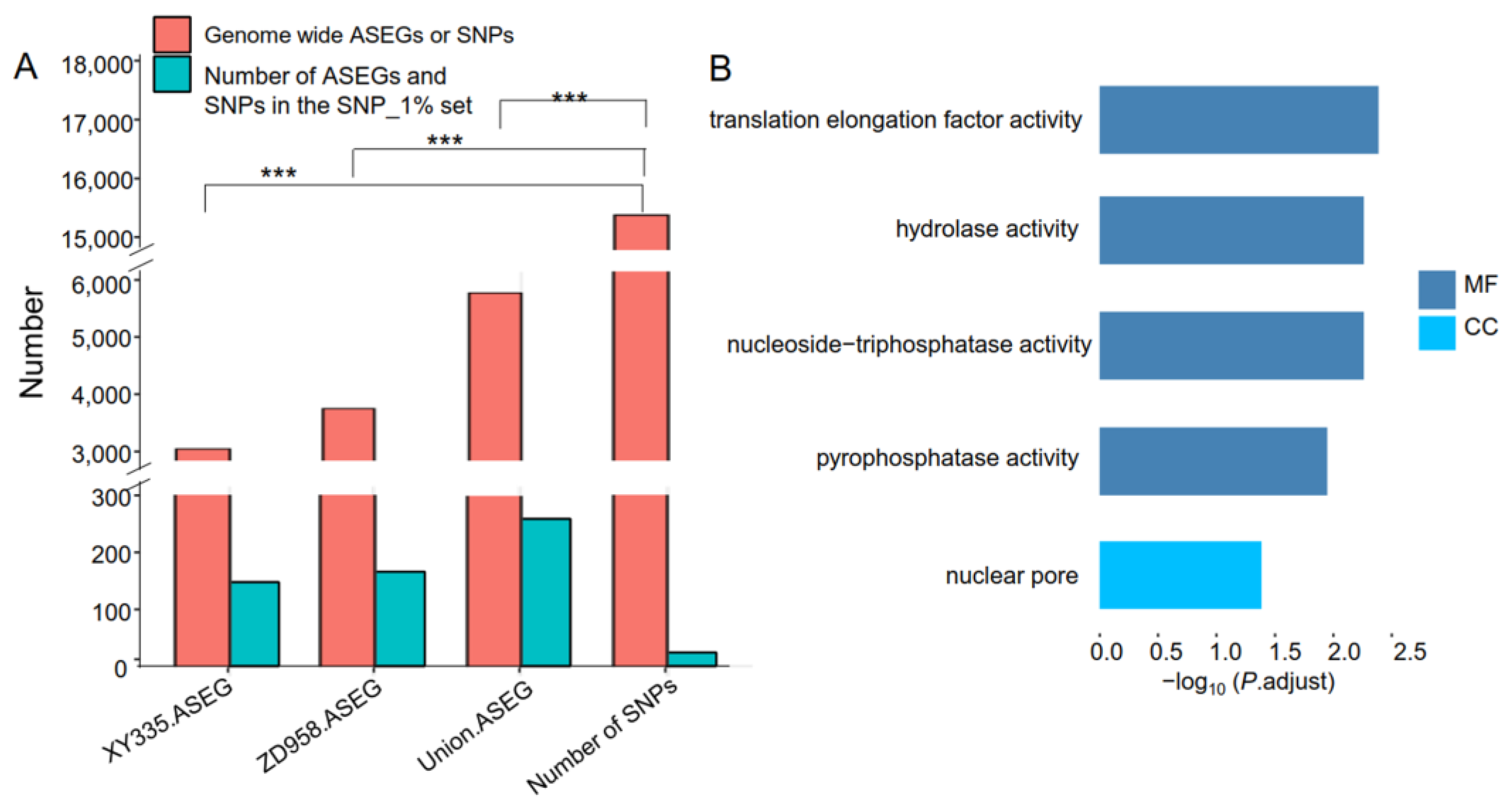
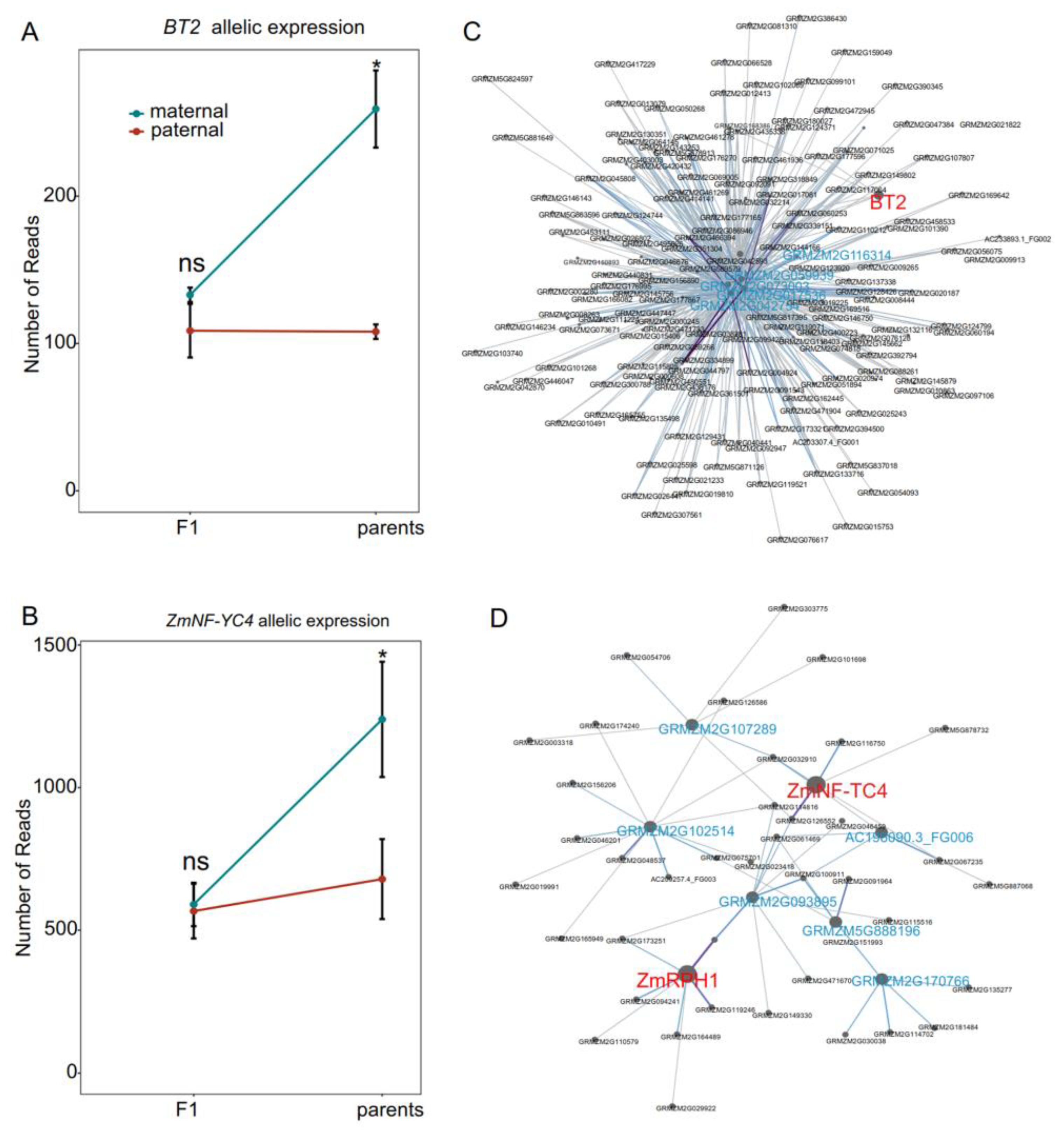
3.3. Identification of Candidate Genes and Their Co-Expression Networks Associated with MPH
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chen, F.C.; Li, W.H. Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. J. Hum. Genet. 2001, 68, 444–456. [Google Scholar] [CrossRef]
- Riedelsheimer, C.; Czedik-Eysenberg, A.; Grieder, C.; Lisec, J.; Technow, F.; Sulpice, R.; Altmann, T.; Stitt, M.; Willmitzer, L.; Melchinger, A.E. Genomic and metabolic prediction of complex heterotic traits in hybrid maize. Nat. Genet. 2012, 44, 217–220. [Google Scholar] [CrossRef]
- Wu, X.; Liu, Y.; Zhang, Y.; Gu, R. Advances in research on the mechanism of heterosis in plants. Front. Plant Sci. 2021, 12, 745726. [Google Scholar] [CrossRef] [PubMed]
- Das, A.K.; Choudhary, M.K.; Kumar, P.; Karjagi, C.G.; Kr, Y.; Kumar, R.; Singh, A.; Kumar, S.; Rakshit, S. Heterosis in genomic era: Advances in the molecular understanding and techniques for rapid exploitation. CRC Crit. Rev. Plant Sci. 2021, 40, 218–242. [Google Scholar] [CrossRef]
- Schnable, P.S.; Springer, N.M. Progress toward understanding heterosis in crop plants. Annu. Rev. Plant Biol. 2013, 64, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Gong, J.; Zhu, Z.; Li, Z.; Feng, Q.; Wang, C.; Zhao, Y.; Zhan, Q.; Zhou, C.; Wang, A.; et al. Structure and function of rice hybrid genomes reveal genetic basis and optimal performance of heterosis. Nat. Genet. 2023, 55, 1745–1756. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yang, S.; Gong, J.; Zhao, Y.; Feng, Q.; Gong, H.; Li, W.; Zhan, Q.; Cheng, B.; Xia, J.; et al. Genomic analysis of hybrid rice varieties reveals numerous superior alleles that contribute to heterosis. Nat. Commun. 2015, 6, 6258. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Jiang, S.; Cheng, Q.; Wang, X.; Yan, J.; Zhang, R.; Qiao, F.; Ma, C.; Luo, J.; Li, W.; et al. The genetic mechanism of heterosis utilization in maize improvement. Genome Biol. 2021, 22, 148. [Google Scholar] [CrossRef]
- Li, C.; Guan, H.; Jing, X.; Li, Y.; Wang, B.; Li, Y.; Liu, X.; Zhang, D.; Liu, C.; Xie, X.; et al. Genomic insights into historical improvement of heterotic groups during modern hybrid maize breeding. Nat. Plants 2022, 8, 750–763. [Google Scholar] [CrossRef]
- Shao, L.; Xing, F.; Xu, C.; Zhang, Q.; Che, J.; Wang, X.; Son, G.J.; Li, X.; Xiao, J.; Chen, L.L.; et al. Patterns of genome-wide allele-specific expression in hybrid rice and the implications on the genetic basis of heterosis. Proc. Natl. Acad. Sci. USA 2019, 116, 5653–5658. [Google Scholar] [CrossRef]
- Zhou, T.; Afzal, R.; Haroon, M.; Ma, Y.; Zhang, H.; Li, L. Dominant complementation of biological pathways in maize hybrid lines is associated with heterosis. Planta 2022, 256, 111. [Google Scholar] [CrossRef]
- Wang, P.; Gu, M.; Yu, X.; Shao, S.; Du, J.; Wang, Y.; Wang, F.; Chen, S.; Liao, Z.; Ye, N.; et al. Allele-specific expression and chromatin accessibility contribute to heterosis in tea plants (Camellia sinensis). Plant J. 2022, 112, 1194–1211. [Google Scholar] [CrossRef]
- Li, D.; Lu, X.; Zhu, Y.; Pan, J.; Zhou, S.; Zhang, X.; Zhu, G.; Shang, Y.; Huang, S.; Zhang, C. The multi-omics basis of potato heterosis. J. Integr. Plant Biol. 2022, 64, 671–687. [Google Scholar] [CrossRef]
- Shi, X.; Li, W.; Guo, Z.; Wu, M.; Zhang, X.; Yuan, L.; Qiu, X.; Xing, Y.; Sun, X.; Xie, H.; et al. Comparative transcriptomic analysis of maize ear heterosis during the inflorescence meristem differentiation stage. BMC Plant Biol. 2022, 22, 348. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, D.; Cao, Y.; Wang, L.; Li, J.; Lübberstedt, T.; Wang, T.; Li, Y.; Li, H. Heterosis-related genes under different planting densities in maize. J. Exp. Bot. 2018, 69, 5077–5087. [Google Scholar] [CrossRef] [PubMed]
- Sang, Z.; Zhang, Z.; Yang, Y.; Li, Z.; Liu, X.; Xu, Y.; Li, W. Heterosis and heterotic patterns of maize germplasm revealed by a multiple-hybrid population under well-watered and drought-stressed conditions. J. Integr. Agric. 2022, 21, 2477–2491. [Google Scholar] [CrossRef]
- Ma, Y.; Li, D.; Xu, Z.; Gu, R.; Wang, P.; Fu, J.; Wang, J.; Du, W.; Zhang, H. Dissection of the genetic basis of yield traits in line per se and testcross populations and identification of candidate genes for hybrid performance in maize. Int. J. Mol. Sci. 2022, 23, 5074. [Google Scholar] [CrossRef]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Jiang, Y.; Schmidt, R.H.; Zhao, Y.; Reif, J.C. A quantitative genetic framework highlights the role of epistatic effects for grain-yield heterosis in bread wheat. Nat. Genet. 2017, 49, 1741–1746. [Google Scholar] [CrossRef] [PubMed]
- Pérez, P.; de los Campos, G. Genome-Wide regression and prediction with the BGLR statistical package. Genetics. 2014, 198, 483–495. [Google Scholar] [CrossRef]
- Gao, X. Multiple testing corrections for imputed SNPs. Genet. Epidemiol. 2011, 35, 154–158. [Google Scholar] [CrossRef]
- Gao, X.; Becker, L.C.; Becker, D.M.; Starmer, J.D.; Province, M.A. Avoiding the high bonferroni penalty in genome-wide association studies. Genet. Epidemiol. 2010, 34, 100–105. [Google Scholar] [CrossRef]
- Gao, X.; Starmer, J.; Martin, E.R. A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet. Epidemiol. 2008, 32, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Peluso, P.; Shi, J.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.; Chin, C.S.; et al. Improved maize reference genome with single-molecule technologies. Nature 2017, 546, 524–527. [Google Scholar] [CrossRef]
- Zhao, K.; Tung, C.W.; Eizenga, G.C.; Wright, M.H.; Ali, M.L.; Price, A.H.; Norton, G.J.; Islam, M.R.; Reynolds, A.; Mezey, J.; et al. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat. Commun. 2011, 2, 467. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lu, Y.; Ma, Y.; Fu, J.; Wang, G. Genetic and molecular control of grain yield in maize. Mol. Breed. 2021, 41, 18. [Google Scholar] [CrossRef] [PubMed]
- Lewin, A.; Grieve, I.C. Grouping gene ontology terms to improve the assessment of gene set enrichment in microarray data. BMC Bioinform. 2006, 7, 426. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Ren, D.; Wang, X.; Yang, M.; Yang, L.; He, G.; Deng, X.W. A new regulator of seed size control in Arabidopsis identified by a genome-wide association study. New Phytol. 2019, 222, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Ren, Y.; Liu, L.; Wang, F.; Zhang, H.; Tian, P.; Pan, T.; Wang, Y.; Jing, R.; Liu, T.; et al. Ubiquitin specific protease 15 has an important role in regulating grain width and size in rice. Plant Physiol. 2019, 180, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Du, L.; Xu, R.; Cui, R.; Hao, J.; Sun, C.; Li, Y. Transcription factors SOD7/NGAL2 and DPA4/NGAL3 act redundantly to regulate seed size by directly repressing KLU expression in Arabidopsis thaliana. Plant Cell. 2015, 27, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, D.; Zhu, H.; Yin, H.; Wang, G.; Yang, A.; Song, Z.; Jing, Q.; Shuai, B.; Xu, N.; et al. Revisiting maize brittle endosperm-2 reveals new insights in BETL development and starchy endosperm filling. Plant Sci. 2023, 332, 111727. [Google Scholar] [CrossRef]
- Wang, H.; Wang, K.; Du, Q.; Wang, Y.; Fu, Z.; Guo, Z.; Kang, D.; Li, W.X.; Tang, J. Maize Urb2 protein is required for kernel development and vegetative growth by affecting pre-ribosomal RNA processing. New Phytol. 2018, 218, 1233–1246. [Google Scholar] [CrossRef]
- Liu, J.; Li, M.; Zhang, Q.; Wei, X.; Huang, X. Exploring the molecular basis of heterosis for plant breeding. J. Integr. Plant Biol. 2020, 62, 287–298. [Google Scholar] [CrossRef]
- Li, D.; Zhou, Z.; Lu, X.; Jiang, Y.; Li, G.; Li, J.; Wang, H.; Chen, S.; Li, X.; Würschum, T.; et al. Genetic dissection of hybrid performance and heterosis for yield-related traits in maize. Front. Plant Sci. 2021, 12, 774478. [Google Scholar] [CrossRef]
- Gogna, A.; Zhang, J.; Jiang, Y.; Schulthess, A.W.; Zhao, Y.; Reif, J.C. Filtering for SNPs with high selective constraint augments mid-parent heterosis predictions in wheat (Triticum aestivum L.). Crop. J. 2023, 11, 166–176. [Google Scholar] [CrossRef]
- Garcia, A.A.; Wang, S.; Melchinger, A.E.; Zeng, Z.B. Quantitative trait loci mapping and the genetic basis of heterosis in maize and rice. Genetics. 2008, 180, 1707–1724. [Google Scholar] [CrossRef]
- Li, Y.; Wang, T.Y. Germplasm base of maize breeding in China and formation of foundation parents. J. Maize Sci. 2010, 18, 1–8. [Google Scholar]
- Sun, Q.; Lu, S.; Li, W.; Yu, Y.; Zhao, M.; Li, W.; Meng, Z. Pedigree analysis on parental inbred lines of Chinese approved maize hybrids released by pioneer corporation. J. Maize Sci. 2020, 28, 8–13. [Google Scholar]
- Liu, Z.; Tang, J.; Wang, Q.; Hu, Y.; Ji, H.; Chen, W. Analysis of heterotic patterns of maize hybrids used in China′s henan province. Sci. Agric. Sin. 2006, 39, 1689–1696. [Google Scholar]
- Wang, B.; Hou, M.; Shi, J.; Ku, L.; Song, W.; Li, C.; Ning, Q.; Li, X.; Li, C.; Zhao, B.; et al. De novo genome assembly and analyses of 12 founder inbred lines provide insights into maize heterosis. Nat. Genet. 2023, 55, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gu, S.; Wen, W.; Lu, X.; Jin, Y.; Zhang, Y.; Guo, X. Disentangling the heterosis in biomass production and radiation use efficiency in maize: A phytomer-based 3D modelling approach. Plants 2023, 12, 1229. [Google Scholar] [CrossRef]
- Ko, D.K.; Rohozinski, D.; Song, Q.; Taylor, S.H.; Juenger, T.E.; Harmon, F.G.; Chen, Z.J. Temporal shift of circadian-mediated gene expression and carbon fixation contributes to biomass heterosis in maize hybrids. PLoS Genet. 2016, 12, e1006197. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, Y.; He, H.; He, G.; Deng, X.W. From hybrid genomes to heterotic trait output: Challenges and opportunities. Curr. Opin. Plant Biol. 2022, 66, 102193. [Google Scholar] [CrossRef]
- Li, W.; Ge, F.; Qiang, Z.; Zhu, L.; Zhang, S.; Chen, L.; Wang, X.; Li, J.; Fu, Y. Maize ZmRPH1 encodes a microtubule-associated protein that controls plant and ear height. J. Plant Biotechnol. 2020, 18, 1345–1347. [Google Scholar] [CrossRef]
- Pan, Z.; Liu, M.; Xiao, Z.; Ren, X.; Zhao, H.; Gong, D.; Liang, K.; Tan, Z.; Shao, Y.; Qiu, F. ZmSMK9, a pentatricopeptide repeat protein, is involved in the cis-splicing of nad5, kernel development and plant architecture in maize. Plant Sci. 2019, 288, 110205. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y.; Yang, W.; Zhang, H.; Wang, P.; Liu, Q.; Du, W. Identification of Allele-Specific Expression Genes Associated with Maize Heterosis. Agronomy 2023, 13, 2722. https://doi.org/10.3390/agronomy13112722
Ma Y, Yang W, Zhang H, Wang P, Liu Q, Du W. Identification of Allele-Specific Expression Genes Associated with Maize Heterosis. Agronomy. 2023; 13(11):2722. https://doi.org/10.3390/agronomy13112722
Chicago/Turabian StyleMa, Yuting, Wenyan Yang, Hongwei Zhang, Pingxi Wang, Qian Liu, and Wanli Du. 2023. "Identification of Allele-Specific Expression Genes Associated with Maize Heterosis" Agronomy 13, no. 11: 2722. https://doi.org/10.3390/agronomy13112722
APA StyleMa, Y., Yang, W., Zhang, H., Wang, P., Liu, Q., & Du, W. (2023). Identification of Allele-Specific Expression Genes Associated with Maize Heterosis. Agronomy, 13(11), 2722. https://doi.org/10.3390/agronomy13112722