
Culturomics and Amplicon-Based Metagenomic Insights into the Bacteria of Soils with High Yield of Oryza sativa L. subsp. Japonica

Abstract
:1. Introduction
2. Materials and Methods
2.1. Material Source
2.2. Isolation and Purification of Strains
2.3. Analysis of Bacterial Diversity
2.4. Species Identification
2.5. Rice Growth Promotion Experiment in the Laboratory
3. Results
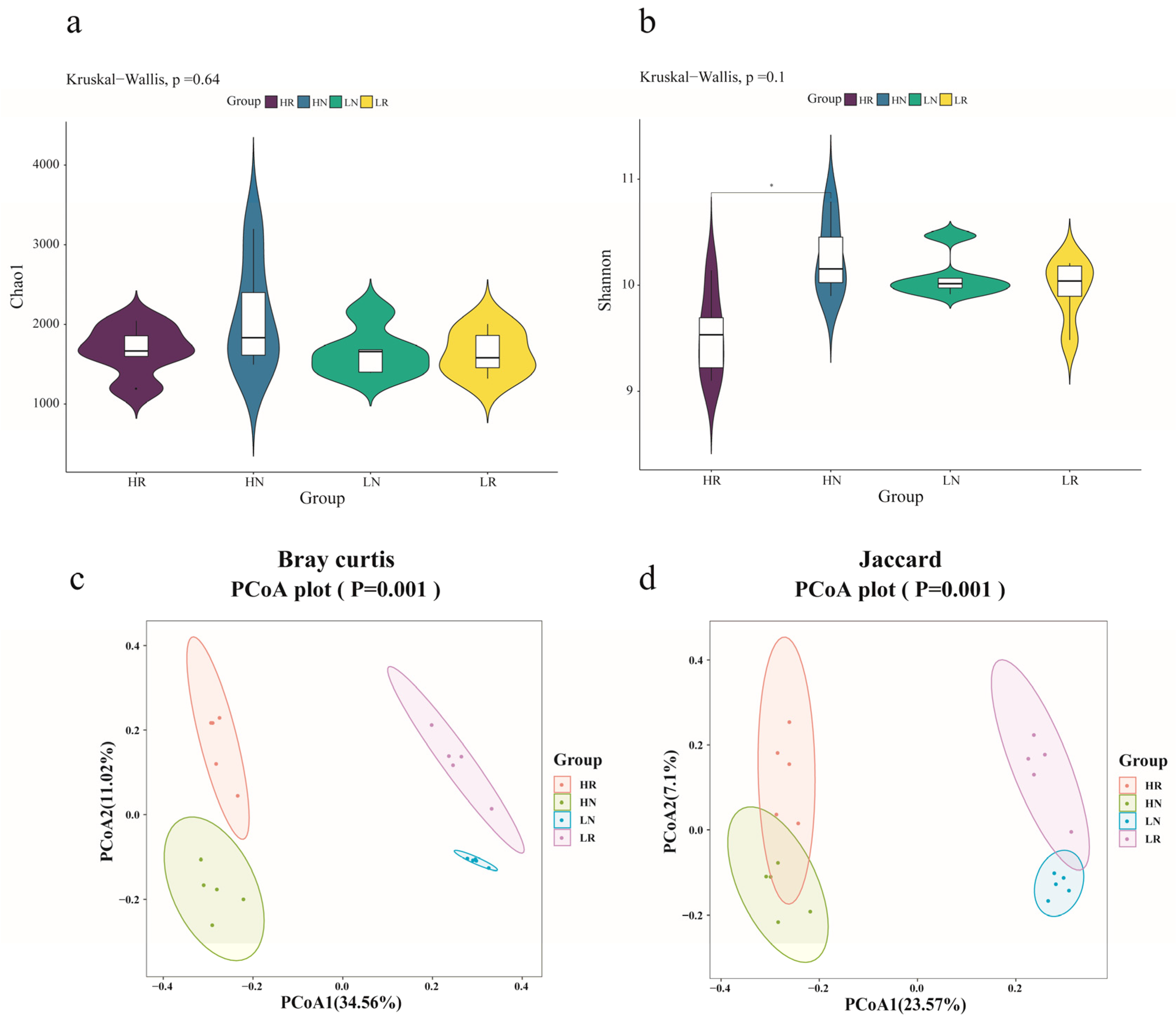
3.1. Analysis of Bacterial Diversity Based on Amplicon Sequencing
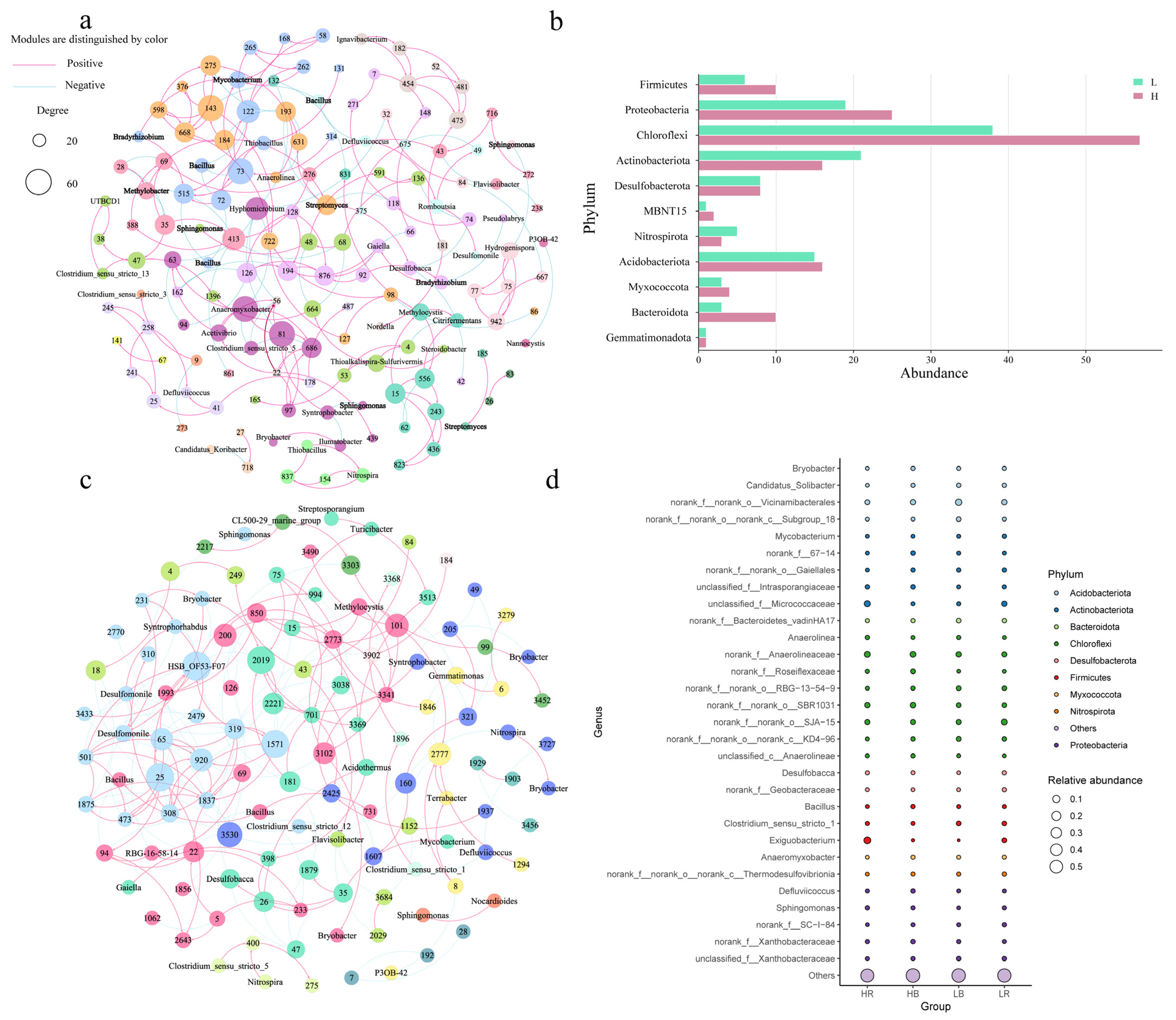
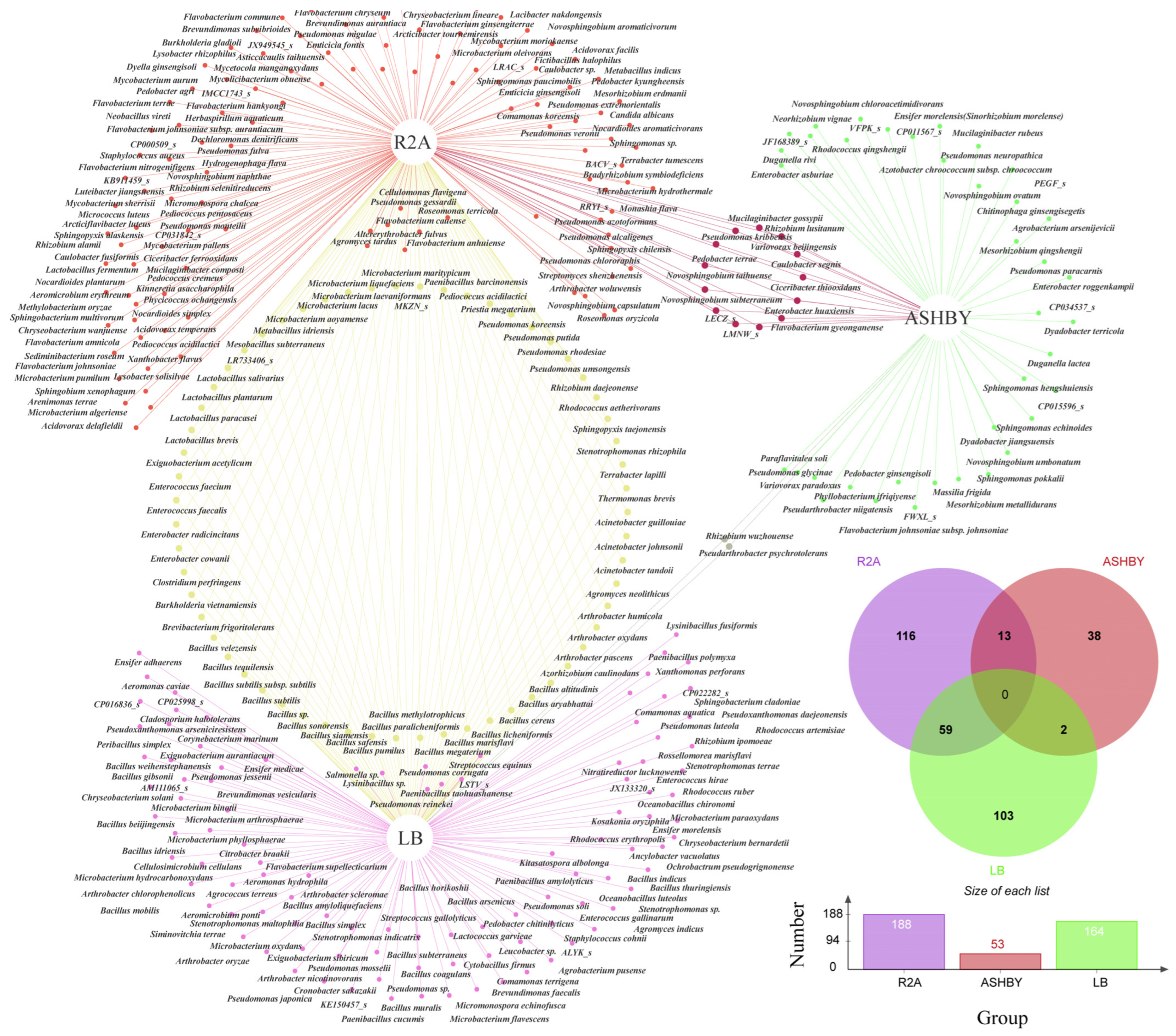
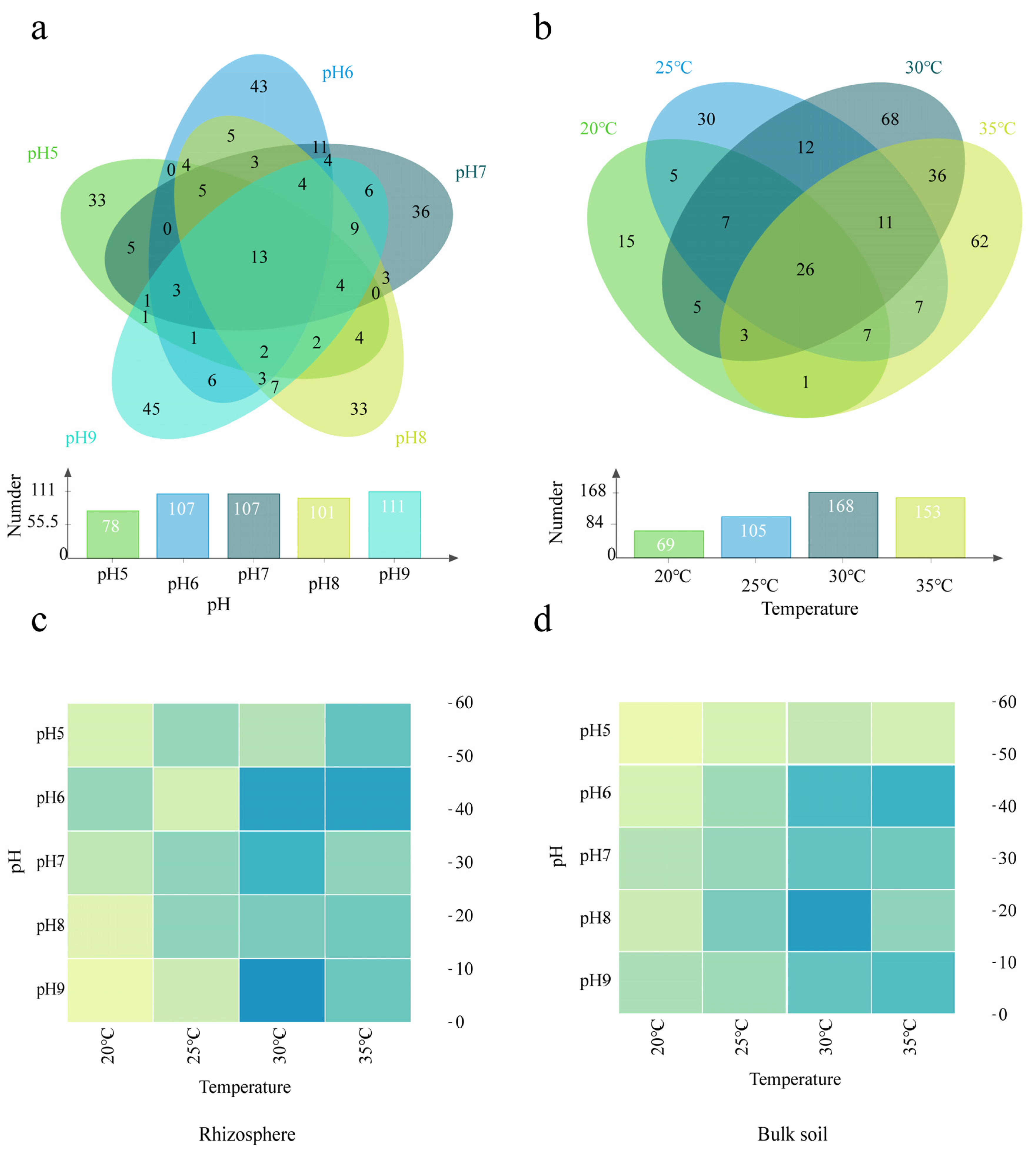
3.2. Bacterial Diversity Based on Culturomics
3.3. Culturomics and Amplicon Sequencing Combined Analysis
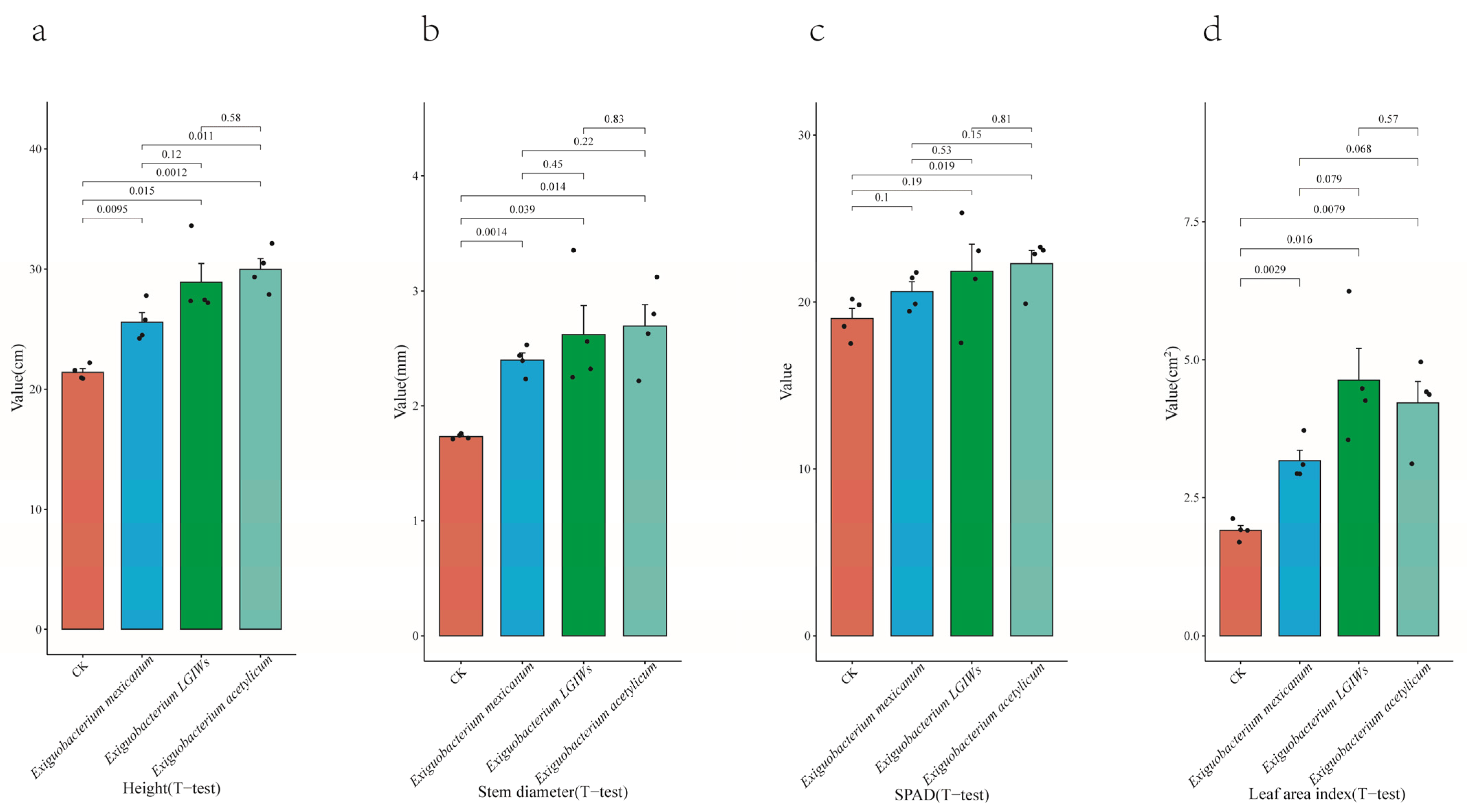
3.4. Rice Growth Promotion Experiment in the Laboratory
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Food and Agriculture Organization of the United Nations. Available online: https://www.fao.org/faostat/en/#data/QCL/visualize (accessed on 5 September 2023).
- Ren, M.; Huang, C.; Wu, Y.; Deppermann, A.; Frank, S.; Havlík, P.; Zhu, Y.; Fang, C.; Ma, X.; Liu, Y.; et al. Enhanced Food System Efficiency Is the Key to China’s 2060 Carbon Neutrality Target. Nat. Food 2023, 4, 552–564, Correction in Nat. Food 2023, 4, 625. [Google Scholar] [CrossRef]
- Busby, P.E.; Soman, C.; Wagner, M.R.; Friesen, M.L.; Kremer, J.; Bennett, A.; Morsy, M.; Eisen, J.A.; Leach, J.E.; Dangl, J.L. Research Priorities for Harnessing Plant Microbiomes in Sustainable Agriculture. PLoS Biol. 2017, 15, e2001793. [Google Scholar] [CrossRef]
- Bai, Y.; Cotrufo, M. Francesca Grassland Soil Carbon Sequestration: Current Understanding, Challenges, and Solutions. Science 2022, 377, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.; Johnson, C.; Santos-Medellín, C.; Lurie, E.; Podishetty, N.K.; Bhatnagar, S.; Eisen, J.A.; Sundaresan, V.; Jeffery, L.D. Structure, Variation, and Assembly of the Root-Associated Microbiomes of Rice. Proc. Natl. Acad. Sci. USA 2015, 112, E911–E920. [Google Scholar] [CrossRef]
- Moronta-Barrios, F.; Gionechetti, F.; Pallavicini, A.; Marys, E.; Venturi, V. Bacterial Microbiota of Rice Roots: 16s-Based Taxonomic Profiling of Endophytic and Rhizospheric Diversity, Endophytes Isolation and Simplified Endophytic Community. Microorganisms 2018, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Hu, J.; Xia, Q.; Zhang, S.; Li, X.; Pan, X.; Zhao, R.; Wang, R.; Yan, W.; Shangguan, Z.; et al. Soil Microbial Mechanisms Promoting Ultrahigh Rice Yield. Soil Biol. Biochem. 2020, 143, 107741, Correction in Soil Biol. Biochem. 2020, 150, 107894. [Google Scholar] [CrossRef]
- Pang, Z.; Xu, P.; Yu, D. Environmental Adaptation of the Root Microbiome in Two Rice Ecotypes. Microbiol. Res. 2020, 241, 126588. [Google Scholar] [CrossRef]
- Arunrat, N.; Sansupa, C.; Kongsurakan, P.; Sereenonchai, S.; Hatano, R. Soil Microbial Diversity and Community Composition in Rice–Fish Co-Culture and Rice Monoculture Farming System. Biology 2022, 11, 1242. [Google Scholar] [CrossRef]
- Feng, X.; Wang, Z.; Li, X.; Wang, W.; Gu, A.; Liu, Y. Analysis of Endophytic Bacterial Community Structure and Diversity of Characteristic Rice Germplasm Resources in Yunnan Province, China. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.X.; Zhang, N.; Hu, B.; Jin, T.; Xu, H.; Qin, Y.; Yan, P.; Zhang, X.; Guo, X.; et al. NRT1.1B Is Associated with Root Microbiota Composition and Nitrogen Use in Field-Grown Rice. Nat. Biotechnol. 2019, 37, 676–684. [Google Scholar] [CrossRef]
- Masuda, Y.; Satoh, S.; Miyamoto, R.; Takano, R.; Ishii, K.; Ohba, H.; Shiratori, Y.; Senoo, K. Biological Nitrogen Fixation in the Long-Term Nitrogen-Fertilized and Unfertilized Paddy Fields, with Special Reference to Diazotrophic Iron-Reducing Bacteria. Arch. Microbiol. 2023, 205, 291. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Xu, J.; Peng, L.; Cheng, Z.; Kuang, X.; Li, D.; Peng, C.; Song, H. Cadmium Accumulation in Rice Grains Is Mitigated by Duckweed-like Hydrophyte through Adsorption and Increased Ammonia Nitrogen. Sci. Total Environ. 2023, 890, 164510. [Google Scholar] [CrossRef]
- Mahmud, F.M.A.; Islam, M.A.; Rubel, M.H.; Mukharjee, S.K.; Kumar, M.; Bhattacharya, P.; Ahmed, F. Effects of Halotolerant Rhizobacteria on Rice Seedlings under Salinity Stress. Sci. Total Environ. 2023, 892, 163774. [Google Scholar] [CrossRef] [PubMed]
- Shan, S.; Wei, Z.; Cheng, W.; Du, D.; Zheng, D.; Ma, G. Biofertilizer Based on Halotolerant Microorganisms Promotes the Growth of Rice Plants and Alleviates the Effects of Saline Stress. Front. Microbiol. 2023, 14, 1165631. [Google Scholar] [CrossRef]
- Hernández, I.; Taulé, C.; Pérez-Pérez, R.; Battistoni, F.; Fabiano, E.; Villanueva-Guerrero, A.; Nápoles, M.C.; Herrera, H. Endophytic Seed-Associated Bacteria as Plant Growth Promoters of Cuban Rice (Oryza sativa L.). Microorganisms 2023, 11, 2317. [Google Scholar] [CrossRef]
- Chaganti, C.; Phule, A.S.; Chandran, L.P.; Sonth, B.; Kavuru, V.P.B.; Govindannagari, R.; Sundaram, R.M. Silicate Solubilizing and Plant Growth Promoting Bacteria Interact with Biogenic Silica to Impart Heat Stress Tolerance in Rice by Modulating Physiology and Gene Expression. Front. Microbiol. 2023, 14, 1168415. [Google Scholar] [CrossRef] [PubMed]
- Islam, T.; Fatema; Hoque, M.N.; Gupta, D.R.; Mahmud, N.U.; Sakif, T.I.; Sharpe, A.G. Improvement of Growth, Yield and Associated Bacteriome of Rice by the Application of Probiotic Paraburkholderia and Delftia. Front. Microbiol. 2023, 14, 1212505. [Google Scholar] [CrossRef]
- Prodhan, M.Y.; Rahman, M.B.; Rahman, A.; Akbor, M.A.; Ghosh, S.; Nahar, M.N.-E.-N.; Simo; Shamsuzzoha, M.; Cho, K.M.; Haque, M.A. Characterization of Growth-Promoting Activities of Consortia of Chlorpyrifos Mineralizing Endophytic Bacteria Naturally Harboring in Rice Plants—A Potential Bio-Stimulant to Develop a Safe and Sustainable Agriculture. Microorganisms 2023, 11, 1821. [Google Scholar] [CrossRef]
- Shivaji, S.; Suresh, K.; Chaturvedi, P.; Dube, S.; Sengupta, S. Bacillus arsenicus Sp. Nov., an Arsenic-Resistant Bacterium Isolated from a Siderite Concretion in West Bengal, India. Int. J. Syst. Evol. Microbiol. 2005, 55, 1123–1127. [Google Scholar] [CrossRef]
- Nichols, D.; Cahoon, N.; Trakhtenberg, E.M.; Pham, L.; Mehta, A.; Belanger, A.; Kanigan, T.; Lewis, K.; Epstein, S.S. Use of Ichip for High-Throughput in Situ Cultivation of “uncultivable Microbial Species. Appl. Environ. Microbiol. 2010, 76, 2445–2450. [Google Scholar] [CrossRef]
- Aoi, Y.; Kinoshita, T.; Hata, T.; Ohta, H.; Obokata, H.; Tsuneda, S. Hollow-Fiber Membrane Chamber as a Device for in Situ Environmental Cultivation. Appl. Environ. Microbiol. 2009, 75, 3826–3833. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, D.K.; Khulan, A.; Kim, J. Development of a Novel Cultivation Technique for Uncultured Soil Bacteria. Sci. Rep. 2019, 9, 6666. [Google Scholar] [CrossRef] [PubMed]
- Svenning, M.M.; Wartiainen, I.; Hestnes, A.G.; Binnerup, S.J. Isolation of Methane Oxidising Bacteria from Soil by Use of a Soil Substrate Membrane System. FEMS Microbiol. Ecol. 2003, 44, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Girguis, P.R.; Buie, C.R. Nanoporous Microscale Microbial Incubators. Lab Chip 2016, 16, 480–488. [Google Scholar] [CrossRef]
- Kaminski, T.S.; Scheler, O.; Garstecki, P. Droplet Microfluidics for Microbiology: Techniques, Applications and Challenges. Lab Chip 2016, 16, 2168–2187. [Google Scholar] [CrossRef]
- Du, W.; Li, L.; Nichols, K.P.; Ismagilov, R.F. SlipChip. Lab Chip 2009, 9, 2286–2292. [Google Scholar] [CrossRef]
- Lau, A.Y.; Lee, L.P.; Chan, J.W. An Integrated Optofluidic Platform for Raman-Activated Cell Sorting. Lab Chip 2008, 8, 1116–1120. [Google Scholar] [CrossRef]
- Batani, G.; Bayer, K.; Böge, J.; Hentschel, U.; Thomas, T. Fluorescence in Situ Hybridization (FISH) and Cell Sorting of Living Bacteria. Sci. Rep. 2019, 9, 18618. [Google Scholar] [CrossRef]
- Cross, K.L.; Campbell, J.H.; Balachandran, M.; Campbell, A.G.; Cooper, S.J.; Griffen, A.; Heaton, M.; Joshi, S.; Klingeman, D.; Leys, E.; et al. Targeted Isolation and Cultivation of Uncultivated Bacteria by Reverse Genomics. Nat. Biotechnol. 2019, 37, 1314–1321. [Google Scholar] [CrossRef]
- Lewis, W.H.; Tahon, G.; Geesink, P.; Sousa, D.Z.; Ettema, T.J.G. Innovations to Culturing the Uncultured Microbial Majority. Nat. Rev. Microbiol. 2021, 19, 225–240. [Google Scholar] [CrossRef]
- Lagier, J.C.; Million, M.; Hugon, P.; Armougom, F.; Raoult, D. Human Gut Microbiota: Repertoire and Variations. Front. Cell Infect. Microbiol. 2012, 2, 136. [Google Scholar] [CrossRef] [PubMed]
- Fournier, P.E.; Lagier, J.C.; Dubourg, G.; Raoult, D. From Culturomics to Taxonomogenomics: A Need to Change the Taxonomy of Prokaryotes in Clinical Microbiology. Anaerobe 2015, 36, 73–78. [Google Scholar] [CrossRef]
- Kambouris, M.E.; Pavlidis, C.; Skoufas, E.; Arabatzis, M.; Kantzanou, M.; Velegraki, A.; Patrinos, G.P. Culturomics: A New Kid on the Block of OMICS to Enable Personalized Medicine. OMICS 2018, 22, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Sheth, R.U.; Zhao, S.; Cohen, L.A.; Dabaghi, K.; Moody, T.; Sun, Y.; Ricaurte, D.; Richardson, M.; Velez-Cortes, F.; et al. High-Throughput Microbial Culturomics Using Automation and Machine Learning. Nat. Biotechnol. 2023, 41, 1424–1433. [Google Scholar] [CrossRef]
- Dubourg, G.; Morand, A.; Mekhalif, F.; Godefroy, R.; Corthier, A.; Yacouba, A.; Diakite, A.; Cornu, F.; Cresci, M.; Brahimi, S.; et al. Deciphering the Urinary Microbiota Repertoire by Culturomics Reveals Mostly Anaerobic Bacteria From the Gut. Front. Microbiol. 2020, 11, 513305. [Google Scholar] [CrossRef] [PubMed]
- Hamad, I.; Ranque, S.; Azhar, E.I.; Yasir, M.; Jiman-Fatani, A.A.; Tissot-Dupont, H.; Raoult, D.; Bittar, F. Culturomics and Amplicon-Based Metagenomic Approaches for the Study of Fungal Population in Human Gut Microbiota. Sci. Rep. 2017, 7, 16788. [Google Scholar] [CrossRef]
- Dubourg, G.; Lagier, J.C.; Robert, C.; Armougom, F.; Hugon, P.; Metidji, S.; Dione, N.; Dangui, N.P.M.; Pfleiderer, A.; Abrahao, J.; et al. Culturomics and Pyrosequencing Evidence of the Reduction in Gut Microbiota Diversity in Patients with Broad-Spectrum Antibiotics. Int. J. Antimicrob. Agents 2014, 44, 117–124. [Google Scholar] [CrossRef]
- Pfleiderer, A.; Lagier, J.C.; Armougom, F.; Robert, C.; Vialettes, B.; Raoult, D. Culturomics Identified 11 New Bacterial Species from a Single Anorexia Nervosa Stool Sample. Eur. J. Clin. Microbiol. Infect. Dis. 2013, 32, 1471–1481. [Google Scholar] [CrossRef]
- Greub, G. Culturomics: A New Approach to Study the Human Microbiome. Clin. Microbiol. Infect. 2012, 18, 1157–1159. [Google Scholar] [CrossRef]
- Haney, C.H.; Samuel, B.S.; Bush, J.; Ausubel, F.M. Associations with Rhizosphere Bacteria Can Confer an Adaptive Advantage to Plants. Nat. Plants 2015, 1, 15051. [Google Scholar] [CrossRef]
- Hu, Y.L.; Dai, R.; Liu, Y.X.; Zhang, J.Y.; Hu, B.; Chu, C.C.; Yuan, H.B.; Bai, Y. Analysis of Rice Root Bacterial Microbiota of Nipponbare and IR24. Yi Chuan 2020, 42, 506–518. [Google Scholar] [CrossRef]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; Del Rio, T.G.; et al. Defining the Core Arabidopsis Thaliana Root Microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Tkacz, A.; Cheema, J.; Chandra, G.; Grant, A.; Poole, P.S. Stability and Succession of the Rhizosphere Microbiota Depends upon Plant Type and Soil Composition. ISME J. 2015, 9, 2349–2359. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Lyu, F.; Han, F.; Ge, C.; Mao, W.; Chen, L.; Hu, H.; Chen, G.; Lang, Q.; Fang, C. OmicStudio: A Composable Bioinformatics Cloud Platform with Real-Time Feedback That Can Generate High-Quality Graphs for Publication. iMeta 2023, 2, e85. [Google Scholar] [CrossRef]
- OmicStudio. Available online: https://www.omicstudio.cn/home (accessed on 10 November 2023).
- Somerfield, P.J. Identification of the Bray-Curtis Similarity Index: Comment on Yoshioka. Mar. Ecol. Prog. Ser. 2008, 372, 303–306. [Google Scholar] [CrossRef]
- Chung, N.C.; Miasojedow, B.Z.; Startek, M.; Gambin, A. Jaccard/Tanimoto Similarity Test and Estimation Methods for Biological Presence-Absence Data. BMC Bioinform. 2019, 20, 644. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Jiang, Y.H.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular Ecological Network Analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- MENA. Available online: http://ieg4.rccc.ou.edu/mena/main.cgi (accessed on 20 August 2023).
- Chen, T.; Zhang, H.; Liu, Y.; Liu, Y.X.; Huang, L. EVenn: Easy to Create Repeatable and Editable Venn Diagrams and Venn Networks Online. J. Genet. Genom. 2021, 48, 863–866. [Google Scholar] [CrossRef]
- Evenn. Available online: http://www.ehbio.com/test/venn/#/ (accessed on 10 July 2023).
- Bioinformatics. Available online: https://www.bioinformatics.com.cn (accessed on 10 July 2023).
- Lagier, J.C.; Dubourg, G.; Million, M.; Cadoret, F.; Bilen, M.; Fenollar, F.; Levasseur, A.; Rolain, J.M.; Fournier, P.E.; Raoult, D. Culturing the Human Microbiota and Culturomics. Nat. Rev. Microbiol. 2018, 16, 540–550. [Google Scholar] [CrossRef]
- Xu, J.; Sun, L.; Sun, Z.; Xing, X.; Li, J.; Lu, X.; Huang, Y.; Ren, Q. Pontibacter beigongshangensis Sp. Nov., Isolated from the Mash of Wine. Curr. Microbiol. 2019, 76, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Ni, C.; Sugimoto, C.R.; Jiang, J. Degree, Closeness, and Betweenness: Application of Group Centrality Measurements to Explore Macro-Disciplinary Evolution Diachronically. In Proceedings of the ISSI 2011—13th Conference of the International Society for Scientometrics and Informetrics, Durban, South Africa, 4–7 July 2011; Volume 2. [Google Scholar]
- Evans, T.S.; Chen, B. Linking the Network Centrality Measures Closeness and Degree. Commun. Phys. 2022, 5, 172. [Google Scholar] [CrossRef]
- Sanford, R.A.; Cole, J.R.; Tiedje, J.M. Characterization and Description of Anaeromyxobacter dehalogenans Gen. Nov., Sp. Nov., an Aryl-Halorespiring Facultative Anaerobic Myxobacterium. Appl. Environ. Microbiol. 2002, 68, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, I.; Bindschedler, S.; Junier, P. Chapter 18—Firmicutes. In Beneficial Microbes in Agro-Ecology; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar]
- Kersters, K.; De Vos, P.; Gillis, M.; Swings, J.; Vandamme, P.; Stackebrandt, E. Introduction to the Proteobacteria. In The Prokaryotes; Springer: New York, NY, USA, 2006. [Google Scholar]
- Hanada, S. The Phylum Chloroflexi, the Family Chloroflexaceae, and the Related Phototrophic Families Oscillochloridaceae and Roseiflexaceae. In The Prokaryotes: Other Major Lineages of Bacteria and the Archaea; Springer: Berlin/Heidelberg, Germany, 2014; Volume 9783642389542. [Google Scholar]
- Ul-Hassan, A.; Wellington, E.M. Actinobacteria. In Encyclopedia of Microbiology, 3rd ed.; Springer: University of Warwick, Coventry, UK, 2009; pp. 25–44. [Google Scholar] [CrossRef]
- Kielak, A.M.; Barreto, C.C.; Kowalchuk, G.A.; van Veen, J.A.; Kuramae, E.E. The Ecology of Acidobacteria: Moving beyond Genes and Genomes. Front. Microbiol. 2016, 7, 74. [Google Scholar] [CrossRef]
- Orizola, J.; Ríos-Silva, M.; Muñoz-Villagrán, C.; Vargas, E.; Vásquez, C.; Arenas, F. In Vitro Biosynthesis of Ag, Au and Te-Containing Nanostructures by Exiguobacterium Cell-Free Extracts. BMC Biotechnol. 2020, 20, 29. [Google Scholar] [CrossRef]
- Ali, M.; Ahmed, I.; Tariq, H.; Abbas, S.; Zia, M.H.; Mumtaz, A.; Sharif, M. Growth Improvement of Wheat (Triticum Aestivum) and Zinc Biofortification Using Potent Zinc-Solubilizing Bacteria. Front. Plant Sci. 2023, 14, 1140454. [Google Scholar] [CrossRef]
- Marfetán, J.A.; Gallo, A.L.; Farias, M.E.; Vélez, M.L.; Pescuma, M.; Ordoñez, O.F. Exiguobacterium Sp. as a Bioinoculant for Plant-Growth Promotion and Selenium Biofortification Strategies in Horticultural Plants. World J. Microbiol. Biotechnol. 2023, 39, 134. [Google Scholar] [CrossRef]
- Wu, J.; Song, Q.; Wu, Y.; Liu, J.; Wu, Z.; Zhou, J.; Wang, Y.; Wu, W. Application of Phosphorus Amendments Reduces Metal Uptake and Increases Yield of Oryza Saliva L. (Rice) in Cd/Cu-Contaminated Paddy Field. Chemosphere 2023, 318, 137875. [Google Scholar] [CrossRef]
- Selvakumar, G.; Kundu, S.; Joshi, P.; Nazim, S.; Gupta, A.D.; Gupta, H.S. Growth Promotion of Wheat Seedlings by Exiguobacterium Acetylicum 1P (MTCC 8707) a Cold Tolerant Bacterial Strain from the Uttarakhand Himalayas. Indian J. Microbiol. 2010, 50, 50–56. [Google Scholar] [CrossRef]
- Cui, Y.; Cui, Y.W.; Huang, J.L. A Novel Halophilic Exiguobacterium mexicanum Strain Removes Nitrogen from Saline Wastewater via Heterotrophic Nitrification and Aerobic Denitrification. Bioresour. Technol. 2021, 333, 125189. [Google Scholar] [CrossRef]
- Zhang, D.; Zhu, Z.; Li, Y.; Li, X.; Guan, Z.; Zheng, J. Comparative Genomics of Exiguobacterium Reveals What Makes a Cosmopolitan Bacterium. mSystems 2021, 6, e0038321. [Google Scholar] [CrossRef]
- Lagier, J.C.; Armougom, F.; Million, M.; Hugon, P.; Pagnier, I.; Robert, C.; Bittar, F.; Fournous, G.; Gimenez, G.; Maraninchi, M.; et al. Microbial Culturomics: Paradigm Shift in the Human Gut Microbiome Study. Clin. Microbiol. Infect. 2012, 18, 1185–1193. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.W.; Lian, Z.H.; Liu, L.; Fang, B.Z.; Li, W.J.; Jiao, J.Y. Cultivation Strategies for Prokaryotes from Extreme Environments. iMeta 2023, 2, e123. [Google Scholar] [CrossRef]
- Agler, M.T.; Ruhe, J.; Kroll, S.; Morhenn, C.; Kim, S.T.; Weigel, D.; Kemen, E.M. Microbial Hub Taxa Link Host and Abiotic Factors to Plant Microbiome Variation. PLoS Biol 2016, 14, e1002352. [Google Scholar] [CrossRef]
- Wang, H.Q.; Zhao, X.Y.; Xuan, W.; Wang, P.; Zhao, F.J. Rice Roots Avoid Asymmetric Heavy Metal and Salinity Stress via an RBOH-ROS-Auxin Signaling Cascade. Mol. Plant 2023, 16, 1678–1694. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Tang, L.H.; Nie, J.W.; Zhang, C.R.; Han, X.; Li, Q.Y.; Qin, L.; Wang, M.H.; Huang, X.; Yu, F.; et al. Structure and Activation Mechanism of the Rice Salt Overly Sensitive 1 (SOS1) Na+/H+ Antiporter. Nat. Plants 2023, 9, 1924–1936. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yao, Z.; Zhan, Y.; Zheng, X.; Zhou, M.; Yan, G.; Wang, L.; Werner, C.; Butterbach-Bahl, K. Potential Benefits of Liming to Acid Soils on Climate Change Mitigation and Food Security. Glob. Chang. Biol. 2021, 27, 2807–2821. [Google Scholar] [CrossRef]
- Liu, R.; Wang, Y.; Hong, Y.; Wang, F.; Mao, X.; Yi, J. Controlled-Release Urea Application and Optimized Nitrogen Applied Strategy Reduced Nitrogen Leaching and Maintained Grain Yield of Paddy Fields in Northwest China. Front. Plant Sci. 2023, 14, 1033506. [Google Scholar] [CrossRef]
- Zhou, W.; Guo, Z.; Chen, J.; Jiang, J.; Hui, D.; Wang, X.; Sheng, J.; Chen, L.; Luo, Y.; Zheng, J.; et al. Direct Seeding for Rice Production Increased Soil Erosion and Phosphorus Runoff Losses in Subtropical China. Sci. Total Environ. 2019, 695, 133845. [Google Scholar] [CrossRef]
- Zuo, W.; Song, B.; Shi, Y.; Zupanic, A.; Guo, S.; Huang, H.; Jiang, L.; Yu, Y. Using Bacillus Thuringiensis HM-311@hydroxyapatite@biochar Beads to Remediate Pb and Cd Contaminated Farmland Soil. Chemosphere 2022, 307, 135797. [Google Scholar] [CrossRef]
- Zou, W.; Zhao, C.; Chen, J.; Wang, Y.; Jin, C.; Zhang, X. Systematic Stress Persistence and Recovery Patterns of Rice (Oryza sativa L.) Roots in Response to Molybdenum Disulfide Nanosheets. Chemosphere 2023, 321, 138166. [Google Scholar] [CrossRef]
- Zou, M.; Zhou, S.; Zhou, Y.; Jia, Z.; Guo, T.; Wang, J. Cadmium Pollution of Soil-Rice Ecosystems in Rice Cultivation Dominated Regions in China: A Review. Environ. Pollut. 2021, 280, 116965. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.K.; Du, P.P.; Zeng, L.J.; Lü, H.; Zhao, H.M.; Li, Y.W.; Mo, C.H.; Cai, Q.Y. Variation in Metabolism and Degradation of Di-n-Butyl Phthalate (DBP) by High- and Low-DBP Accumulating Cultivars of Rice (Oryza sativa L.) and Crude Enzyme Extracts. Sci. Total Environ. 2019, 668, 1117–1127. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, W.; Shao, S.; Zhang, S.; Cheng, X.; Ye, Q. Fate Characteristics of the Chiral Pesticide Dufulin in Flooded Anaerobic Soils and Its Interaction with Soil Microorganisms. Sci. Total Environ. 2023, 878, 162983. [Google Scholar] [CrossRef] [PubMed]
- Philippot, L.; Chenu, C.; Kappler, A.; Rillig, M.C.; Fierer, N. The Interplay between Microbial Communities and Soil Properties. Nat. Rev. Microbiol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Russ, D.; Fitzpatrick, C.R.; Teixeira, P.J.P.L.; Dangl, J.L. Deep Discovery Informs Difficult Deployment in Plant Microbiome Science. Cell 2023, 186, 4496–4513. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phylum | Number of Species Effectively Published | Number of Potential New Species | Increase in Percentage |
|---|---|---|---|
| Proteobacteria | 7484 | 17 | 0.23% |
| Actinobacteria | 4298 | 9 | 0.21% |
| Firmicutes | 3364 | 2 | 0.06% |
| Bacteroidetes | 2463 | 18 | 0.73% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Cao, Q.; Ruan, W.; Guo, Y.; Zhuang, Y.; Li, Y.; Ruan, Z. Culturomics and Amplicon-Based Metagenomic Insights into the Bacteria of Soils with High Yield of Oryza sativa L. subsp. Japonica. Agronomy 2023, 13, 2867. https://doi.org/10.3390/agronomy13122867
Zhang L, Cao Q, Ruan W, Guo Y, Zhuang Y, Li Y, Ruan Z. Culturomics and Amplicon-Based Metagenomic Insights into the Bacteria of Soils with High Yield of Oryza sativa L. subsp. Japonica. Agronomy. 2023; 13(12):2867. https://doi.org/10.3390/agronomy13122867
Chicago/Turabian StyleZhang, Li, Qingmei Cao, Wenzhong Ruan, Yapeng Guo, Yan Zhuang, Yan Li, and Zhiyong Ruan. 2023. "Culturomics and Amplicon-Based Metagenomic Insights into the Bacteria of Soils with High Yield of Oryza sativa L. subsp. Japonica" Agronomy 13, no. 12: 2867. https://doi.org/10.3390/agronomy13122867