
Proteomics Analysis Reveals Hormone Metabolic Process Involved in the Regulation of Kernel Water Content Induced by Exogenous Abscisic Acid in Maize

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Field Design and Plant Materials
2.2. Grain Yield Determinations
2.3. Grain-Filling Characteristics and Kernel Water Content
2.4. Protein Extraction
2.5. Proteomics Sample Pretreatment
2.6. LC-MS/MS-Based Proteomic Analysis
2.7. Data Processing for Proteomics
2.8. Bioinformatic Analysis
2.9. Quantitative Real-Time (q-RT) PCR Analysis
2.10. Statistical Analysis
3. Results
3.1. Effects of Exogenous ABA on Yield in Two Maize Genotypes
3.2. Effects of Exogenous ABA on Dynamic Changes in the Grain Water Content
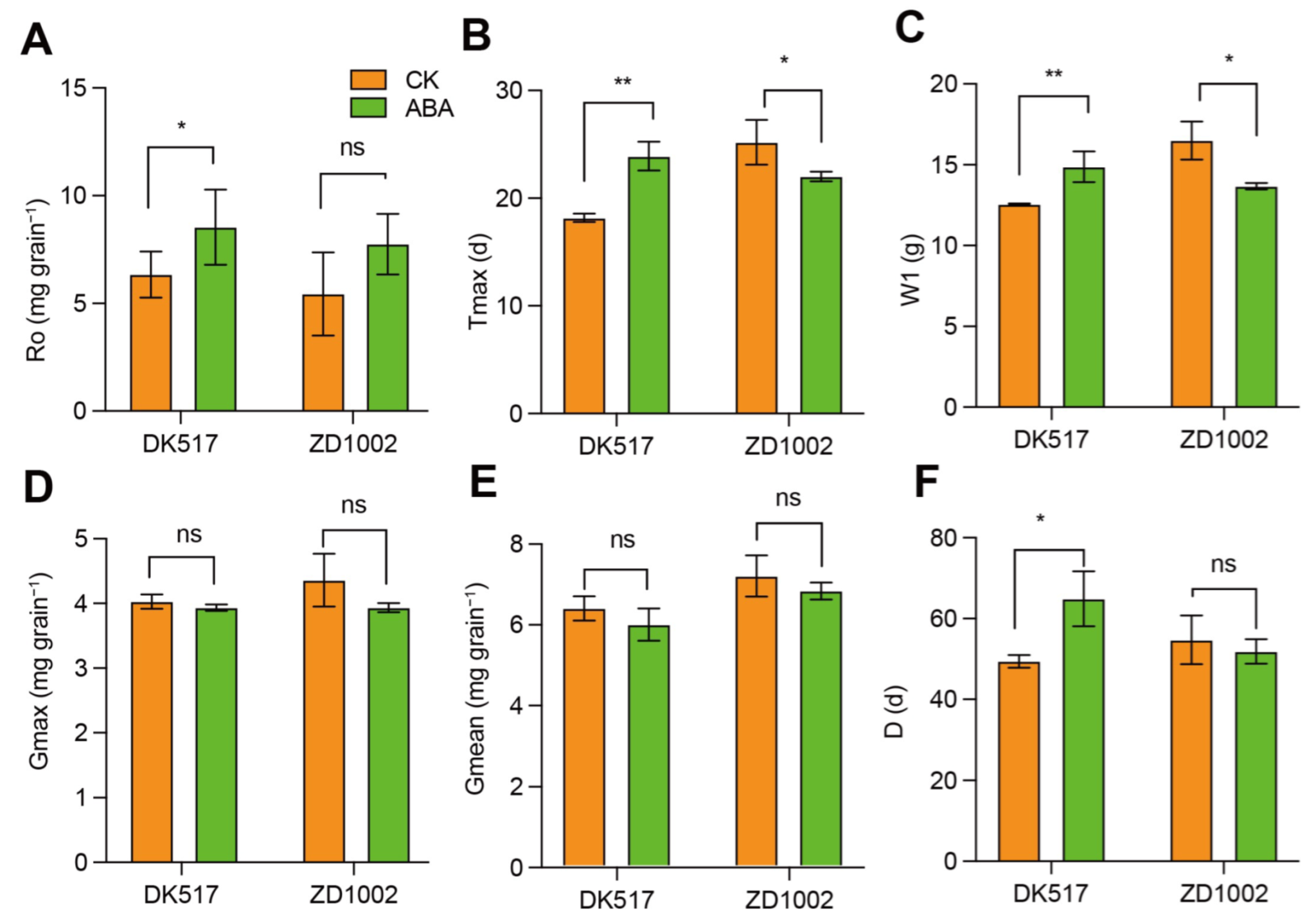
3.3. Effect of Exogenous ABA on Grain-Filling Characteristics
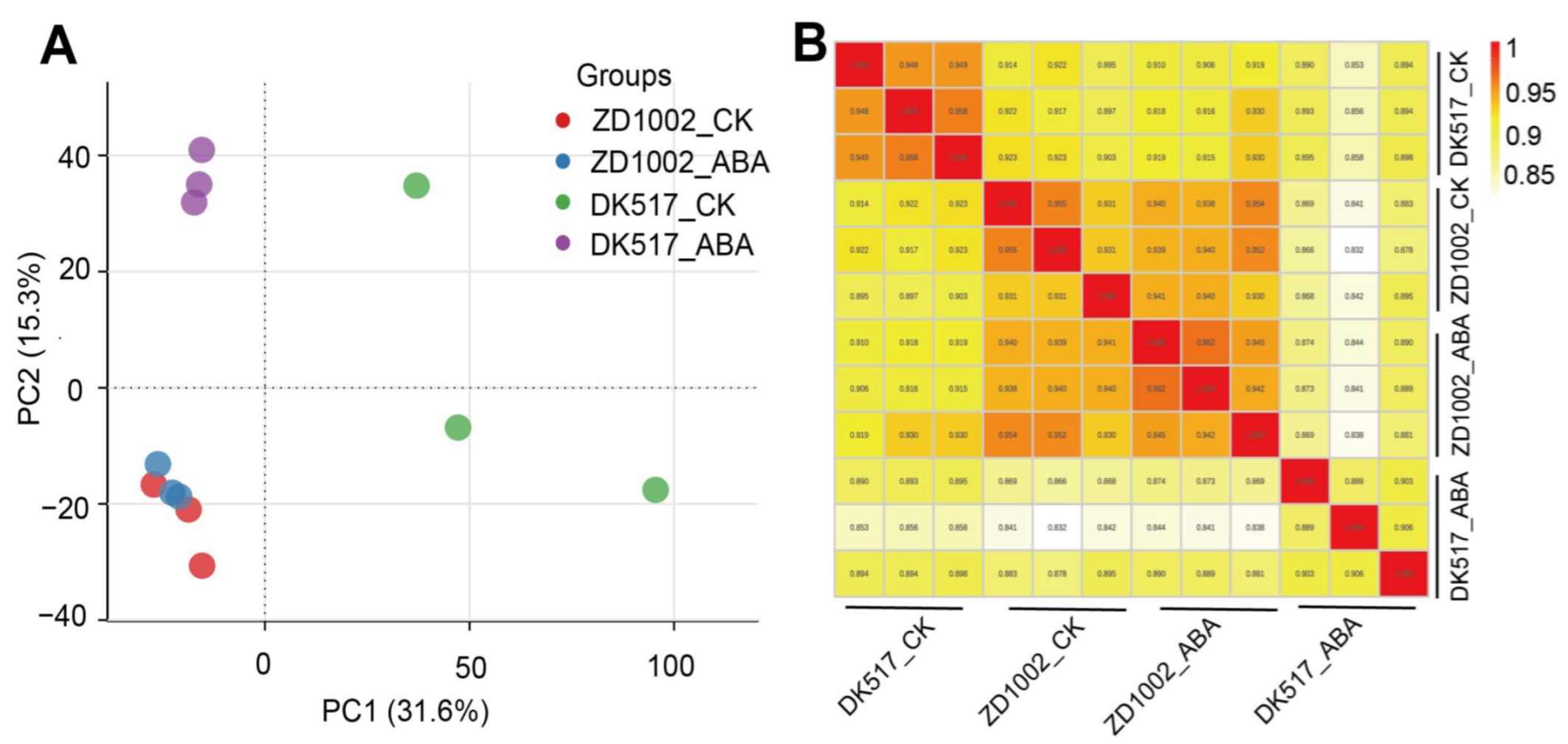
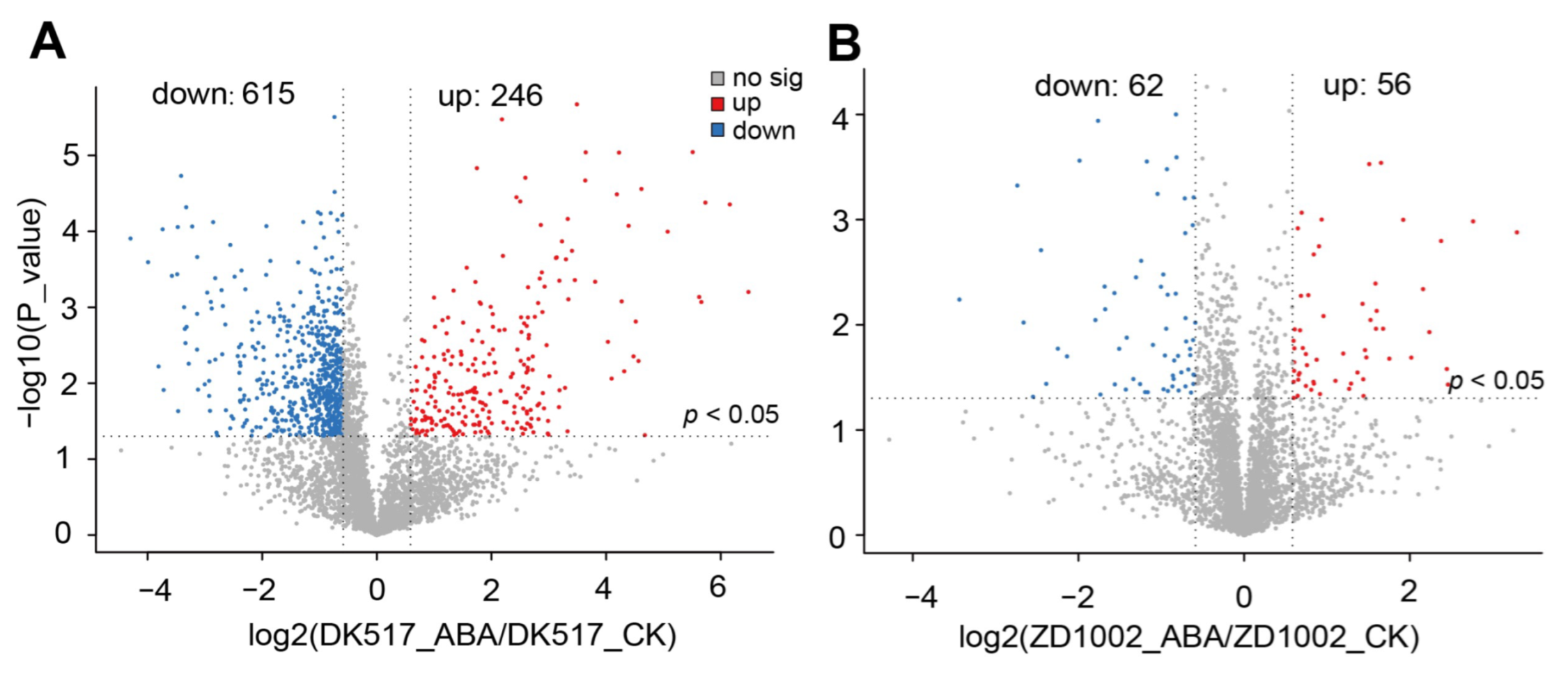
3.4. Screening of Differential Proteins in Each Maize Variety
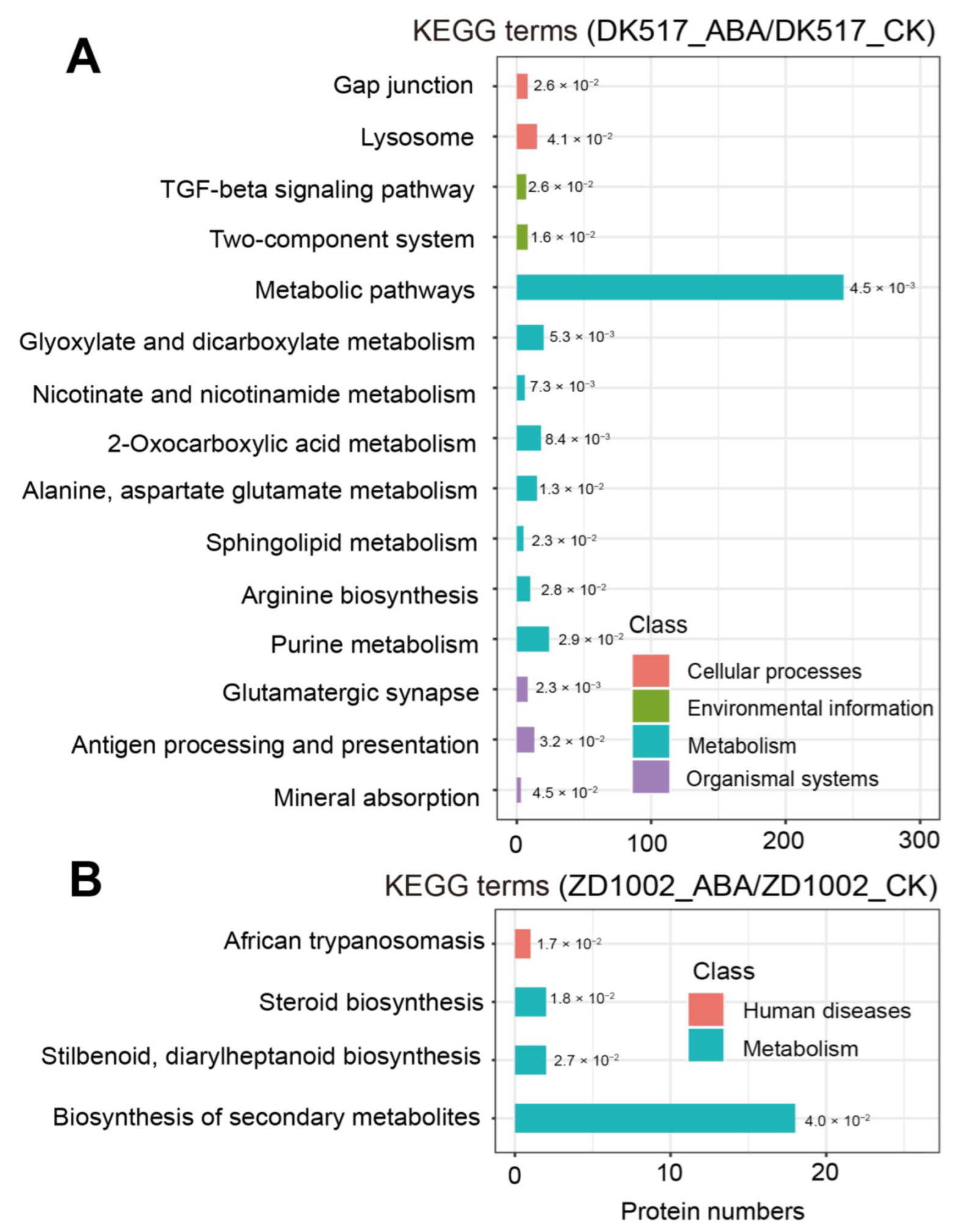
3.5. Proteomic Analysis of DK517 Treated with Exogenous ABA
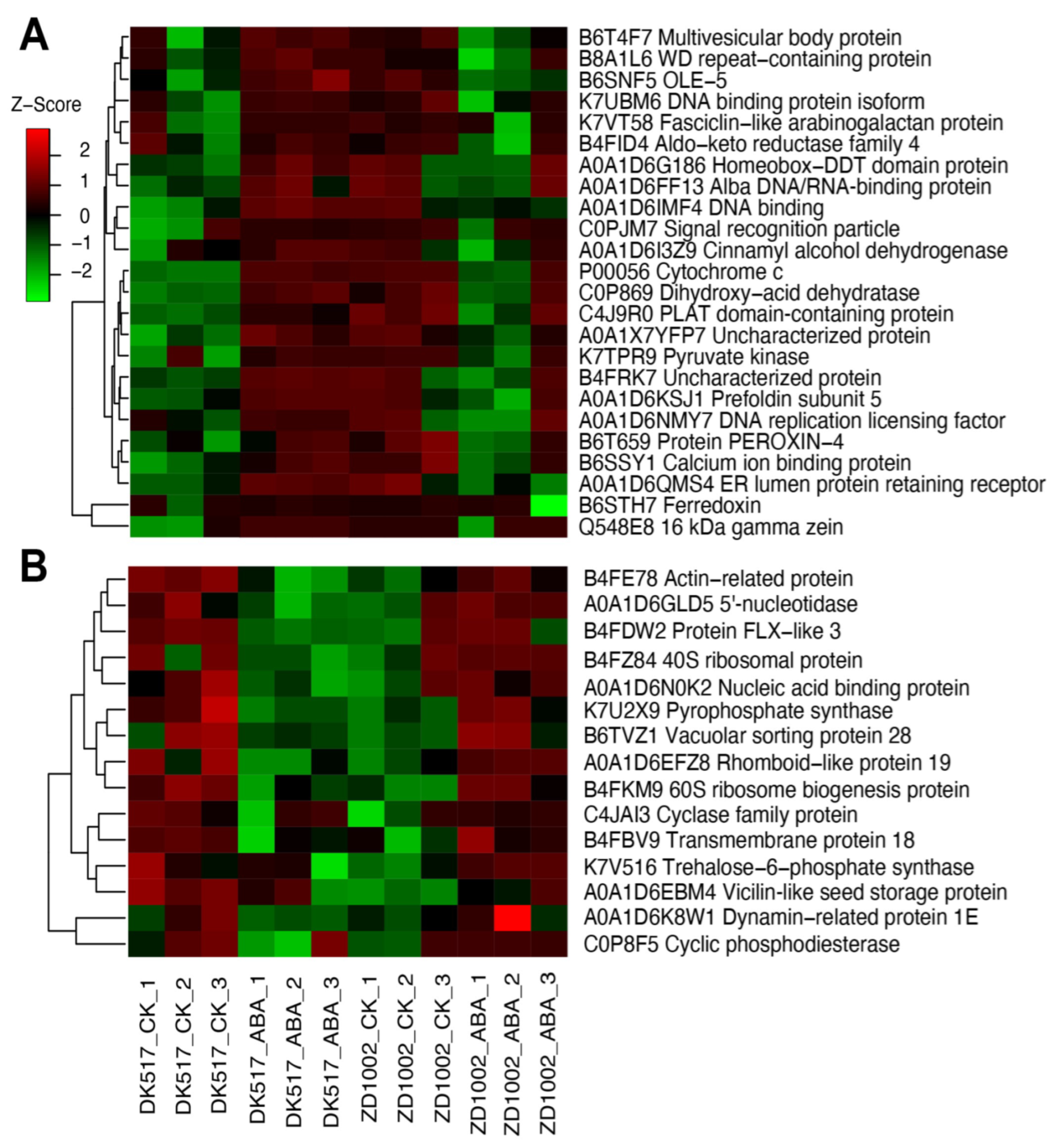
3.6. Key DAPs Associated with Kernel Water Contents Induced by ABA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Revilla, P.; Alves, M.L.; Andelković, V.; Balconi, C.; Dinis, I.; Mendes-Moreira, P.; Redaelli, R.; Ruiz de Galarreta, J.I.; Vaz Patto, M.C.; Žilić, S. Traditional foods from maize (Zea mays L.) in Europe. Front. Nutr. 2022, 8, 683399. [Google Scholar] [CrossRef] [PubMed]
- Hasan, K.; Tanaka, T.S.; Alam, M.; Ali, R.; Saha, C.K. Impact of modern rice harvesting practices over traditional ones. Rev. Agric. Sci. 2020, 8, 89–108. [Google Scholar] [CrossRef] [PubMed]
- Isaak, M.; Yahya, A.; Razif, M.; Mat, N. Mechanization status based on machinery utilization and workers’ workload in sweet corn cultivation in Malaysia. Comput. Electron. Agric. 2020, 169, 105208. [Google Scholar] [CrossRef]
- Jetté-Nantel, S.; Hu, W.; Liu, Y. Economies of scale and mechanization in Chinese corn and wheat production. Appl. Econ. 2020, 52, 2751–2765. [Google Scholar] [CrossRef]
- Bamboriya, S.; Jat, S.; Shreelatha, D.; Mahala, D.; Rakshit, S. Mechanized maize production for enhanced productivity and profitability. IIMR Tech. Bull. 2020, 1, 4–46. [Google Scholar]
- Chazarreta, Y.D.; Carcedo, A.J.; Prado, S.A.; Massigoge, I.; Amas, J.I.; Fernandez, J.A.; Ciampitti, I.A.; Otegui, M.E. Enhancing maize grain dry-down predictive models. Agric. For. Meteorol. 2023, 334, 109427. [Google Scholar] [CrossRef]
- Shahini, E.; Shehu, D.; Kovalenko, O.; Nikonchuk, N. Comparative analysis of the main economic and biological parameters of maize hybrids that determine their productivity. Sci. Horiz. 2023, 26, 86–96. [Google Scholar] [CrossRef]
- Shuqi, S.; Jiasheng, W.; Dongwei, W. Development of Agricultural Mechanization in China and Its Current Strategic Focus. Agric. Mach. Technol. 2019, 13, 4–7. [Google Scholar] [CrossRef]
- Alam, M.R.; Nakasathien, S.; Molla, M.S.H.; Islam, M.A.; Maniruzzaman, M.; Ali, M.A.; Sarobol, E.; Vichukit, V.; Hassan, M.M.; Dessoky, E.S. Kernel water relations and kernel filling traits in maize (Zea mays L.) are influenced by water-deficit condition in a tropical environment. Front. Plant Sci. 2021, 12, 717178. [Google Scholar] [CrossRef]
- Becerra-Sanchez, F.; Taylor, G. Reducing post-harvest losses and improving quality in sweet corn (Zea mays L.): Challenges and solutions for less food waste and improved food security. Food Energy Secur. 2021, 10, e277. [Google Scholar] [CrossRef]
- Gambin, B.L.; Di Salvo, J.I.; Sciarresi, C.; Trifunovic, S.; Narvel, J.; Zhou, X.; Lamkey, K.; Archontoulis, S.V. Maize kernel weight genetic gain is achieved through different mechanisms depending on the hybrid maturity. Field Crops Res. 2023, 303, 109123. [Google Scholar] [CrossRef]
- Li, W.; Yu, Y.; Wang, L.; Luo, Y.; Peng, Y.; Xu, Y.; Liu, X.; Wu, S.; Jian, L.; Xu, J. The genetic architecture of the dynamic changes in grain moisture in maize. Plant Biotechnol. J. 2021, 19, 1195–1205. [Google Scholar] [CrossRef]
- Maiorano, A.; Fanchini, D.; Donatelli, M. MIMYCS.Moisture, a process-based model of moisture content in developing maize kernels. Eur. J. Agron. 2014, 59, 86–95. [Google Scholar] [CrossRef]
- Suganya, A.; Saravanan, A.; Manivannan, N. Role of zinc nutrition for increasing zinc availability, uptake, yield, and quality of maize (Zea mays L.) grains: An overview. Commun. Soil Sci. Plant Anal. 2020, 51, 2001–2021. [Google Scholar]
- Ordóñez, R.A.; Savin, R.; Cossani, C.M.; Slafer, G.A. Maize grain weight sensitivity to source–sink manipulations under a wide range of field conditions. Crop Sci. 2018, 58, 2542–2557. [Google Scholar] [CrossRef]
- Ramos-Fuentes, I.A.; Elamri, Y.; Cheviron, B.; Dejean, C.; Belaud, G.; Fumey, D. Effects of shade and deficit irrigation on maize growth and development in fixed and dynamic agrivoltaic systems. Agric. Water Manag. 2023, 280, 108187. [Google Scholar] [CrossRef]
- Paul, S.; Roychoudhury, A. Comparative analysis of the expression of candidate genes governing salt tolerance and yield attributes in two contrasting rice genotypes, encountering salt stress during grain development. J. Plant Growth Regul. 2019, 38, 539–556. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, Y.; Chen, T.; Zhang, H.; Yang, J.; Zhang, J. Abscisic acid and the key enzymes and genes in sucrose-to-starch conversion in rice spikelets in response to soil drying during grain filling. Planta 2015, 241, 1091–1107. [Google Scholar] [CrossRef]
- Li, Y.; Yu, G.; Lv, Y.; Long, T.; Li, P.; Hu, Y.; Liu, H.; Zhang, J.; Liu, Y.; Li, W.-C. Combinatorial interaction of two adjacent cis-active promoter regions mediates the synergistic induction of Bt2 gene by sucrose and ABA in maize endosperm. Plant Sci. 2018, 274, 332–340. [Google Scholar] [CrossRef]
- Zhang, L.; Liang, X.-G.; Shen, S.; Yin, H.; Zhou, L.-L.; Gao, Z.; Lv, X.-Y.; Zhou, S.-L. Increasing the abscisic acid level in maize grains induces precocious maturation by accelerating grain filling and dehydration. Plant Growth Regul. 2018, 86, 65–79. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, M.; Wang, Y.; Liang, J. RGB1 regulates rice panicle architecture and grain filling through monitoring cytokinin level in inflorescence meristem and grain abscisic acid level during filling stage. Rice Sci. 2021, 28, 317. [Google Scholar]
- Ahmad, S.; Kamran, M.; Zhou, X.; Ahmad, I.; Meng, X.; Javed, T.; Iqbal, A.; Wang, G.; Su, W.; Wu, X. Melatonin improves the seed filling rate and endogenous hormonal mechanism in grains of summer maize. Physiol. Plant. 2021, 172, 1059–1072. [Google Scholar] [CrossRef]
- Tsukaguchi, T.; Tanaka, R.; Inoue, H.; Nakagawa, H. Effects of high temperature and shading on grain abscisic acid content and grain filling pattern in rice (Oryza sativa L.). Plant Prod. Sci. 2018, 21, 407–412. [Google Scholar] [CrossRef]
- Perveen, S.; Qu, M.; Chen, F.; Essemine, J.; Khan, N.; Lyu, M.-J.A.; Chang, T.; Song, Q.; Chen, G.-Y.; Zhu, X.-G. Overexpression of maize transcription factor mEmBP-1 increases photosynthesis, biomass, and yield in rice. J. Exp. Bot. 2020, 71, 4944–4957. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.-x.; Li, J. The effects of exogenous ABA applied to maize (Zea mays L.) roots on plant responses to chilling stress. Acta Physiol. Plant. 2018, 40, 1–13. [Google Scholar] [CrossRef]
- Zhang, L.; Li, X.-H.; Gao, Z.; Shen, S.; Liang, X.-G.; Zhao, X.; Lin, S.; Zhou, S.-L. Regulation of maize kernel weight and carbohydrate metabolism by abscisic acid applied at the early and middle post-pollination stages in vitro. J. Plant Physiol. 2017, 216, 1–10. [Google Scholar] [CrossRef]
- Hu, J.; Ren, B.; Dong, S.; Liu, P.; Zhao, B.; Zhang, J. Comparative proteomic analysis reveals that exogenous 6-benzyladenine (6-BA) improves the defense system activity of waterlogged summer maize. BMC Plant Biol. 2020, 20, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.-F.; Shen, Z.; Chao, Q.; Yan, Z.; Ge, X.-L.; Lu, T.; Zheng, H.; Qian, C.-R.; Wang, B.-C. Large-scale proteomic and phosphoproteomic analyses of maize seedling leaves during de-etiolation. Genom. Proteom. Bioinform. 2020, 18, 397–414. [Google Scholar] [CrossRef]
- Labuschagne, M. A review of cereal grain proteomics and its potential for sorghum improvement. J. Cereal Sci. 2018, 84, 151–158. [Google Scholar] [CrossRef]
- Kim, J.-T.; Yi, G.; Kim, M.-J.; Son, B.-Y.; Bae, H.-H.; Go, Y.S.; Kim, S.-L.; Baek, S.-B.; Kim, S.-H.; Chung, I.-M. Glycolysis stimulation and storage protein accumulation are hallmarks of maize (Zea mays L.) grain filling. Appl. Biol. Chem. 2020, 63, 1–10. [Google Scholar] [CrossRef]
- Landolfi, V.; D’Auria, G.; Nicolai, M.A.; Nitride, C.; Blandino, M.; Ferranti, P. The effect of nitrogen fertilization on the expression of protein in wheat and tritordeum varieties using a proteomic approach. Food Res. Int. 2021, 148, 110617. [Google Scholar] [CrossRef]
- Yang, G.; Dong, Y.; Li, Y.; Wang, Q.; Shi, Q.; Zhou, Q. QTL verification of grain protein content and its correlation with oil content by using connected RIL populations of high-oil maize. Genet. Mol. Res. 2014, 13, 881–894. [Google Scholar] [CrossRef]
- Zhao, Q.; Lin, J.; Yousaf, L.; Xue, Y.; Shen, Q. Protein structural properties and proteomic analysis of rice during storage at different temperatures. Food Chem. 2021, 361, 130028. [Google Scholar] [CrossRef]
- Li, H.; Wang, Y.; Xue, J.; Xie, R.; Wang, K.; Zhao, R.; Liu, W.; Ming, B.; Hou, P.; Zhang, Z. Allocation of maize varieties according to temperature for use in mechanical kernel harvesting in Ningxia, China. Int. J. Agric. Biol. Eng. 2021, 14, 20–28. [Google Scholar] [CrossRef]
- Yang, D.; Luo, Y.; Ni, Y.; Yin, Y.; Yang, W.; Peng, D.; Cui, Z.; Wang, Z. Effects of exogenous ABA application on post-anthesis dry matter redistribution and grain starch accumulation of winter wheat with different staygreen characteristics. Crop J. 2014, 2, 144–153. [Google Scholar] [CrossRef]
- Bakry, M.; Abdel-Baky, N.F. Impact of the fall armyworm, Spodoptera frugiperda (Lepidoptera: Noctuidae) infestation on maize growth characteristics and yield loss. Braz. J. Biol. 2023, 84, e274602. [Google Scholar] [CrossRef]
- Liang, C.; Song, Y.; Guo, X.; Kong, D.; Wang, Y.; Zhao, Q.; Huang, K. Characteristics of the grain-filling process and starch accumulation of high-yield common buckwheat ‘cv. Fengtian 1’and tartary buckwheat ‘cv. Jingqiao 2’. Cereal Res. Commun. 2016, 44, 393–403. [Google Scholar] [CrossRef]
- Li, J.; Essemine, J.; Shang, C.; Zhang, H.; Zhu, X.; Yu, J.; Chen, G.; Qu, M.; Sun, D. Combined proteomics and metabolism analysis unravels prominent roles of antioxidant system in the prevention of alfalfa (Medicago sativa L.) against salt stress. Int. J. Mol. Sci. 2020, 21, 909. [Google Scholar] [CrossRef]
- Jiang, C.; Bi, Y.; Mo, J.; Zhang, R.; Qu, M.; Feng, S.; Essemine, J. Proteome and transcriptome reveal the involvement of heat shock proteins and antioxidant system in thermotolerance of Clematis florida. Sci. Rep. 2020, 10, 8883. [Google Scholar] [CrossRef]
- Karimi, F.; Hamidian, Y.; Behrouzifar, F.; Mostafazadeh, R.; Ghorbani-HasanSaraei, A.; Alizadeh, M.; Mortazavi, S.-M.; Janbazi, M.; Asrami, P.N. An applicable method for extraction of whole seeds protein and its determination through Bradford’s method. Food Chem. Toxicol. 2022, 164, 113053. [Google Scholar] [CrossRef] [PubMed]
- Välikangas, T.; Suomi, T.; Elo, L.L. A comprehensive evaluation of popular proteomics software workflows for label-free proteome quantification and imputation. Brief. Bioinform. 2018, 19, 1344–1355. [Google Scholar] [CrossRef]
- Yan, S.; Bhawal, R.; Yin, Z.; Thannhauser, T.W.; Zhang, S. Recent advances in proteomics and metabolomics in plants. Mol. Hortic. 2022, 2, 17. [Google Scholar] [CrossRef] [PubMed]
- Coudert, E.; Gehant, S.; de Castro, E.; Pozzato, M.; Baratin, D.; Neto, T.; Sigrist, C.J.; Redaschi, N.; Bridge, A. Annotation of biologically relevant ligands in UniProtKB using ChEBI. Bioinformatics 2023, 39, btac793. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; Von Mering, C.; Bork, P. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef]
- Sajulga, R.; Easterly, C.; Riffle, M.; Mesuere, B.; Muth, T.; Mehta, S.; Kumar, P.; Johnson, J.; Gruening, B.A.; Schiebenhoefer, H. Survey of metaproteomics software tools for functional microbiome analysis. PLoS ONE 2020, 15, e0241503. [Google Scholar] [CrossRef]
- Song, J.; Lu, D.; Niu, Y.; Sun, H.; Zhang, P.; Dong, W.; Li, Y.; Zhang, Y.; Lu, L.; Men, Q. Label-free quantitative proteomics of maize roots from different root zones provides insight into proteins associated with enhance water uptake. BMC Genom. 2022, 23, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Hamdani, S.; Wang, H.; Zheng, G.; Perveen, S.; Qu, M.; Khan, N.; Khan, W.; Jiang, J.; Li, M.; Liu, X. Genome-wide association study identifies variation of glucosidase being linked to natural variation of the maximal quantum yield of photosystem II. Physiol. Plant. 2019, 166, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Li, L.-L.; Bo, M.; Jun, X.; Shang, G.; Wang, K.-R.; Xie, R.-Z.; Peng, H.; Li, S.-K. Difference in corn kernel moisture content between pre-and post-harvest. J. Integr. Agric. 2021, 20, 1775–1782. [Google Scholar] [CrossRef]
- Minghui, D.; Junrong, G.; Peifeng, C. Difference of abscisic acid content in grains at different spikelet positions of rice and its relationship with grain filling. Plant Physiol. J. 2013, 49, 729–737. [Google Scholar]
- Zhu, G.; Ye, N.; Yang, J.; Peng, X.; Zhang, J. Regulation of expression of starch synthesis genes by ethylene and ABA in relation to the development of rice inferior and superior spikelets. J. Exp. Bot. 2011, 62, 3907–3916. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Zhai, H.; Wang, P.; Zhang, X.; Wu, X.; Zhang, H.; Chen, S.; Huang, Z. Physiological and omics analysis of maize inbred lines during late grain development. Genes Genom. 2022, 44, 993–1006. [Google Scholar] [CrossRef]
- Soma, F.; Takahashi, F.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Cellular phosphorylation signaling and gene expression in drought stress responses: ABA-dependent and ABA-independent regulatory systems. Plants 2021, 10, 756. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Mogami, J.; Yamaguchi-Shinozaki, K. ABA-dependent and ABA-independent signaling in response to osmotic stress in plants. Curr. Opin. Plant Biol. 2014, 21, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; He, J.; Zhao, Y.; Wu, T.; Zhou, X.; Ding, Y.; Kong, L.; Wang, X.; Wang, Y.; Li, J. EAR1 negatively regulates ABA signaling by enhancing 2C protein phosphatase activity. Plant Cell 2018, 30, 815–834. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, J.; Li, C.; Iqbal, Z.; Zhang, M.; Zhang, P.; Niu, J.; Shao, R.; Guo, H.; Mu, W.; Qiao, J. Proteomics Analysis Reveals Hormone Metabolic Process Involved in the Regulation of Kernel Water Content Induced by Exogenous Abscisic Acid in Maize. Agronomy 2023, 13, 2897. https://doi.org/10.3390/agronomy13122897
He J, Li C, Iqbal Z, Zhang M, Zhang P, Niu J, Shao R, Guo H, Mu W, Qiao J. Proteomics Analysis Reveals Hormone Metabolic Process Involved in the Regulation of Kernel Water Content Induced by Exogenous Abscisic Acid in Maize. Agronomy. 2023; 13(12):2897. https://doi.org/10.3390/agronomy13122897
Chicago/Turabian StyleHe, Jiawen, Chuan Li, Zubair Iqbal, Meiwei Zhang, Panpan Zhang, Jun Niu, Ruixin Shao, Hanxiao Guo, Weilin Mu, and Jiangfang Qiao. 2023. "Proteomics Analysis Reveals Hormone Metabolic Process Involved in the Regulation of Kernel Water Content Induced by Exogenous Abscisic Acid in Maize" Agronomy 13, no. 12: 2897. https://doi.org/10.3390/agronomy13122897