Abstract
Modern sugarcane cultivars (Saccharum spp. hybrids) are the major contributors to sucrose and bioenergy in the world. The global changes in gene expression and the molecular mechanism of heterosis between modern sugarcane hybrids and their parents remain to be elucidated. In this study, we performed a comparative transcriptome analysis between hybrids and their parents using the Illumina RNA-Seq method to understand the differences in transcript expression after hybridization. The results show that (1) introduction of the S. spontaneum lineage resulted in significant upregulation of biotic and abiotic stress resistance genes in S. hybrids, including hexokinase (HXK) genes, pathogenesis-related protein (PR1) genes, coronatine-insensitive protein (COI-1), jasmonate ZIM domain-containing protein (JAZ) genes, and serine/threonine protein kinase 2 (SnRK2) genes. (2) Transgressive genes in hybrids were mainly concentrated in the synthesis pathways of biotin and vitamin B6, helping establish advantages in terms of stress resistance, antioxidant activity, and growth. (3) Glutathione-S-transferase (GST) was likely to enhance stress resistance in hybrids, and corresponding genes were key positive selection genes in processes, including round-robin selection and other adaptations. In this study, we propose explanations for heterosis in sugarcane hybrids from a transcriptomic perspective, in addition to identifying candidate genes to aid in the improvement of sugarcane cultivars.
1. Introduction
Sugarcane hybrids are typical polyploid plants and have undergone an improvement process in which S. officinarum (noble cane) were crossed with S. spontaneum (wild cane) to generate hybrid offspring; then, these sugarcane hybrids were backcrossed with S. officinarum successively to obtain high sugar content and production features [1]. Modern sugarcane hybrids combine the high sugar content of S. officinarum and the excellent traits of S. spontaneum, such as high resistance to stress, high tillering, and perennial root, which are of great value for the crop to satisfy worldwide increasing demand for table sugar. Regardless of its importance in sugarcane production, a limited number of studies have been conducted on how hybridization affects robust phenotypes in sugarcane hybrids, and the contribution of S. spontaneum to these superior traits in hybrids remains to be elucidated.
An increasing number of reports have been published on the genome structure and evolution of polyploid hybrids, and the trend seems to be that one of the parental genomes has gradually become dominant, whereas the other genome has been suppressed in subsequent generations [2]. The abovementioned genomic changes often result in the loss or silencing of alleles, which induces a series of changes in gene expression levels [3,4]. As a result, hybridization and subsequent evolution can cause global changes of gene expression in hybrid offspring, which is collectively referred to as “transcriptome shock” [5]. One signature feature of transcriptome shock is that genes are not additive, which means the gene expression in the progeny of the cross is not equal to the average parental expression [6]. Some of these genes are expressed close to one parent but significantly differ from the other parent, which is known as “parental expression dominance”, whereas other genes exhibit expression variations beyond their parental expression range, which is referred to as “transgressive expression” [7]. These expression phenomena are observed in allopolyploids in nature. For example, interspecific hybridization analysis of cotton (Gossypium L.) showed that the expression of most genes in allopolyploid cotton is intermediate between the expression levels of the parents, with some gene expression being upregulated or downregulated [8]. Analysis of the copepod species Tigriopus californicus showed that 1.2% of the transcriptome was transgressive in F3+ intergroup hybrids, of which approximately 80% were upregulated [9]. The transgressively expressed genes reflect an interesting pattern that may be the key factor for new hybrid offspring to survive in new environments to which their parental species are not adapted. Some arguments suggest that the pattern of transgressive gene expression may be the result of directional selection or drift, leading to cis and trans regulation [10,11,12]. However, further research is needed to verify these speculations.
To date, a limited number of sugarcane studies have reported global transcriptomic changes in response to “hybridization” shocks [13,14,15,16]. Therefore, further understanding of how hybridization events between two ancestral parents produce global changes in the transcriptome, as well as the long-term selection and adaptation mechanisms within hybrid species following hybridization, is an important aspect of understanding the molecular basis of interspecific hybridization-mediated biological evolution. In this study, we performed a comparative transcriptome analysis among two ancestral parents, two ancient hybrids, and one modern hybrid in the genus of Saccharum. All three hybrids showed consistent robust agronomic traits relative to their ancestral parents. We searched for convergent information from these differences to mine the key genes in the process of “hybridization” and “adaptation” of sugarcane hybrids.
2. Materials and Methods
2.1. Plant Material and RNA Sequencing
Five representative germplasms of sugarcane were used for sequencing: S. spontaneum (Y83), S. officinarum (NJ), S. sinense (YBA), S. barberi (KAT), and S. hybrid (ZZ1). S. spontaneum (Y83) and S. officinarum (NJ) are the ancestral germplasm of the sugarcane hybrids. S. sinense (YBA) and S. barberi (KAT) are natural sugarcane hybrids. S. hybrid (ZZ1) is a modern sugarcane hybrid. All experimental materials were grown in the experimental field of Guangxi University (108°17′ E, 22°50′ N), and disease-free tissue samples of leaves and roots were collected at the seedling stage. We used an RNAprep Pure Plant Plus Kit (polysaccharide- and polyphenolic-rich) (Tiangen, Beijing, China) to extract RNA following the manufacturer’s manual.
RNA integrity was assessed using an RNA Nano 6000 assay kit with a Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA). A total of 1 μg RNA per sample was used for library preparation for transcriptome sequencing. Briefly, mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. Fragmentation was carried out using divalent cations under an elevated temperature in first-strand synthesis reaction buffer (5X). First-strand cDNA was synthesized using a random hexamer primer and M-MuLV reverse transcriptase (RNase H-). Second-strand cDNA synthesis was subsequently performed using DNA polymerase I and RNase H. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of 3′ ends of DNA fragments, adaptors with a hairpin loop structure were ligated to prepare for hybridization. In order to select cDNA fragments of preferentially 370~420 bp in length, the library fragments were purified with an AMPure XP system (Beckman Coulter, Brea, CA, USA). Then, PCR was performed with Phusion High-Fidelity DNA polymerase, universal PCR primers and index (X) primer. Finally, PCR products were purified (AMPure XP system), and library quality was assessed on an Agilent Bioanalyzer 2100 system. Index-coded samples were clustered on a cBot Cluster Generation System using a TruSeq PE Cluster Kit v3-cBot-HS (Illumina Inc., San Diego, CA, USA) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina Novaseq platform, and 150 bp paired-end reads were generated.
2.2. Assembly, Construction and Annotation of Transcriptomes
The raw data generated by RNA-seq sequencing was quality-controlled by fastp (v0.23.1) [17] with adapter trimming, quality filtering, and per-read quality pruning. Clean reads were mapped to the S. spontaneum genome AP85-441 (2n = 4x = 14) (http://sugarcane.zhangjisenlab.cn/sgd/html/download.html, accessed on 18 January 2022) using hisat2 (v2.2.1) [18]. Trinity (v2.1.1) [19] was used for genome-guided transcript assembly, and unmapped reads were used to assemble with the Trinity de novo strategy. The transcript dataset of S. spontaneum (Y83) was used as a reference for other germplasm accessions, and this process used GMAP (v2021-08-25) [20] (parameter-min-identity 0.9) and CD-HIT-EST (v4.8.1) [21] to remove the redundant sequences (parameter-c 0.8) to ultimately obtain the representative transcript datasets of five germplasm accessions.
The clean reads were compared to the reference transcripts for comparison rate assessment. BUSCO (benchmarking universal single-copy orthologs) was used for transcriptome assembly integrity assessment (mbryophyta-odb10) [22]. GC content, Contig N50, and mean size were used for assembly quality assessment. Functional annotations were performed using multiple public databases, and reference transcripts were mapped to the Non-Redundant Protein Sequence Database (NR: http://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Protein), Protein Data Bank (PBD: https://www.rcsb.org/), clusters of orthologous groups for eukaryotic complete genomes (KOG: http://ftp.ncbi.nih.gov/pub/COG/KOG/kyva), and the Swiss-Prot Protein Database (http://www.gpmaw.com/html/swiss-prot.html) with e-values < 10−5.
2.3. Differential Expression and KEGG Enrichment Analysis
The clean reads were aligned to reference transcripts of each species by bowtie2 (v2.2.5) [23], and the uniquely mapped reads were extracted and quantified by salmon (v1.6.0) [24]. Expression quantification was performed for genes shared by five species, and a target species was defined as a species-specific expressed gene when its gene expression TPM > 1 and other species’ TPM = 0. TMM normalization of TPM by edgeR [25] was performed to screen for differential genes with |log2 Fold Change| ≥ 2, FDR < 0.01 condition. Specific and differentially expressed genes were enriched and annotated by the Kyoto Encyclopedia of Genes and Genomes(KEGG) pathway database [26].
2.4. Orthologous Analysis and Selection Pressure Analysis
The longest open reading frame (ORF) was predicted and translated into protein sequences using TransDecoder (5.5.0) (http://transdecoder.github.io, accessed on 18 January 2022). All-vs.-all blast-based alignment was performed using Orthofinder (2.3.8) [27], multiple sequence concatenation was performed using MSA [28], and tree building was performed using RAxML [29]. Protein sequence alignment was performed using the muscle function, and Ka/Ks was calculated by KaKs_Calculator (2.0) [30] based on the backtranslation of protein alignment sequences into codon counterparts to account for alignment results. Enrichment annotation of positively selected genes was performed using the KEGG pathway database.
3. Results
3.1. Transcriptomic Data Processing and Assembly
Transcriptomes of leaf and root tissues of four sugarcane species (S. spontaneum, S. officinarum, S. barberi, and S. sinense) and one modern sugarcane cultivar (S. hybrid) were sequenced in three biological replicates. A total of 30 sequencing samples generated 2.0 billion clean reads and 298.3 G bases, with an average of 66.6 million clean reads and 9.9 G bases per sequenced sample and average Q20 and GC content of 98.18% and 54.56%, respectively (Table S1). The above results show that sequencing data were deep and that the quality of sequencing data was high, meeting the requirements of subsequent transcriptome analysis.
After transcript assembly and removal of redundant transcripts, the number of unigenes generated from the five sugarcane materials ranged from 255,387 to 471,251 (Table 1). The average length of the unigenes of the S. spontaneum, S. officinarum, S. barberi, S. sinense, and S. hybrid ranged from 586 to 745 base pairs (bp), with N50 values of 829, 1080, 995, 1168, and 1045 bp and GC contents of 49.37%, 49.59%, 49.82%, 49.36%, and 49.99%, respectively (Table S2). Annotation was performed using the NR, SWISS, KOG, and PDB public databases, and unigenes with significant matches in at least one of the four databases were considered to be successfully annotated. The annotation rate of the five sugarcane accessions ranged from 52.42% to 56.02%. The unigenes were further assessed for assembly quality using BUSCO, and the results showed that the unigenes of the five accessions could cover an average of 72.7% of the conserved plant orthologous single-copy genes (n = 1614), in which only single-copy coverage and multi-copy coverage were considered complete coverage. The highest complete coverage was found in S. spontaneum, which covered 1241 conserved plant orthologous single-copy genes (76.89%), whereas the lowest complete coverage was found in S. officinarum, which covered 1113 conserved plant orthologous single-copy genes (68.96%). The average fragmentation coverage (18.24%) and the average loss coverage (9.06%) of the five germplasm accessions were both lower than the average complete coverage (72.7%) (Table S3).

Table 1.
Assembly and annotation results of reference sequences.
The unigenes of the five sugarcane accessions were mapped to the NR database, and the percentage of target sequences matching the closest species was counted. The five sugarcane accessions had the closest species match with Sorghum bicolor, with an average match rate of 45.7%, followed by Miscanthus lutarioriparius (15.8%) and Zea mays (13.7%); the average match rate of all other species was less than 3.0% (Table 2). S. bicolor and M. lutarioriparius were found to be close relatives of Saccharum spp. [31], so it was not unexpected to have a high match rate. In contrast, sugarcane hybrid R570 had an average match rate of less than 3.0%, which may be related to its incomplete genome assembly.
Table 2.
Comparison of the unigene rates of five sugarcane species with protein sequences from various plants in the NR database.
3.2. Shared and Unique Unigenes and Characterization of Specifically Expressed Genes of the Five Sugarcane Transcriptomes
The five sugarcane transcriptomes had 75,221 shared unigenes (Figure 1a). The number of unique unigenes was relatively high, specifically: S. spontaneum Y83, 283,694); S. officinarum NJ, 137,528; S. barberi KAT, 192,954; S. sinense YBA, 142,815; and S. hybrid ZZ1, 166,530. The remaining unigenes existed in more than two but not all sugarcane accessions. With respect to the gene ontology (GO) annotation results of shared genes, “cellular processes”, “metabolic processes”, and “single-organism processes” were most enriched in biological process for unigenes, whereas “cell” and “cell part” were most enriched in cellular components for unigenes, and “binding” and “catalytic activity” were most enriched in molecular function for unigenes (Figure S1). The annotation results of the KEGG pathway for shared genes show that the top three KEGG metabolic categories were “global and overview maps”, “carbohydrate metabolism”, and “amino acid metabolism” (Figure 1b). From the perspective of genetic information processing, the number of unigenes was distributed as “translation”; “folding, sorting, and degradation”; “transcription”; and “replication and repair”. From the perspective of environmental information processing, unigenes were mainly distributed in “signal transduction” and “membrane transport”. From the perspective of cellular processes, unigenes were mainly distributed in “transport and catabolism”. From the perspective of tissue systems, unigenes were mainly distributed in “environmental adaptation”.
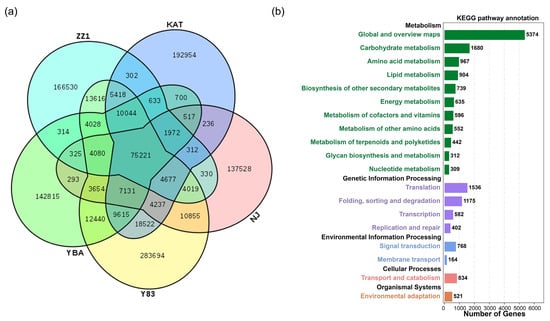
Figure 1.
Functional annotations of shared and unique unigenes in five sugarcane transcriptomes. (a) The intersection of shared and unique unigenes among the five species; (b) annotation of the KEGG pathway for shared unigenes.
Specifically expressed genes from five sugarcane materials were identified. In S. spontaneum, 91 leaf-specific expressed genes and 60 root-specific expressed genes were characterized (Table 3), which were enriched in “proteasome” and “glycosaminoglycan degradation” pathways, respectively. In S. officinarum, 123 leaf-specific expressed genes and 90 root-specific expressed genes were characterized, which were enriched in “flavonoid biosynthesis” and “biosynthesis of amino acids and carbon metabolism” pathways, respectively. In S. barberi, 77 leaf-specific expressed genes were characterized, which were enriched in “the biosynthesis of secondary metabolites”; “histidine metabolism”; “biosynthesis of amino acids”; “tropane, piperidine, and pyridine alkaloid biosynthesis”; and “pantothenate and CoA metabolite” pathways, and 72 root-specific expressed genes were characterized, which were enriched in the “taurine and hypotaurine metabolism” pathway. In S. sinense, 89 leaf-specific expressed genes were characterized, which were enriched in the “stilbenoid, diarylheptanoid, and gingerol biosynthesis” pathways, and 64 root-specific expressed genes were characterized, which were enriched in “phenylpropanoid biosynthesis”, “ubiquinone and other terpenoid-quinone biosynthesis”, and “glutathione metabolism” pathways. In the S. hybrid, 116 leaf-specific expressed genes were characterized, which were enriched in “phenylpropanoid biosynthesis” and “linoleic acid metabolism” pathways, and 83 root-specific expressed genes were characterized, which were enriched in “pyruvate metabolism” and “linoleic acid metabolism” pathways. The number of specifically expressed genes in both leaf and root for S. spontaneum, S. officinarum, S. barberi, S. sinense, and S. hybrid was 19, 28, 16, 27, and 29, respectively. KEGG enrichment analysis showed that these genes were significantly enriched in “glycosaminoglycan degradation” and “cellular autophagy” pathways in S. spontaneum, in the “flavonoid biosynthesis” pathway in S. officinarum, in the “ubiquitin-mediated protein hydrolysis” pathway in both S. barberi and S. sinense, and in the “linoleic acid metabolism” pathway in the S. hybrid.
Table 3.
Number of tissue-specific expressed genes in five sugarcane species.
3.3. Differential Gene Expression Analysis of the Five Sugarcane Transcriptomes
Using transcriptome data from leaf tissues, S. barberi, S. sinense, and S. hybrid were first compared with their ancestral parent S. officinarum separately for differential expression analysis (Figure 2). We identified 6673 upregulated and 7026 downregulated differential genes in S. barberi vs. S. officinarum (KAT vs. NJ), 6541 upregulated and 6756 downregulated differential genes in S. sinense vs. S. officinarum (YBA vs. NJ), and 6354 upregulated and 6939 downregulated differential genes in S. hybrid vs. S. officinarum (ZZ1 vs. NJ). KEGG enrichment analysis showed that the upregulated genes in the three comparisons were not significantly enriched to any common pathway, whereas the downregulated genes were significantly enriched in the “spliceosome” pathway. Using transcriptome data from root tissues, we identified 6189 upregulated and 6606 downregulated differential genes in S. barberi vs. S. officinarum (KAT vs. NJ), 6119 upregulated and 6935 downregulated differential genes in S. sinense vs. S. officinarum (ZZ1 vs. NJ), and 5895 upregulated and 6300 downregulated differential genes in S. hybrid vs. S. officinarum (ZZ1 vs. NJ). KEGG enrichment analysis showed that the upregulated genes in the three comparison groups were significantly enriched in “the neomycin, kanamycin, and gentamicin biosynthesis” and “plant hormone signal transduction” pathways, whereas the downregulated genes in the three comparison groups were significantly enriched in the “carbon metabolism” pathway.
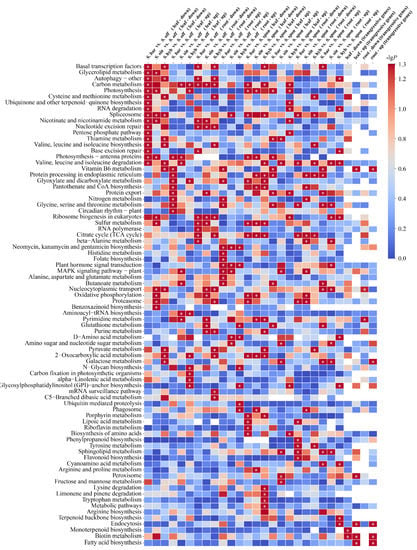
Figure 2.
KEGG pathway enrichment analysis of differentially expressed genes based on sugarcane species and tissue. The significance of the most represented pathway in each comparison is shown by log-transformed P (red) for a total of 24 comparison groups. “Up” indicates upregulation, “down” indicates downregulation, “leaf” indicates leaf tissue, and “root” indicates root tissue. “Transgressive genes” indicates that the expression of genes in the S. hybrids are higher or lower than those in both ancestral parents. Intersections of transgressive genes among three S. hybrids were included in the KEGG pathway enrichment in four cases: leaf-up, leaf-down, root-up, and root-down. Note: White cells indicate a missing value, and * indicates significant or extremely significant enrichment.
Using transcriptome data from leaf tissues, S. barberi, S. sinense, and S. hybrid were also compared with the other ancestral parent S. spontaneum separately for differential expression analysis (Figure 2). We identified 7369 upregulated and 6163 downregulated differential genes in S. barberi vs. S. spontaneum (KAT vs. Y83), 7320 upregulated and 6051 downregulated differential genes in S. sinense vs. S. spontaneum (YBA vs. Y83), and 7160 upregulated and 6374 downregulated differential genes in S. hybrid vs. S. spontaneum (ZZ1 vs. Y83). KEGG enrichment analysis showed that the upregulated genes in the three comparisons were not significantly enriched in any common pathway, whereas the downregulated genes were significantly enriched in the “sulfur metabolism” and “2-oxocarboxylic acid metabolism” pathways and the “acid metabolism” pathway. Using transcriptomic data from root tissues, we identified 6532 upregulated and 5928 downregulated differential genes in S. barberi vs. S. spontaneum (KAT vs. Y83), 6220 upregulated and 6282 downregulated differential genes in S. sinense vs. S. spontaneum (ZZ1 vs. Y83), and 6220 upregulated and 6197 downregulated differential genes in S. hybrid vs. S. spontaneum (ZZ1 vs. Y83). KEGG enrichment analysis showed that upregulated genes or downregulated genes in the three comparisons were not significantly enriched to any common pathway.
To understand heterosis of sugarcane cultivars, the above 24 comparisons were regrouped to investigate genes that were convergently expressed in sugarcane hybrids compared with their ancestral parents, yielding four intersections of transgressive genes (Figure 2). First, we screened genes that were upregulated in the three sugarcane hybrids (S. barberi, S. sinense, and S. hybrid) relative to their ancestral parents (S. officinarum and S. spontaneum), and these transgressive expression genes likely promote superior traits over their parents. A total of 406 convergent upregulated genes were identified in leaf tissues, which were significantly enriched in “biotin metabolism”, “vitamin B6 metabolism”, “endocytosis and fatty acid biosynthesis”, and “peroxisome” pathways, whereas a total of 333 convergent upregulated genes were identified in the root tissue and significantly enriched in “biotin metabolism”, “endocytosis”, “vitamin B6 metabolism”, “fatty acid biosynthesis”, and “galactose metabolism” pathways. Secondly, we screened genes that were downregulated in three sugarcane hybrids relative to both ancestral parents, which may be adapting to the “transcriptome shock” generated by the crossbreeding process. As a result, a total of 376 convergent downregulated genes were identified in the leaf tissue and significantly enriched in “monoterpenoid biosynthesis” and “biotin metabolism” pathways, whereas a total of 360 convergent downregulated genes were identified in root tissue and significantly enriched in the “pyrimidine metabolism” pathway.
3.4. Selection Pressure Analysis
Based on the 3843 orthologous gene pairs identified in the five sugarcane accessions, S. officinarum and S. spontaneum were separately grouped with S. barberi, S. sinense, and the S. hybrid for selection pressure analysis. Then, Ka/Ks ratios were calculated in the six groups (S. officinarum vs. S. barberi; S. officinarum vs. S. sinense; S. officinarum vs. S. hybrid; S. spontaneum vs. S. barberi; S. spontaneum vs. S. sinense; S. spontaneum vs. S. hybrid). The results show that in the three groups of S. officinarum vs. sugarcane hybrids (S. officinarum vs. S. barberi, S. officinarum vs. S. sinense, S. officinarum vs. S. hybrid), there were 693 (18.0%), 688 (17.9%), and 723 (18.8%) positively selected genes with Ka/Ks > 1, respectively. In the three comparison groups of S. spontaneum vs. sugarcane hybrids (S. spontaneum vs. S. barberi, S. spontaneum vs. S. sinense, and S. spontaneum vs. S. hybrid), there were 628 (16.3%), 631 (16.4%), and 588 (15.3%) positively selected genes with Ka/Ks > 1, respectively. Obviously, the proportion of positive selection genes in S. officinarum vs. sugarcane hybrids was higher than the proportion of positive selection genes in S. spontaneum vs. sugarcane hybrids (Figure 3).
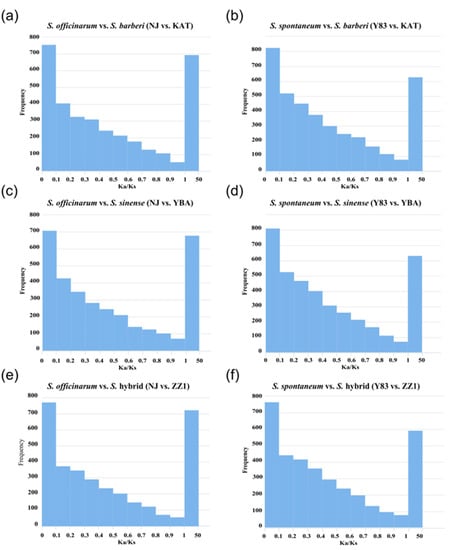
Figure 3.
Distribution of selection pressure on orthologous genes between ancestral parents (S. officinarum and S. spontaneum) and hybrids (S. barberi, S. sinense, and S. hybrid). (a) S. officinarum vs. S. barberi (NJ vs. KAT); (b) S. spontaneum vs. S. barberi (Y83 vs. KAT); (c) S. officinarum vs. S. sinense (NJ vs. YBA); (d) S. spontaneum vs. S. sinense (Y83 vs. YBA); (e) S. officinarum vs. S. hybrid (NJ vs. ZZ1); (f) S. spontaneum vs. S. hybrid (Y83 vs. ZZ1).
KEGG enrichment analysis of the group S. officinarum vs. S. barberi showed that the positive selection genes were significantly enriched in the “N-glycan biosynthesis” pathway; the positive selection genes of the group S. officinarum vs. S. sinense were significantly enriched in “homologous recombination”, “proteasome”, “glutathione metabolism”, “inositol phosphate metabolism”, and “mRNA surveillance” pathways; and the positive selection genes of the group S. officinarum vs. S. hybrid were significantly enriched in the “ubiquitin-mediated proteolysis” pathway. KEGG enrichment analysis of the group S. spontaneum vs. S. barberi showed that the positive selection genes were significantly enriched in “glutathione metabolism” and “tropane, piperidine, and pyridine alkaloid biosynthesis” pathways; the positive selection genes of the group S. spontaneum vs. S. sinense were significantly enriched in “tropane, piperidine and pyridine alkaloid biosynthesis”, “SNARE interactions in vesicular transport”, and “glutathione metabolism” pathways; and the positive selection genes of the group S. spontaneum vs. S. hybrid were significantly enriched in the “glutathione metabolism” pathway. The positive selection genes in five of the six abovementioned groups were significantly enriched in the “glutathione metabolism” pathway, which relatively reliably indicated that genes related to the “glutathione metabolism” pathway were under positive selection pressure during long-term hybridization.
Interestingly, we found some intersections of positively selected genes with tissue-specific expressed genes, representing mutant genes used to adapt to the environment expressed in specific tissues. No genes were found that met the above conditions among all groups in leaf tissues. However, we still identified four genes that met the above conditions from the group S. officinarum vs. S. Hybrid in root tissues (Table 4). With NR protein database annotation, we found that the gene Y83_TRINITY_GG_44371_c184_g1_i1 was annotated as a putative lipid-transfer protein. We also identified five genes that met the above conditions from the group S. spontaneum vs. S. hybrid in root tissues (Table 4). The gene Y83_TRINITY_DN181546_c0_g1_i4 was annotated as cysteine-rich receptor-like protein kinase, and Y83_TRINITY_GG_13372_c64_g1_i1 and Y83_TRINITY_GG_50524_c0_g1_i1 genes were annotated as glycine-rich cell wall structural protein.
Table 4.
Annotation of the intersection of positive selection and tissue-specific expressed genes.
4. Discussion
Modern sugarcane cultivars are the main contributors to sugar and bioenergy worldwide, with the combined high sugar traits of S. officinarum and the strong stress resistance traits of S. spontaneum [32,33]. Mining information on the convergence of S. barberi, S. sinense, and S. hybrid at the transcriptional level can elucidate the molecular mechanisms underlying high sugar content, high yield, and high stress resistance in sugarcane hybrids, which can guide future sugarcane variety selection and genetic improvement. Preliminary investigations on the genome and transcriptome of sugarcane hybrids have been conducted, but the molecular-level effects of the hybridization process are still unclear. In this study, we performed comparative transcriptome analysis of three sugarcane hybrids with their two ancestral parents one by one to explore (1) global alterations in gene expression in the hybrid progeny, (2) transgressively expressed genes in the hybrid progeny, and (3) positive selection of genes in subgenomes.
4.1. Global Alterations in Gene Expression in Hybrid Progeny
Interspecific hybridization is a driving force in evolution and species formation of higher plants and often causes global changes in gene expression, which are collectively referred to as “transcriptome shock” [11,34,35]. Transcriptome shock affects phenotypic changes and contributes to better survival of nascent hybrids in the face of complex environmental conditions. It has been suggested that altered gene expression after hybridization may be associated with incompatible regulatory networks between parents [36], and DNA methylation and histone methylation may also maintain genomic stability after hybridization [37]. During the course of sugarcane crosses, it is reasonable to infer, based on the apparent trait alterations between hybrid progeny and parents, that although S. officinarum often serves as parent of successive backcrosses and occupies most of the genomic lineage in hybrids, the smaller percentage of S. spontaneum genomes that is admixed still has a non-negligible impact. In this study, based on differential expression analysis of three sugarcane hybrids with one of the ancestral parents, the transcriptome shock caused by the other ancestral parent was assessed. The impact of the S. spontaneum lineage showed some common trends in different hybrids, most likely as an important pattern of sugarcane hybridization.
Within root tissues, the upregulated genes in groups, whether S. barberi vs. S. officinarum, S. sinense vs. S. officinarum, or S. hybrid vs. S. officinarum, were all significantly enriched in the “neomycin, kanamycin, and gentamicin biosynthetic” pathway, where the gene encoding hexokinase (HXK) was common to the three groups; thus, the gene encoding HXK may be a key gene for S. spontaneum impact. HXK has the function of activating the substrate hexose to produce hexose-6-phosphate to help with the synthesis of downstream metabolites. Studies in tobacco have shown that the NbHXK1 gene was upregulated upon infection by pathogens, HXK-overexpressing plants showed greater resistance to necrotrophic fungal pathogens, and HXK overexpression provided mild protection against the bacterial pathogen Pseudomonas syringae pv. tomato DC3000 [38], indicating that hexokinase is critical for plant disease resistance. The analysis conducted in the present study showed that the impact of S. spontaneum is highly likely to increase the expression level of hexokinase-encoding genes in root tissues of sugarcane hybrids, thus improving the biotic stress resistance of sugarcane.
Within root tissues, the upregulated genes in groups, whether S. barberi vs. S. officinarum, S. sinense vs. S. officinarum, or S. hybrid vs. S. officinarum, were also all significantly enriched in the “plant hormone signal transduction” pathway, with common corresponding coding genes, including pathogenesis-related protein (PR1), coronatine-insensitive protein (COI-1), jasmonate ZIM domain-containing protein (JAZ), auxin influx carrier (AUX1 LAX family), transcription factor TGA (TGA), auxin-responsive protein IAA (IAA), auxin response factor (ARF), DELLA protein (DELLA), serine/threonine protein kinase 2 (SnRK2), ethylene-insensitive protein (EIN3), and EIN3-binding F-box protein (EBF). Among the abovementioned genes, some of the common corresponding genes have clear associations with biotic stress resistance. It has been shown that members of the PR1 family are among the most abundant proteins produced in plants upon pathogen attack, and PR1 gene expression has long been used as a marker of salicylic-acid-mediated disease resistance [39]. COI-1 is a receptor for JAZ, and both COI-1 and JAZ are associated with jasmonic acid signaling. Stresses such as insect feeding or pathogen infection prompt plants to synthesize jasmonyl-L-isoleucine (JA-Ile), and increased levels of JA-Ile promote the interaction of JAZ blocker proteins with F-box protein encoded by COI-1, which ubiquitinates JAZ and releases DNA-binding transcription factors (e.g., MYC2) to induce the expression of JA-responsive genes and the deletion of the COI-1 locus or its functional defect, which leads to the disappearance or weakening of the plant response to JA, indicating that COI-1 and JAZ are of considerable importance in JA signaling [40]. Overexpression of the SnRK2 gene in Arabidopsis thaliana significantly enhances tolerance to drought, salinity, and freezing stresses; does not retard the growth of transgenic plants under water conditions; and is considered an important gene for improving crop response to abiotic stresses [41]. In addition, compared to the abovementioned hormones, other hormones are relatively indirectly associated with stress resistance; however, related genes encoding these hormones are also differentially expressed; for example, genes encoding auxin influx carrier (AUX1 LAX family), auxin-responsive protein (IAA), and auxin response factor (ARF) are auxin-related, and genes encoding DELLA protein (DELLA), ethylene-insensitive protein (EIN3), and EIN3-binding F-box protein (EBF) are-gibberellin related. We speculated that crosstalk between hormones led to such a result, and the result suggests that the impact of S. spontaneum genomes not only brought about changes in stress resistance but also may indirectly affect plant development.
4.2. Features of Transgressive Expression of Genes in Hybrids
The recombination and regulation of parental genes have a complex impact on the transcriptome so that the gene expression of hybrids may appear different from either parent. For example, there is a phenomenon at the transcript level whereby the expression level of the hybrid progeny is not equal to the average of parental expression but beyond the parental expression range, which is called a transgressive expression pattern [5]. Our study was devoted to discovering the commonalities presented by three hybrids (S. barberi, S. sinense, and S. hybrid) by identifying the transgressive expression genes of the hybrids vs. both parents.
Target genes in both leaves and roots were enriched for “biotin metabolism”, “vitamin B6 metabolism”, and “fatty acid biosynthesis”, suggesting that these pathways may encompass key factors that allow hybrids to outcompete their parents. Previous studies have found that biotin mitigates the effects of carbonate stress on plants and helps to reduce the accumulation of reactive oxygen species [42]. A study of A. thaliana showed that intron insertion mutants of AtPDX2 (resulting in B6 deficiency) produced seeds with delayed development compared to their wild-type or heterozygous counterparts, suggesting that vitamin B6 was essential for plant viability in higher plants [43]. Based on this information, we suggested that the overexpressed genes in sugarcane hybrids were mainly concentrated in the biosynthetic pathways of important vitamins (biotin and vitamin B6) and manipulated the hybrid progeny to establish advantages in stress resistance, antioxidant activity, and growth by encoding genes that exceed the expression of the parents.
4.3. Positive Selection of Genes in Subgenomes
A study in allopolyploid Gossypium showed that higher transgressive rate and gene silencing were observed in natural allopolyploids compared to heterozygous polyploids of the hybrid generation, suggesting that the regulation induced by the initial gene combination was further reconciled during selective adaptation [44]. Adaptation is inextricably linked to positive selection, in which genes favorable to individual adaptation are preserved. In contrast to S. officinarum and S. spontaneum, sugarcane hybrids (S. barberi, S. sinense, and S. hybrid) are undergoing a long adaptation process after crossing, so there are reasons to believe that positive selection exists in sugarcane hybrids and has some influence on adaptation processes. It has been shown that both reproduction and stress-tolerance-related genes are among the important genes for adaptation to the environment in terrestrial plants. For example, Corre et al. studied the FRI sequences of 25 wild-type Arabidopsis thaliana flowering-related genes in Western Europe and found that these sequences contained an excess of nonsynonymous substitution sites, indicating that the polymorphism of FRI in Western European A. thaliana is caused by positive selection in a series of isolated populations [45]. Studies related to plant resistance genes (R genes) have shown that a favorable mutation in the R gene allows the host to recognize antigenic determinants that were previously unrecognized and is therefore preserved by selection pressure and acquires high gene frequencies [46].
Among the 3843 orthologous gene pairs identified in our study, the proportion of positively selected gene pairs from the S. officinarum lineage was higher than that of the S. spontaneum lineage, which showed the same patterns in two ancient hybrids and one modern hybrid, indicating that genes from the S. officinarum lineage were more positively selected. Further KEGG enrichment analysis of these positively selected genes revealed that these genes were identified in five of the six comparison groups and were significantly enriched to the “glutathione metabolism” pathway, and the common of positively selected genes in the above comparison groups were genes encoding glutathione-S-transferase (GST), indicating that the “glutathione metabolism” pathway was most likely one of the target genes strongly selected by the environment after hybridization. Active glutathione has effects on intracellular antioxidant activities, plant stress resistance, and GST is its corresponding key binding enzyme. Some studies have shown that GST is involved in the detoxification of toxic xenobiotic compounds in plants [47,48,49]. Recent studies have reported GST plays a key role in the defense of plants against fungi, bacteria, and viruses [50]. Based on the above discussion, combined with the results of our study, in which different sugarcane hybrids were consistently subjected to positive selection for the GST coding genes, GST is highly likely to help hybrids enhance resistance and that its encoding genes are a key positive selection genes after hybridization, including breeding selection and other adaptation processes.
5. Conclusions
We conducted a comparative transcriptome analysis between sugarcane hybrids and their ancestral parents and summarized the results in terms of the convergent information displayed by the three hybrids. By exploring the expression of “transcriptome shock” in the progeny of the crosses, we analyzed the molecular mechanisms by which crosses affect global changes in the transcriptome from different perspectives and obtained candidate genes that can be used to improve sugarcane cultivars, although the complex regulatory networks involved still require further study. First, the S. spontaneum genome lineage had profound effects on the expression of biotic and abiotic resistance-related genes in sugarcane hybrids and may further influence growth through hormone crosstalk. Secondly, we noted that the relevant coding genes in the biotin and vitamin B6 synthesis pathways established the growth advantage of hybrid progeny through transgressive dominance. Finally, the screened GST-encoding genes were most likely key positive selection genes in processes including round-robin selection and other adaptations.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/agronomy13020348/s1; Table S1. Basic information about transcriptome sequencing; Table S2. Statistics of reference quality assessment of the five species; Table S3: Statistics for reference quality assessment by BUSCO. Figure S1. Annotation of GO terms for shared Unigenes.
Author Contributions
Conceptualization, M.F. and X.Y.; data curation, M.F. and J.Z.; writing—original draft preparation, M.F. and J.Z.; writing—review and editing, M.F., J.Z., S.L., N.W., B.K. and X.Y.; funding acquisition, X.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Guangxi Natural Science Foundation (GK AD20297064) and the ‘One Hundred Person’ Project of Guangxi Province.
Data Availability Statement
The sequencing data for this project have been deposited at the NCBI Sequence Read Archive (SRA) under project PRJNA 910660.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wang, K.; Cheng, H.; Han, J.; Esh, A.; Liu, J.; Zhang, Y.; Wang, B. A comprehensive molecular cytogenetic analysis of the genome architecture in modern sugarcane cultivars. Chromosome Res. 2022, 30, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.C.; Pedersen, B.; Freeling, M. Following tetraploidy in an Arabidopsis ancestor, genes were removed preferentially from one homeolog leaving clusters enriched in dose-sensitive genes. Genome Res. 2006, 16, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Comai, L. Genetic and epigenetic interactions in allopolyploid plants. In Plant Gene Silencing; Springer: Berlin/Heidelberg, Germany, 2000; pp. 267–279. [Google Scholar]
- Adams, K.L.; Wendel, J.F. Polyploidy and genome evolution in plants. Curr. Opin. Plant Biol. 2005, 8, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Sun, Y.; Wang, X.; Lin, X.; Sun, S.; Shen, K.; Wang, J.; Jiang, T.; Zhong, S.; Xu, C. Transcriptome shock in an interspecific F1 triploid hybrid of Oryza revealed by RNA sequencing. J. Integr. Plant Biol. 2016, 58, 150–164. [Google Scholar] [CrossRef]
- Jackson, S.; Chen, Z.J. Genomic and expression plasticity of polyploidy. Curr. Opin. Plant Biol. 2010, 13, 153–159. [Google Scholar] [CrossRef]
- Grover, C.; Gallagher, J.; Szadkowski, E.; Yoo, M.; Flagel, L.; Wendel, J. Homoeolog expression bias and expression level dominance in allopolyploids. New Phytol. 2012, 196, 966–971. [Google Scholar] [CrossRef]
- Rapp, R.A.; Udall, J.A.; Wendel, J.F. Genomic expression dominance in allopolyploids. BMC Biol. 2009, 7, 18. [Google Scholar] [CrossRef]
- Barreto, F.S.; Pereira, R.J.; Burton, R.S. Hybrid dysfunction and physiological compensation in gene expression. Mol. Biol. Evol. 2015, 32, 613–622. [Google Scholar] [CrossRef]
- Go, A.C.; Civetta, A. Hybrid incompatibilities and transgressive gene expression between two closely related subspecies of Drosophila. Front. Genet. 2020, 11, 599292. [Google Scholar] [CrossRef]
- Fu, J.; Zhang, Y.; Yan, T.; Li, Y.; Jiang, N.; Zhou, Y.; Zhou, Q.; Qin, P.; Fu, C.; Lin, H. Transcriptome profiling of two super hybrid rice provides insights into the genetic basis of heterosis. BMC Plant Biol. 2022, 22, 314. [Google Scholar] [CrossRef]
- Wang, M.; Wang, J. Transcriptome and DNA methylome analyses provide insight into the heterosis in flag leaf of inter-subspecific hybrid rice. Plant Mol. Biol. 2022, 108, 105–125. [Google Scholar] [CrossRef]
- Serba, D.D.; Meng, X.; Schnable, J.; Bashir, E.; Michaud, J.; Prasad, P.V.; Perumal, R. Comparative Transcriptome analysis reveals genetic mechanisms of sugarcane aphid resistance in grain Sorghum. Int. J. Mol. Sci. 2021, 22, 7129. [Google Scholar] [CrossRef]
- Zeng, Z.; Liu, X.; Deng, Q.; Ashraf, U.; Chen, J.; Shen, W. Transcriptome analysis revealed mechanisms involved in improved germination and growth of sugarcane by ultrasonic treatment. Ind. Crops Prod. 2023, 192, 116104. [Google Scholar] [CrossRef]
- Huang, X.; Liang, Y.; Zhang, B.; Song, X.; Li, Y.; Li, C.; Qin, Z.; Li, D.; Wei, J.; Wu, J. Comparative transcriptome analysis reveals potential gene modules associated with cold tolerance in sugarcane (Saccharum officinarum L.). J. Plant Growth Regul. 2022, 41, 2614–2628. [Google Scholar] [CrossRef]
- Vignesh, P.; Mahadevaiah, C.; Parimalan, R.; Valarmathi, R.; Dharshini, S.; Nisha, S.; Suresha, G.; Swathi, S.; Swamy, H.M.; Sreenivasa, V. Comparative de novo transcriptome analysis identifies salinity stress responsive genes and metabolic pathways in sugarcane and its wild relative Erianthus arundinaceus [Retzius] Jeswiet. Sci. Rep. 2021, 11, 24514. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar]
- Bodenhofer, U.; Bonatesta, E.; Horejš-Kainrath, C.; Hochreiter, S. msa: An R package for multiple sequence alignment. Bioinformatics 2015, 31, 3997–3999. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef]
- Hoang, N.V.; Furtado, A.; Mason, P.J.; Marquardt, A.; Kasirajan, L.; Thirugnanasambandam, P.P.; Botha, F.C.; Henry, R.J. A survey of the complex transcriptome from the highly polyploid sugarcane genome using full-length isoform sequencing and de novo assembly from short read sequencing. BMC Genom. 2017, 18, 395. [Google Scholar] [CrossRef]
- Yadav, S.; Jackson, P.; Wei, X.; Ross, E.M.; Aitken, K.; Deomano, E.; Atkin, F.; Hayes, B.J.; Voss-Fels, K.P. Accelerating genetic gain in sugarcane breeding using genomic selection. Agronomy 2020, 10, 585. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Tang, H.; Zhang, Q.; Hua, X.; Ma, X.; Zhu, F.; Jones, T.; Zhu, X.; Bowers, J.; et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 2018, 50, 1565–1573. [Google Scholar] [CrossRef]
- Quan, C.; Li, Y.; Chen, G.; Tian, X.; Jia, Z.; Tu, J.; Shen, J.; Yi, B.; Fu, T.; Ma, C.; et al. The dynamics of lncRNAs transcription in interspecific F1 allotriploid hybrids between Brassica species. Genomics 2022, 114, 110505. [Google Scholar] [CrossRef]
- Quan, C.; Chen, G.; Li, S.; Jia, Z.; Yu, P.; Tu, J.; Shen, J.; Yi, B.; Fu, T.; Dai, C.; et al. Transcriptome shock in interspecific F1 allotriploid hybrids between Brassica species. J. Exp. Bot. 2022, 73, 2336–2353. [Google Scholar] [CrossRef]
- Chen, Z.J. Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu. Rev. Plant Biol. 2007, 58, 377–406. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, X.K.; Fan, W.; Yan, D.F.; Zhong, N.S.; Gao, J.Y.; Zhang, W.J. Transcriptome analysis reveals hybridization-induced genome shock in an interspecific F1 hybrid from Camellia. Genome 2018, 61, 477–485. [Google Scholar] [CrossRef]
- Sarowar, S.; Lee, J.Y.; Ahn, E.R.; Pai, H.S. A role of hexokinases in plant resistance to oxidative stress and pathogen infection. J. Plant Biol. 2008, 51, 341–346. [Google Scholar] [CrossRef]
- Breen, S.; Williams, S.J.; Outram, M.; Kobe, B.; Solomon, P.S. Emerging insights into the functions of pathogenesis-related protein 1. Trends Plant Sci. 2017, 22, 871–879. [Google Scholar] [CrossRef]
- Sun, T.; Meng, Y.; Cen, G.; Feng, A.; Su, W.; Chen, Y.; You, C.; Que, Y.; Su, Y. Genome-wide identification and expression analysis of the coronatine-insensitive 1 (COI1) gene family in response to biotic and abiotic stresses in Saccharum. BMC Genom. 2022, 23, 38. [Google Scholar] [CrossRef]
- Mao, X.; Zhang, H.; Tian, S.; Chang, X.; Jing, R. TaSnRK2. 4, an SNF1-type serine/threonine protein kinase of wheat (Triticum aestivum L.), confers enhanced multistress tolerance in Arabidopsis. J. Exp. Bot. 2010, 61, 683–696. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, M.; Ye, X.; Liu, H.; Takano, T.; Tsugama, D.; Liu, S.; Bu, Y. Biotin plays an important role in Arabidopsis thaliana seedlings under carbonate stress. Plant Sci. 2020, 300, 110639. [Google Scholar] [CrossRef] [PubMed]
- Tambasco-Studart, M.; Titiz, O.; Raschle, T.; Forster, G.; Amrhein, N.; Fitzpatrick, T.B. Vitamin B6 biosynthesis in higher plants. Proc. Natl. Acad. Sci. USA 2005, 102, 13687–13692. [Google Scholar] [CrossRef] [PubMed]
- Yoo, M.; Szadkowski, E.; Wendel, J. Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity 2013, 110, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Le Corre, V.; Roux, F.; Reboud, X. DNA polymorphism at the FRIGIDA gene in Arabidopsis thaliana: Extensive nonsynonymous variation is consistent with local selection for flowering time. Mol. Biol. Evol. 2002, 19, 1261–1271. [Google Scholar] [CrossRef]
- Jiang, N.; Yan, J.; Liang, Y.; Shi, Y.; He, Z.; Wu, Y.; Zeng, Q.; Liu, X.; Peng, J. Resistance genes and their interactions with bacterial blight/leaf streak pathogens (Xanthomonas oryzae) in rice (Oryza sativa L.)—An updated review. Rice 2020, 13, 3. [Google Scholar] [CrossRef]
- Lamoureux, G.L. The role of glutathione and glutathione S-transferases in pesticide metabolism, selectivity, and mode of action in plants and insects. Coenzymes Cofactors 1989, 3, 153–196. [Google Scholar]
- Dixon, D.P.; Cummins, I.; Cole, D.J.; Edwards, R. Glutathione-mediated detoxification systems in plants. Curr. Opin. Plant Biol. 1998, 1, 258–266. [Google Scholar] [CrossRef]
- Liu, N.; Li, J.; Lv, J.; Yu, J.; Xie, J.; Wu, Y.; Tang, Z. Melatonin alleviates imidacloprid phytotoxicity to cucumber (Cucumis sativus L.) through modulating redox homeostasis in plants and promoting its metabolism by enhancing glutathione dependent detoxification. Ecotoxicol. Environ. Saf. 2021, 217, 112248. [Google Scholar] [CrossRef]
- Gullner, G.; Komives, T.; Király, L.; Schröder, P. Glutathione S-transferase enzymes in plant-pathogen interactions. Front. Plant Sci. 2018, 9, 1836. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).