
Revealing Genetic Variations Associated with Chip-Processing Properties in Potato (Solanum tuberosum L.)

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Phenotyping
2.2. Genome-Wide Association Study
2.3. Principal Component Analysis
2.4. Selective Sweeps Analysis
2.5. Transcriptome Analysis
2.6. KASP Assay
3. Results
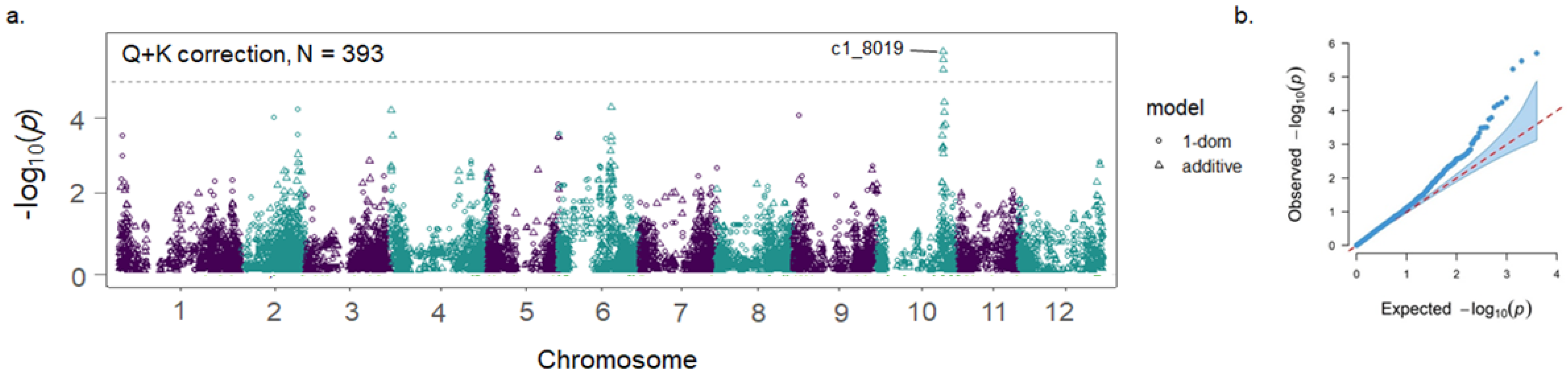
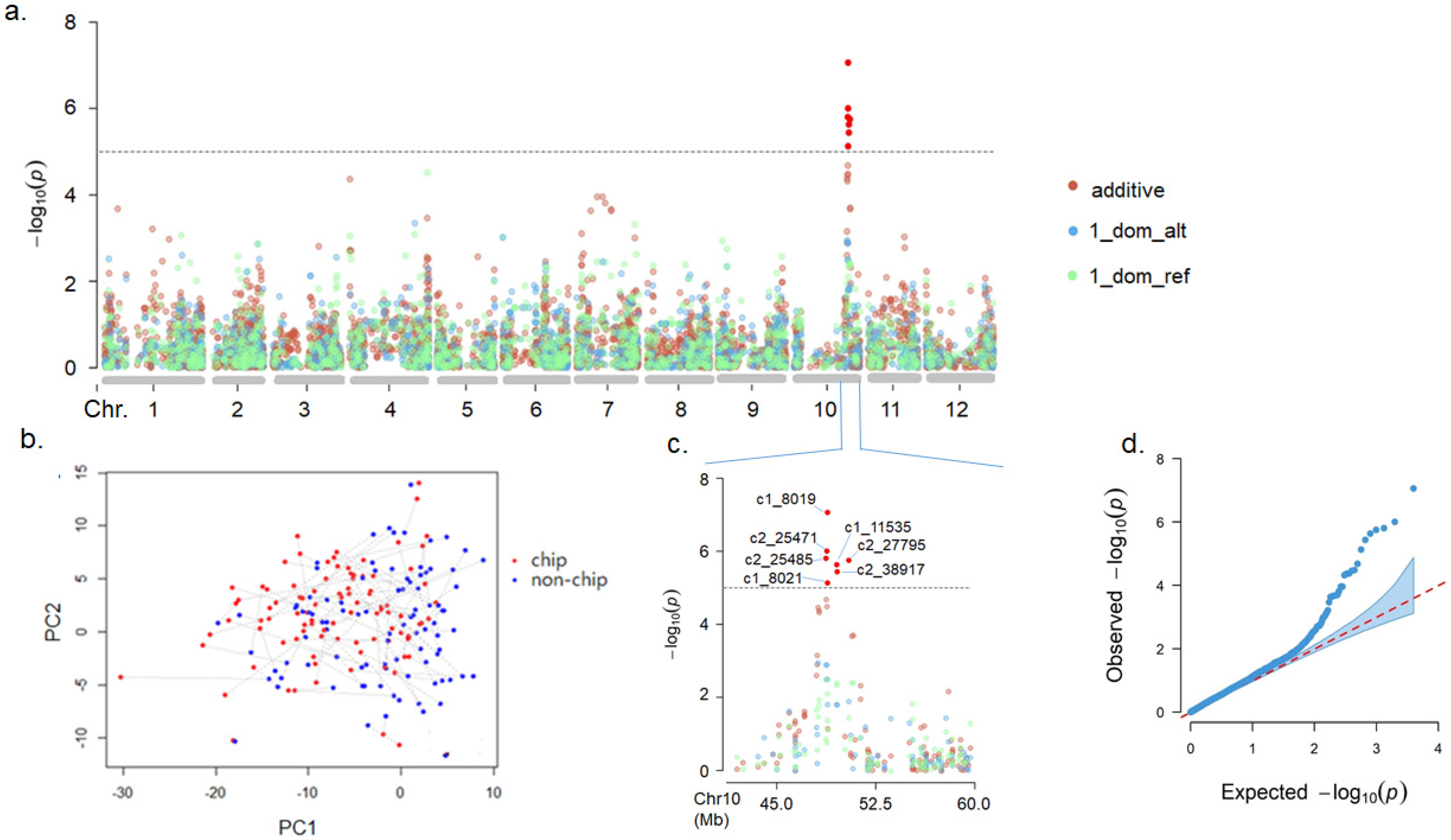
3.1. Genome-Wide Association Study
3.2. Selective Sweeps
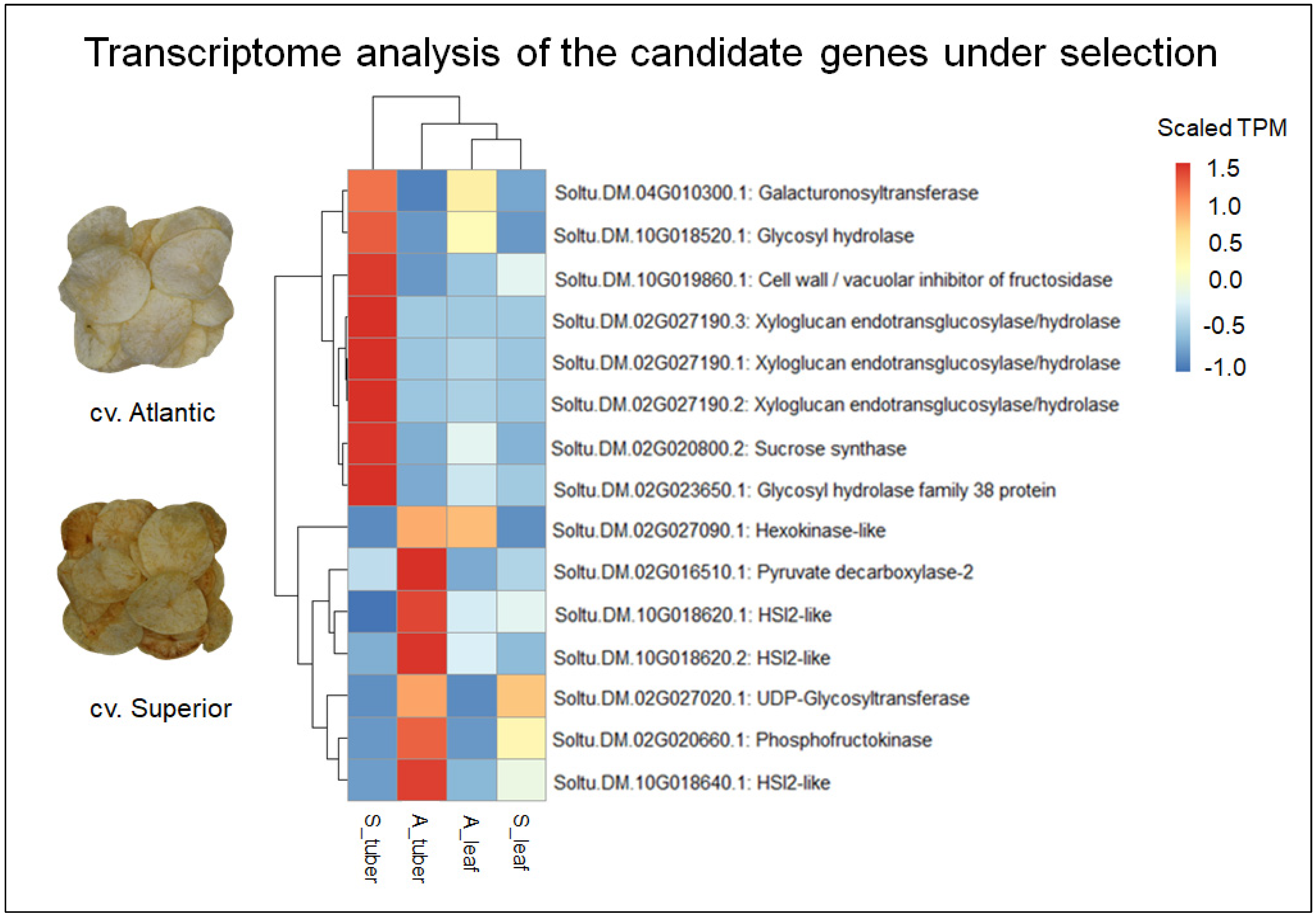
3.3. Transcriptome Analysis
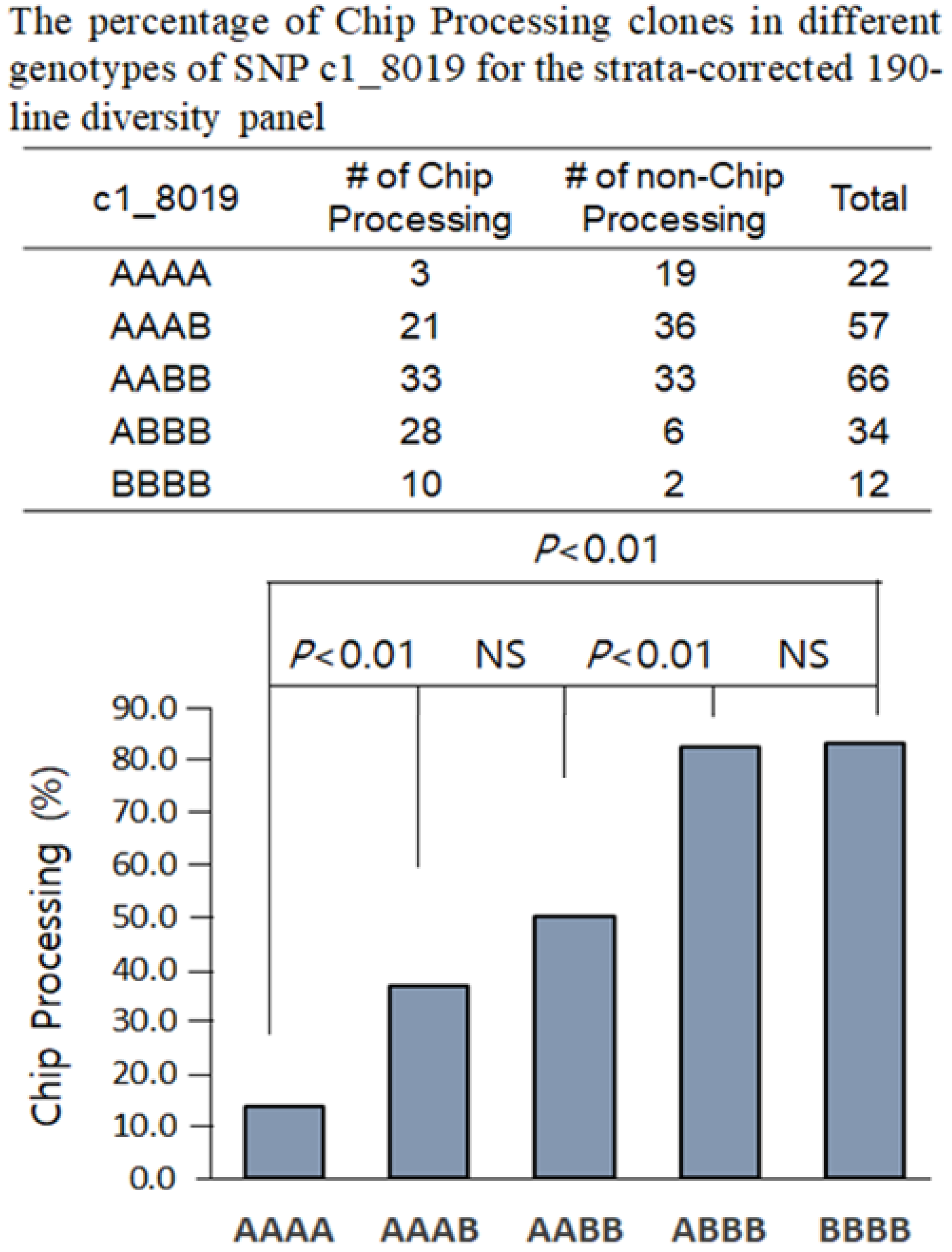
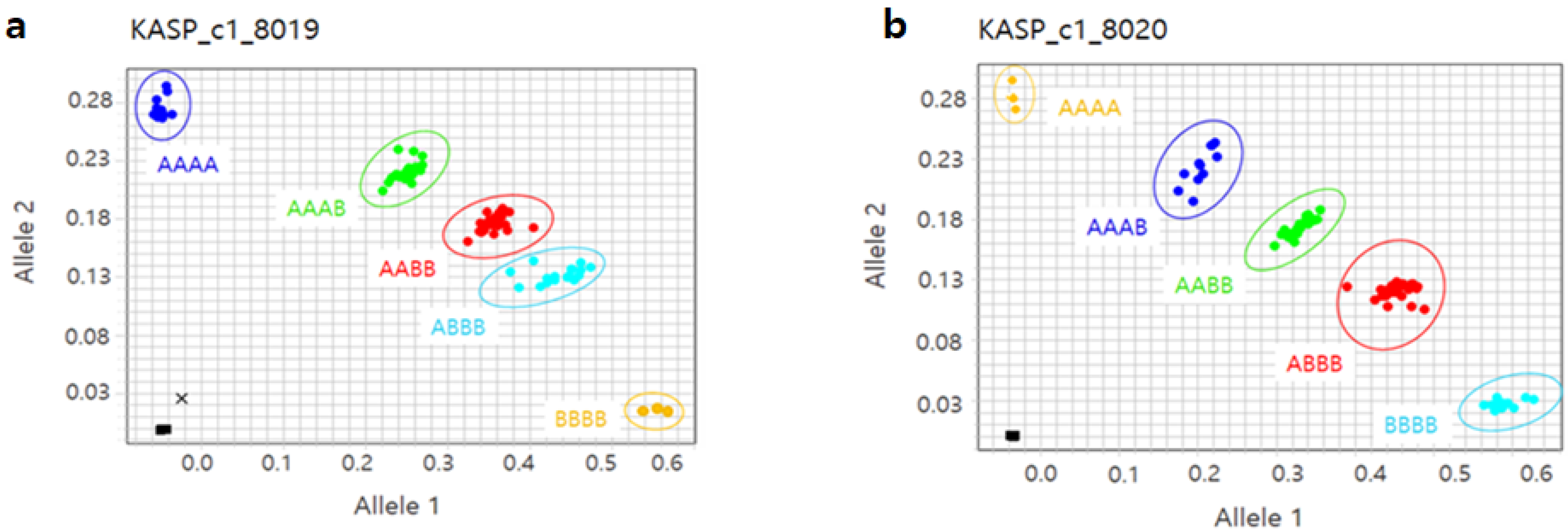
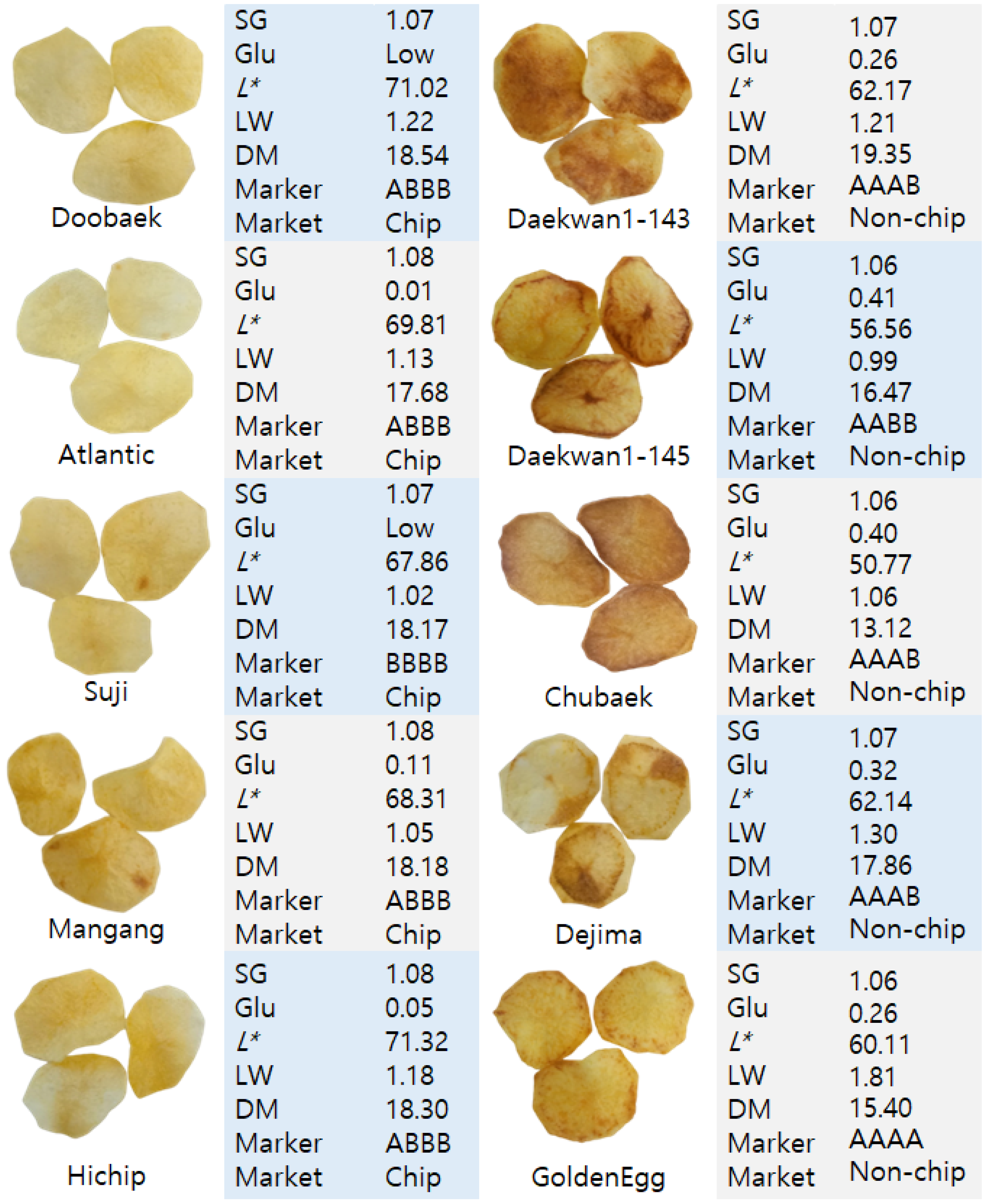
3.4. Kompetitive Allele-Specific PCR (KASP) Assays and Association with Phenotype Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Riaz, M.N. Snack Foods: Processing. In Encyclopedia of Food Grains, 2nd ed.; Wrigley, C., Corke, H., Seetharaman, K., Faubion, J., Eds.; Academic Press: Cambridge, MA, USA, 2016; Volume 3, pp. 414–422. ISBN 9780123947864. [Google Scholar] [CrossRef]
- Hirsch, C.N.; Hirsch, C.D.; Felcher, K.; Coombs, J.; Zarka, D.; Van Deynze, A.; De Jong, W.; Veilleux, R.E.; Jansky, S.; Bethke, P.; et al. Retrospective view of North American potato (Solanum tuberosum L.) breeding in the 20th and 21st centuries. G3 2013, 3, 1003–1013. [Google Scholar] [CrossRef] [Green Version]
- Igarashi, T.; Tsuyama, M.; Ogawa, K.; Koizumi, E.; Sanetomo, R.; Hosaka, K. Evaluation of Japanese potatoes using single nucleotide polymorphisms (SNPs). Mol. Breed. 2019, 39, 9. [Google Scholar] [CrossRef]
- Jo, K.R.; Cho, S.; Cho, J.H.; Park, H.J.; Choi, J.G.; Park, Y.E.; Cho, K.S. Analysis of genetic diversity and population structure among cultivated potato clones from Korea and global breeding programs. Sci. Rep. 2022, 12, 10462. [Google Scholar] [CrossRef] [PubMed]
- Bonierbale, M.W.; Amoros, W.R.; Salas, E.; de Jong, W. Potato Breeding. In The Potato Crop: Its Agricultural, Nutritional and Social Contribution to Humankind; Campos, H., Ortiz, O., Eds.; Springer: Dordrecht, Germany, 2020. [Google Scholar] [CrossRef] [Green Version]
- Jansky, S.H.; Fajardo, D.A. Tuber starch amylose content is associated with cold-induced sweetening in potato. Food Sci. Nutr. 2014, 2, 628–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnel, E. Potato Breeding: A Challenge, as ever! Potato Res. 2008, 51, 327–332. [Google Scholar] [CrossRef]
- D’Hoop, B.B.; Keizer, P.L.C.; Paulo, M.J.; Visser, R.G.F.; van Eeuwijk, F.A.; van Eck, H.J. Identification of agronomically important QTL in tetraploid potato cultivars using a marker–trait association analysis. Theor. Appl. Genet. 2014, 127, 731–748. [Google Scholar] [CrossRef] [PubMed]
- Naeem, M.; Demirel, U.; Yousaf, M.F.; Caliskan, S.; Caliskan, M.E. Overview on domestication, breeding, genetic gain and improvement of tuber quality traits of potato using fast forwarding technique (GWAS): A review. Plant Breed. 2021, 140, 519–542. [Google Scholar] [CrossRef]
- Felcher, K.J.; Coombs, J.J.; Massa, A.N.; Hansey, C.N.; Hamilton, J.P.; Veilleux, R.E.; Bueel, C.R.; Douches, D.S. Integration of two diploid potato linkage maps with the potato genome sequence. PLoS ONE 2012, 7, e36347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vos, P.G.; Uitdewilligen, J.G.; Voorrips, R.E.; Visser, R.G.; van Eck, H.J. Development and analysis of a 20K SNP array for potato (Solanum tuberosum): An insight into the breeding history. Theor. Appl. Genet. 2015, 128, 2387–2401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voorrips, R.E.; Gort, G.; Vosman, B. Genotype calling in tetraploid species from bi-allelic marker data using mixture models. BMC Bioinform. 2011, 12, 172. [Google Scholar] [CrossRef] [Green Version]
- Carley, C.A.S.; Coombs, J.J.; Douches, D.S.; Bethke, P.C.; Palta, J.P.; Novy, R.G.; Endelman, J.B. Automated tetraploid genotype calling by hierarchical clustering. Theor. Appl. Genet. 2017, 130, 717–726. [Google Scholar] [CrossRef]
- Rosyara, U.R.; De Jong, W.S.; Douches, D.S.; Endelman, J.B. Software for genome-wide association studies in autopolyploids and its application to potato. Plant Genome 2016, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endelman, J.B.; Carley, C.A.S.; Bethke, P.C.; Coombs, J.J.; Clough, M.E.; da Silva, W.L.; De Jong, W.S.; Douches, D.S.; Frederick, C.M.; Haynes, K.G.; et al. Genetic Variance Partitioning and Genome-Wide Prediction with Allele Dosage Information in Autotetraploid Potato. Genetics 2018, 209, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Byrne, S.; Meade, F.; Mesiti, F.; Griffin, D.; Kennedy, C.; Milbourne, D. Genome-Wide Association and Genomic Prediction for Fry Color in Potato. Agronomy 2020, 10, 90. [Google Scholar] [CrossRef] [Green Version]
- Sverrisdóttir, E.; Byrne, S.; Sundmark, E.H.R.; Johnsen, H.; Kirk, H.G.; Asp, T.; Janss, L.; Nielsen, K.L. Genomic prediction of starch content and chipping quality in tetraploid potato using genotyping-by-sequencing. Theor. Appl. Genet. 2017, 130, 2091–2108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rak, K.; Bethke, P.C.; Palta, J.P. QTL mapping of potato chip color and tuber traits within an autotetraploid family. Mol. Breed. 2017, 37, 15. [Google Scholar] [CrossRef]
- Park, J.; Massa, A.N.; Douches, D.; Coombs, J.; Akdemir, D.; Yencho, G.C.; Whitworth, J.L.; Novy, R.G. Linkage and QTL mapping for tuber shape and specific gravity in a tetraploid mapping population of potato representing the russet market class. BMC Plant Biol. 2021, 21, 507. [Google Scholar] [CrossRef]
- Van Eck, H.J.; Jacobs, J.M.; Stam, P.; Ton, J.; Stiekema, W.J.; Jacobsen, E. Multiple alleles for tuber shape in diploid potato detected by qualitative and quantitative genetic analysis using RFLPs. Genetics 1994, 137, 303–309. [Google Scholar] [CrossRef]
- Śliwka, J.; Wasilewicz-Flis, I.; Jakuczun, H.; Gebhardt, C. Tagging quantitative trait loci for dormancy, tuber shape, regularity of tuber shape, eye depth and flesh colour in diploid potato originated from six Solanum species. Plant Breed. 2008, 127, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Li, X.Q.; De Jong, H.; De Jong, D.M.; De Jong, W.S. Inheritance and genetic mapping of tuber eye depth in cultivated diploid potatoes. Theor. Appl. Genet. 2005, 110, 1068–1073. [Google Scholar] [CrossRef]
- Prashar, A.; Hornyik, C.; Young, V.; McLean, K.; Sharma, S.K.; Dale, M.F.B.; Bryan, G.J. Construction of a dense SNP map of a highly heterozygous diploid potato population and QTL analysis of tuber shape and eye depth. Theor. Appl. Genet. 2014, 127, 2159–2171. [Google Scholar] [CrossRef]
- Pandey, J.; Scheuring, D.C.; Koym, J.W.; Coombs, J.; Novy, R.G.; Thompson, A.L.; Holm, D.G.; Douches, D.S.; Miller, J.C.; Vales, M.I. Genetic diversity and population structure of advanced clones selected over forty years by a potato breeding program in the USA. Sci. Rep. 2021, 11, 8344. [Google Scholar] [CrossRef]
- Li, L.; Paulo, M.-J.; Strahwald, J.; Lübeck, J.; Hofferbert, H.-R.; Tacke, E.; Junghans, H.; Wunder, J.; Draffehn, A.; van Eeuwijk, F.; et al. Natural DNA variation at candidate loci is associated with potato chip color, tuber starch content, yield and starch yield. Theor Appl Genet. 2008, 116, 1167–1181. [Google Scholar] [CrossRef] [Green Version]
- Schönhals, E.M.; Ortega, F.; Barandalla, L.; Aragones, A.; de Galarreta, J.I.R.; Liao, J.C.; Sanetomo, R.; Walkemeier, B.; Tacke, E.; Ritter, E.; et al. Identification and reproducibility of diagnostic DNA markers for tuber starch and yield optimization in a novel association mapping population of potato (Solanum tuberosum L). Theor. Appl. Genet. 2016, 129, 767–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolini, F.; Servin, B.; Talenti, A.; Rochat, E.; Kim, E.S.; Oget, C.; Palhière, I.; Crisà, A.; Catillo, G.; Steri, R.; et al. Signatures of selection and environmental adaptation across the goat genome post-domestication. Genet. Sel. Evol. 2018, 50, 57. [Google Scholar] [CrossRef]
- Qanbari, S.; Simianer, H. Mapping signatures of positive selection in the genome of livestock. Livest. Sci. 2014, 166, 133–143. [Google Scholar] [CrossRef]
- López, M.E.; Cádiz, M.I.; Rondeau, E.B.; Koop, B.F.; Yáñez, J.M. Detection of selection signatures in farmed coho salmon (Oncorhynchus kisutch) using dense genome-wide information. Sci. Rep. 2021, 11, 9685. [Google Scholar] [CrossRef]
- Oleksyk, T.K.; Smith, M.W.; O’brien, S.J. Genome-wide scans for footprints of natural selection. Philos. Trans. R. Soc. B 2010, 365, 185–205. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.B.; Wang, G.W.; Yuan, M.; Yao, W.; Lv, K.; Zhao, H.; Yang, M. Breeding signatures of rice improvement revealed by a genomic variation map from a large germplasm collection. Proc. Natl. Acad. Sci. USA 2015, 112, 5411–5419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dadshani, S.; Mathew, B.; Ballvora, A.; Mason, A.S.; Léon, J. Detection of breeding signatures in wheat using a linkage disequilibrium-corrected mapping approach. Sci. Rep. 2021, 11, 5527. [Google Scholar] [CrossRef] [PubMed]
- Saleem, A.; Muylle, H.; Aper, J.; Ruttink, T.; Wang, J.; Yu, D.; Roldán-Ruiz, I. A Genome-Wide Genetic Diversity Scan Reveals Multiple Signatures of Selection in a European Soybean Collection Compared to Chinese Collections of Wild and Cultivated Soybean Accessions. Front. Plant Sci. 2021, 12, 631767. [Google Scholar] [CrossRef] [PubMed]
- Bekele, W.A.; Wight, C.P.; Chao, S.; Howarth, C.J.; Tinker, N.A. Haplotype-based genotyping-by-sequencing in oat genome research. Plant Biotechnol. J. 2018, 16, 1452–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean-Rodríguez, F.D.; Costich, D.E.; Camacho-Villa, T.C.; Pè, M.E.; Dell’Acqua, M. Genetic diversity and selection signatures in maize landraces compared across 50 years of in situ and ex situ conservation. Heredity 2021, 126, 913–928. [Google Scholar] [CrossRef] [PubMed]
- Bauchet, G.; Munos, S.; Sauvage, C.; Bonnet, J.; Grivet, L.; Causse, M. Genes involved in floral meristem in tomato exhibit drastically reduced genetic diversity and signature of selection. BMC Plant Biol. 2014, 14, 279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franks, S.J.; Kane, N.C.; O’Hara, N.B.; Tittes, S.; Rest, J.S. Rapid genome-wide evolution in Brassica rapa populations following drought revealed by sequencing of ancestral and descendant gene pools. Mol. Ecol. 2016, 25, 3622–3631. [Google Scholar] [CrossRef]
- Chen, H.; Patterson, N.; Reich, D. Population differentiation as a test for selective sweeps. Genome Res. 2010, 20, 393–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabeti, P.C.; Varilly, P.; Fry, B.; Lohmueller, J.; Hostetter, E.; Cotsapas, C.; Xie, X.; Byrne, E.H.; McCarroll, S.A.; Gaudet, R. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 913–918. [Google Scholar] [CrossRef] [Green Version]
- Tang, K.; Thornton, K.R.; Stoneking, M. A new approach for using genome scans to detect recent positive selection in the human genome. PLoS Biol. 2007, 5, e171. [Google Scholar] [CrossRef]
- Luu, K.; Bazin, E.; Blum, M.G. pcadapt: An R package to perform genome scans for selection based on principal component analysis. Mol. Ecol. Resour. 2017, 17, 67–77. [Google Scholar] [CrossRef]
- Pandey, J.; Scheuring, D.C.; Koym, J.W.; Vales, M.I. Genomic regions associated with tuber traits in tetraploid potatoes and identification of superior clones for breeding purposes. Front. Plant Sci. 2022, 13, 952263. [Google Scholar] [CrossRef]
- Ohara-Takada, A.; Matsuura-Endo, C.; Chuda, Y.; Ono, H.; Yada, H.; Yoshida, M.; Kobayashi, A.; Tsuda, S.; Takigawa, S.; Noda, T.; et al. Change in content of sugars and free amino acids in potato tubers under short-term storage at low temperature and the effect on acrylamide level after frying. Biosci. Biotechnol. Biochem. 2005, 69, 1232–1238. [Google Scholar] [CrossRef] [Green Version]
- Brahmakshatriya, R.D.; Donker, J.D. Five Methods for Determination of Silage Dry Matter1. J. Dairy Sci. 1971, 54, 1470–1474. [Google Scholar] [CrossRef]
- Alvarado, G.; Rodríguez, F.M.; Pacheco, A.; Burgueño, J.; Crossa, J.; Vargas, M.; Pérez-Rodríguez, P.; Lopez-Cruz, M.A. META-R: A software to analyze data from multi-environment plant breeding trials. Crop J. 2020, 8, 745–756. [Google Scholar] [CrossRef]
- Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.W.; Myers, T.A.; Machiela, M.J. PCAmatchR: A flexible R package for optimal case–control matching using weighted principal components. Bioinformatics 2021, 37, 1178–1181. [Google Scholar] [CrossRef]
- da Silva Pereira, G.; Mollinari, M.; Schumann, M.J.; Clough, M.E.; Zeng, Z.-B.; Yencho, G.C. The recombination landscape and multiple QTL mapping in a Solanum tuberosum cv. ‘Atlantic’-derived F1 population. Heredity 2021, 126, 817–830. [Google Scholar] [CrossRef]
- Delaneau, O.; Howie, B.; Cox, A.J.; Zagury, J.; Marchini, J. Haplotype Estimation Using Sequencing Reads. Am. J. Hum. Genet. 2013, 93, 687–696. [Google Scholar] [CrossRef] [Green Version]
- Gautier, M.; Vitalis, R. rehh: An R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics 2012, 28, 1176–1177. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.K.; MacKenzie, K.; McLean, K.; Dale, F.; Daniels, S.; Bryan, G.J. Linkage Disequilibrium and Evaluation of Genome-Wide Association Mapping Models in Tetraploid Potato. G3 Genes Genomes Genet. 2018, 8, 3185–3202. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ji, L. Adjusting multiple testing in multilocus analyses using the eigenvalues of a correlation matrix. Heredity 2005, 95, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Tsukagoshi, H.; Morikami, A.; Nakamura, K. Two B3 domain transcriptional repressors prevent sugar-inducible expression of seed maturation genes in Arabidopsis seedlings. Proc. Natl. Acad. Sci. USA 2007, 104, 2543–2547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, G.M.; Newton, L.; Wiegert-Rininger, K.; Vaillancourt, B.; Douches, D.S.; Buell, C.R. Extensive genome heterogeneity leads to preferential allele expression and copy number-dependent expression in cultivated potato. Plant J. 2017, 92, 624–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uffelmann, E.; Huang, Q.Q.; Munung, N.S.; de Vries, J.; Okada, Y.; Martin, A.R.; Martin, H.C.; Lappalainen, T.; Posthuma, D. Genome-wide association studies. Nat. Rev. Method. Prim. 2021, 1, 59. [Google Scholar] [CrossRef]
- Roberts, G.H.L.; Partha, R.; Rhead, B.; Knight, S.C.; Park, D.S.; Coignet, M.V.; Zhang, M.; Berkowitz, N.; Turrisini, D.A.; Gaddis, M.; et al. Expanded COVID-19 phenotype definitions reveal distinct patterns of genetic association and protective effects. Nat. Genet. 2022, 54, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; He, M.; Fan, Y.; Zhao, H.; Gao, B.; Yang, K.; Li, F.; Tang, Y.; Gao, Q.; Lin, T.; et al. Resequencing of global Tartary buckwheat accessions reveals multiple domestication events and key loci associated with agronomic traits. Genome Biol. 2021, 22, 23. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Veerappan, V.; Abdelmageed, H.; Kang, M.; Allen, R.D. HSI2/VAL1 Silences AGL15 to Regulate the Developmental Transition from Seed Maturation to Vegetative Growth in Arabidopsis. Plant Cell 2018, 30, 600–619. [Google Scholar] [CrossRef] [Green Version]
- Jing, Y.; Guo, Q.; Lin, R. The B3-Domain Transcription Factor VAL1 Regulates the Floral Transition by Repressing FLOWERING LOCUS T. Plant Physiol. 2019, 181, 236–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, G.; Wang, Q.; Xu, J.; Chen, N.; Zhu, W.; Duan, S.; Yang, X.; De Jong, W.S.; Guo, Y.; Jin, L.; et al. Fine Mapping and Candidate Gene Prediction of Tuber Shape Controlling Ro Locus Based on Integrating Genetic and Transcriptomic Analyses in Potato. Int. J. Mol. Sci. 2022, 23, 1470. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Marker | Chr | Position (bp) | Model | MAF | Infomativeness (%) a | −log10(p) |
|---|---|---|---|---|---|---|
| c1_8019 | 10 | 48863165 | additive | 0.39 | 73.0 | 7.06 |
| c2_25471 | 10 | 48808404 | additive | 0.36 | 71.2 | 6.00 |
| c2_25485 | 10 | 48737840 | additive | 0.41 | 72.0 | 5.80 |
| c2_27795 | 10 | 50458044 | additive | 0.38 | 69.5 | 5.76 |
| c1_11535 | 10 | 49553136 | additive | 0.31 | 67.9 | 5.63 |
| c2_38917 | 10 | 49584775 | additive | 0.33 | 69.5 | 5.44 |
| c1_8021 | 10 | 48862950 | additive | 0.32 | 70.0 | 5.13 |
| SNP | Candidate Gene | Selective Sweep Region | Chr | Statistic | Putative Function | ||||
|---|---|---|---|---|---|---|---|---|---|
| a | b | c | d | e | |||||
| c2_52484 | Soltu.DM.01G029570 | - | 1 | 6.64 | 4.47 | Translation initiation factor 3B1 | |||
| c2_37836 | No Hits | - | 1 | 3.90 | 3.55 | - | |||
| c2_52656 | Soltu.DM.02G009060 | - | 2 | 3.67 | 3.08 | Alpha/beta-Hydrolases | |||
| Soltu.DM.02G009030 | chr02:23845672..23949866 (104.2 Kb) | 2 | UDP-Glycosyltransferase | ||||||
| Soltu.DM.02G009040 | chr02:23845672..23949866 (104.3 Kb) | 2 | Transmembrane amino acid transporter | ||||||
| c2_21650 | Soltu.DM.02G009580 | - | 2 | 3.75 | 3.10 | RNA-binding KH domain-containing protein | |||
| Soltu.DM.02G009480 | chr02:24366517..24474785 (108.27 Kb) | 2 | UDP-glucose 6-dehydrogenase | ||||||
| c2_41975 | Soltu.DM.02G011550 | - | 2 | 4.78 | 4.64 | Exostosin | |||
| c2_38935 | Soltu.DM.02G011840 | - | 2 | 4.79 | 3.90 | Glutathione S-transferase | |||
| c1_13186 | Soltu.DM.02G016510 | - | 2 | 4.75 | 3.33 | Pyruvate decarboxylase-2 | |||
| c2_48783 | No Hits | - | 2 | 4.02 | 3.96 | - | |||
| c1_14419 | Soltu.DM.02G017460 | - | 2 | 4.54 | 4.18 | VQ motif-containing protein | |||
| c1_5871 | Soltu.DM.02G020130 | - | 2 | 5.49 | 3.95 | Peroxidase | |||
| Soltu.DM.02G020170.2 | chr02:34333640..34436373 (102.73 Kb) | 2 | Starch synthase | ||||||
| c2_17793 | Soltu.DM.02G020670 | - | 2 | 4.36 | 7.42 | Fibrillin | |||
| Soltu.DM.02G020660 | chr02:34822051..34879350 (57.3 Kb) | 2 | Phosphofructokinase | ||||||
| c2_42085 | Soltu.DM.02G023660.1 | - | 2 | 5.45 | 5.80 | Regulatory particle non-ATPase | |||
| Soltu.DM.02G023650 | chr02:37080051..37137350 (57.3 Kb) | 2 | Glycosyl hydrolase family 38 protein | ||||||
| c1_8129 | Soltu.DM.02G024720 | - | 2 | 6.29 | 6.82 | HOPZ-ACTIVATED RESISTANCE | |||
| Soltu.DM.02G024640 | chr02:37929951..37987250 (57.3 Kb) | 2 | UDP-Glycosyltransferase | ||||||
| c2_7389 | Soltu.DM.02G025590 | - | 2 | 5.70 | 3.15 | Allene oxide cyclase | |||
| Soltu.DM.02G025510 | chr02:38696301..38753600 (57.3 Kb) | 2 | UDP-glucosyl transferase 72E1 | ||||||
| c2_7425 | Soltu.DM.02G027100 | - | 2 | 6.23 | 12.59 | Leucine-rich receptor-like protein kinase | |||
| Soltu.DM.02G027090 | chr02:39894251..39951550 (57.3 Kb) | 2 | Hexokinase-like | ||||||
| c2_54104 | Soltu.DM.02G027250 | - | 2 | 6.19 | 11.17 | Branched-chain amino acid aminotransferase 5 (BCAT5) | |||
| Soltu.DM.02G027190 | chr02:40025601..40082900 (57.3 Kb) | 2 | Xyloglucan endotransglucosylase/hydrolase | ||||||
| c2_52087 | No Hits | - | 2 | 6.19 | 11.26 | - | |||
| c2_43352 | Soltu.DM.02G029880.1 | - | 2 | 6.12 | 4.62 | BEL1-like homeodomain | |||
| c2_54687 | Soltu.DM.03G013510 | - | 3 | 3.26 | 3.86 | Protein kinase | |||
| Soltu.DM.03G013410 | chr03:35810742..35917929 (107.19 Kb) | 3 | Alpha-amylase-like | ||||||
| c2_26799 | Soltu.DM.04G010300 | - | 4 | 3.03 | 3.17 | Galacturonosyltransferase | |||
| Soltu.DM.04G010260.5 | chr04:10700737..10806774 (106.04 Kb) | 4 | Galactosyltransferase | ||||||
| Soltu.DM.04G010350 | chr04:10800737..10906774 (106.04 Kb) | 4 | UDP-glucosyl transferase 73B3 | ||||||
| c2_8295 | Soltu.DM.05G024620 | - | 5 | 6.32 | 3.17 | Nudix hydrolase homolog | |||
| c2_35056 | Soltu.DM.07G020260 | - | 7 | 6.16 | 18.49 | HAESA-like | |||
| Soltu.DM.07G020320 | chr07:50787767..50892909 (105.14 Kb) | 7 | FASCICLIN-like arabinoogalactan | ||||||
| c2_25485 | Soltu.DM.10G018530 | - | 10 | 3.44 | 5.46 | Serine/threonine protein kinase | |||
| Soltu.DM.10G018520 | chr10:49577881..49600800 (22.92 Kb) | 10 | Glycosyl hydrolase | ||||||
| c2_25471 | Soltu.DM.10G018580 | - | 10 | 5.16 | 5.57 | Lung seven transmembrane receptor | |||
| Soltu.DM.10G018520 | chr10:49555549..49663946 (108.4 Kb) | 10 | Glycosyl hydrolase | ||||||
| c1_8019 | Soltu.DM.10G018620.1 | - | 10 | 3.71 | 4.54 | 6.6 | HSI2-like | ||
| Soltu.DM.10G018620.2 | - | 10 | HSI2-like | ||||||
| Soltu.DM.10G018640 | chr10:49709841..49732760 (22.92 Kb) | 10 | HSI2-like | ||||||
| c2_27795 | Soltu.DM.10G019800 | - | 10 | 7.23 | 6.05 | Mediator of RNA polymerase II transcription subunit | |||
| Soltu.DM.10G019820 | chr10:51395638..51504347 (108.71 Kb) | 10 | UDP-Glycosyltransferase | ||||||
| Soltu.DM.10G019860 | chr10:51395638..51504347 (108.71 Kb) | 10 | Cell wall/vacuolar inhibitor of fructosidase | ||||||
| c2_16302 | Soltu.DM.12G019560 | - | 12 | 3.64 | 5.62 | Thioesterase/thiol ester dehydrase-isomerase | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jo, K.R.; Choi, J.-G.; Kwon, D.-H.; Park, Y.-E.; Kim, S.-J. Revealing Genetic Variations Associated with Chip-Processing Properties in Potato (Solanum tuberosum L.). Agronomy 2023, 13, 642. https://doi.org/10.3390/agronomy13030642
Jo KR, Choi J-G, Kwon D-H, Park Y-E, Kim S-J. Revealing Genetic Variations Associated with Chip-Processing Properties in Potato (Solanum tuberosum L.). Agronomy. 2023; 13(3):642. https://doi.org/10.3390/agronomy13030642
Chicago/Turabian StyleJo, Kwang Ryong, Jang-Gyu Choi, Do-Hee Kwon, Young-Eun Park, and Su-Jeong Kim. 2023. "Revealing Genetic Variations Associated with Chip-Processing Properties in Potato (Solanum tuberosum L.)" Agronomy 13, no. 3: 642. https://doi.org/10.3390/agronomy13030642
APA StyleJo, K. R., Choi, J.-G., Kwon, D.-H., Park, Y.-E., & Kim, S.-J. (2023). Revealing Genetic Variations Associated with Chip-Processing Properties in Potato (Solanum tuberosum L.). Agronomy, 13(3), 642. https://doi.org/10.3390/agronomy13030642