
Genome-Wide Identification, Classification and Expression Analyses of MADS-Box Genes Reveal Their Role in Stem Gall Formation and Expansion of Zizania latifolia
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of MADS-Box Family Members in Z. latifolia
2.2. Analysis of Protein Properties, Subcellular Localization and Tertiary Structure of MADS-Box Genes in Z. latifolia
2.3. Phylogenetic Tree Construction of MADS-Box Proteins in Z. latifolia
2.4. Chromosomal Localization, Collinearity Analysis and SSRs Predictions of MADS-Box Genes in Z. latifolia
2.5. Conserved Motif and Gene Structure Analysis of the MADS-Box Family in Z. latifolia
2.6. Cis-Acting Element Analysis of MADS-Box Genes in Z. latifolia
2.7. MicroRNA (miRNA) Target of MADS-Box Genes in Z. latifolia
2.8. Plant Growth and Sample Collection
2.9. Transcriptome Sequencing Analysis
2.10. Gene Expression Analysis by qRT-PCR
2.11. Statistical Analysis
3. Results
3.1. Information on the ZlMADS-Box Gene Family
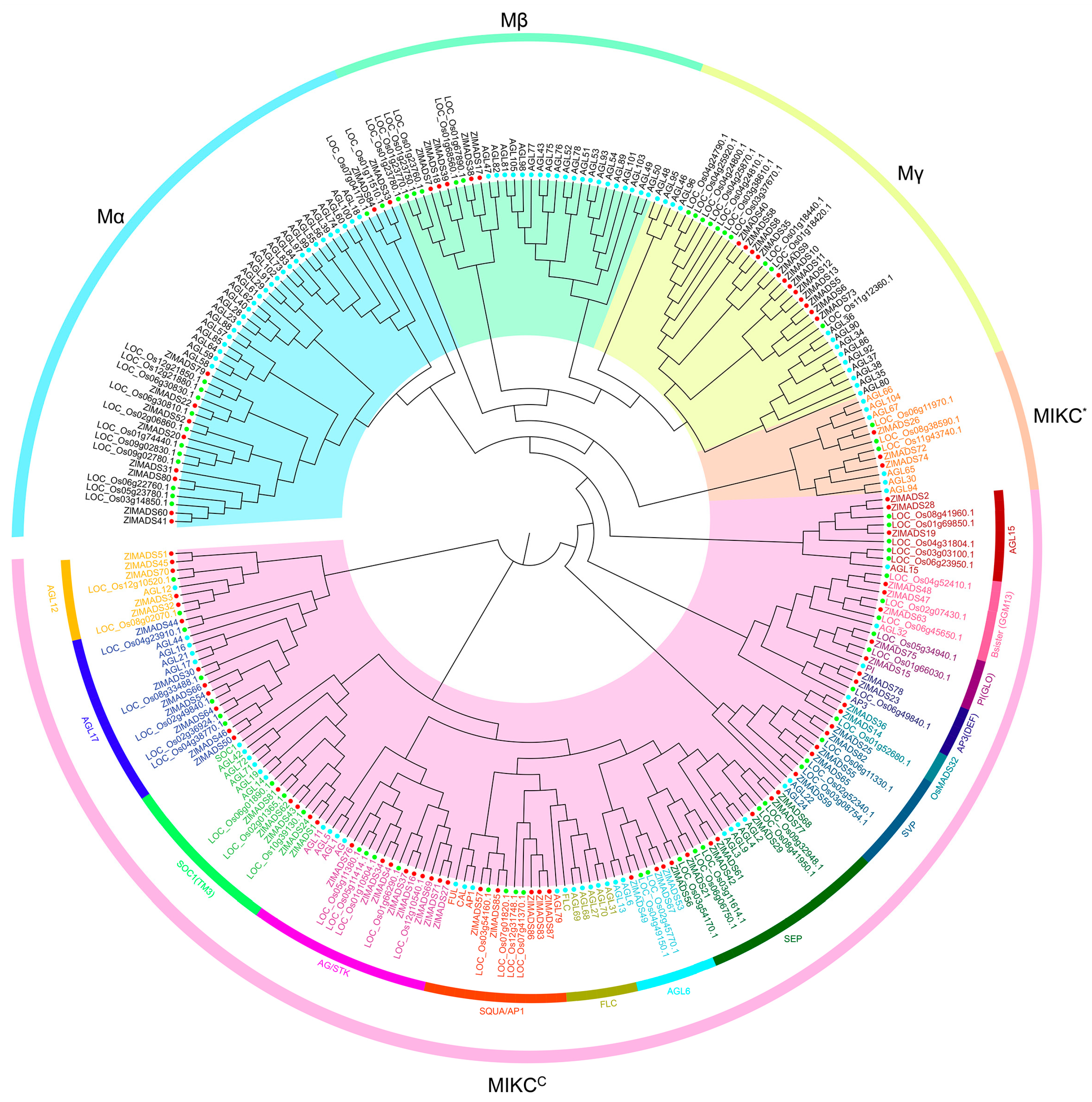
3.2. Phylogenetic Analysis of MADS-Box Genes in Z. latifolia
3.3. Analysis of MADS Protein Conserved Motifs and Gene Structure
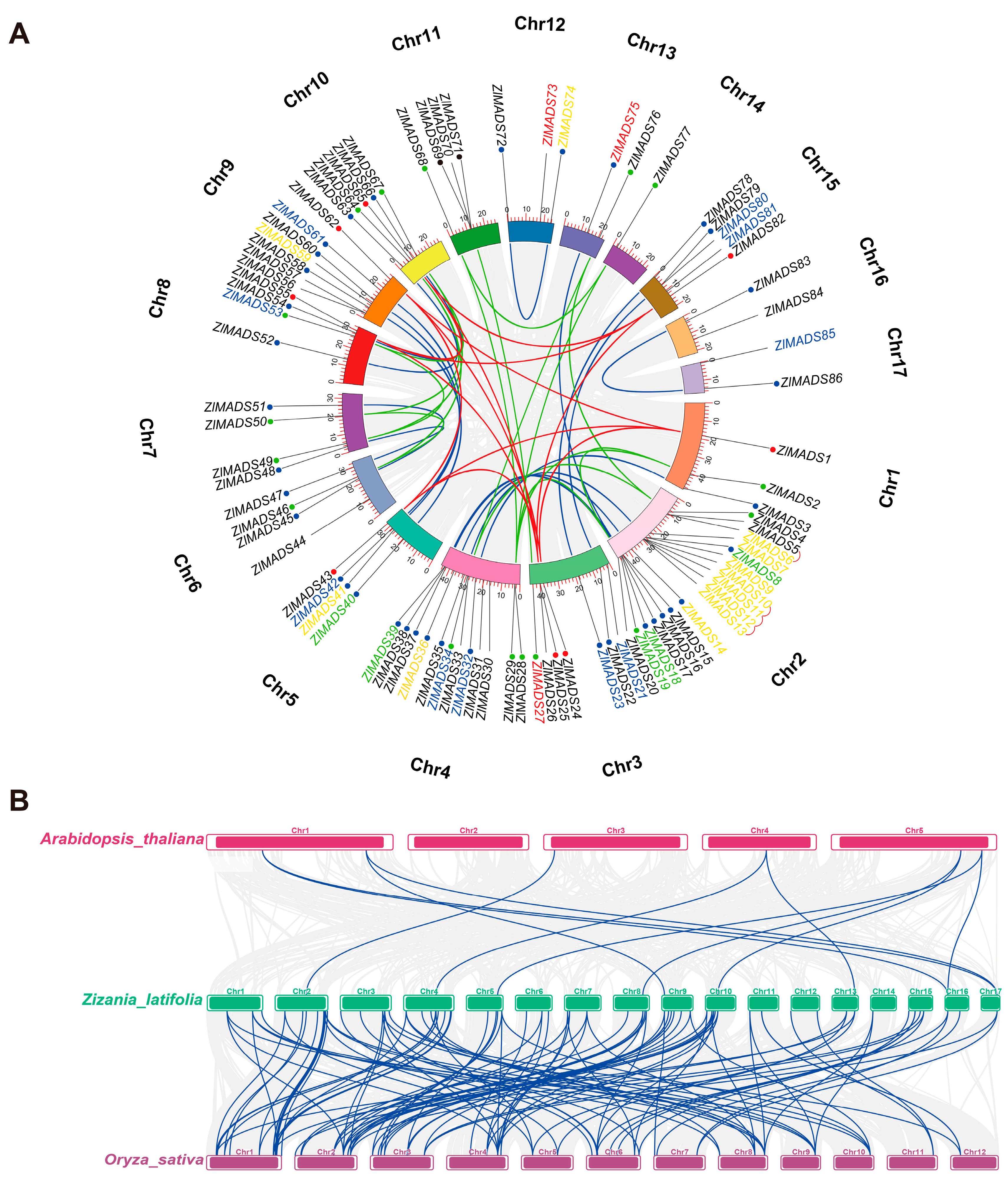
3.4. Chromosome Distribution and Duplication Events of MADS-Box Genes
3.5. Analysis of Cis-Acting Elements of MADS-Box Gene Promoter Regions in Z. latifolia
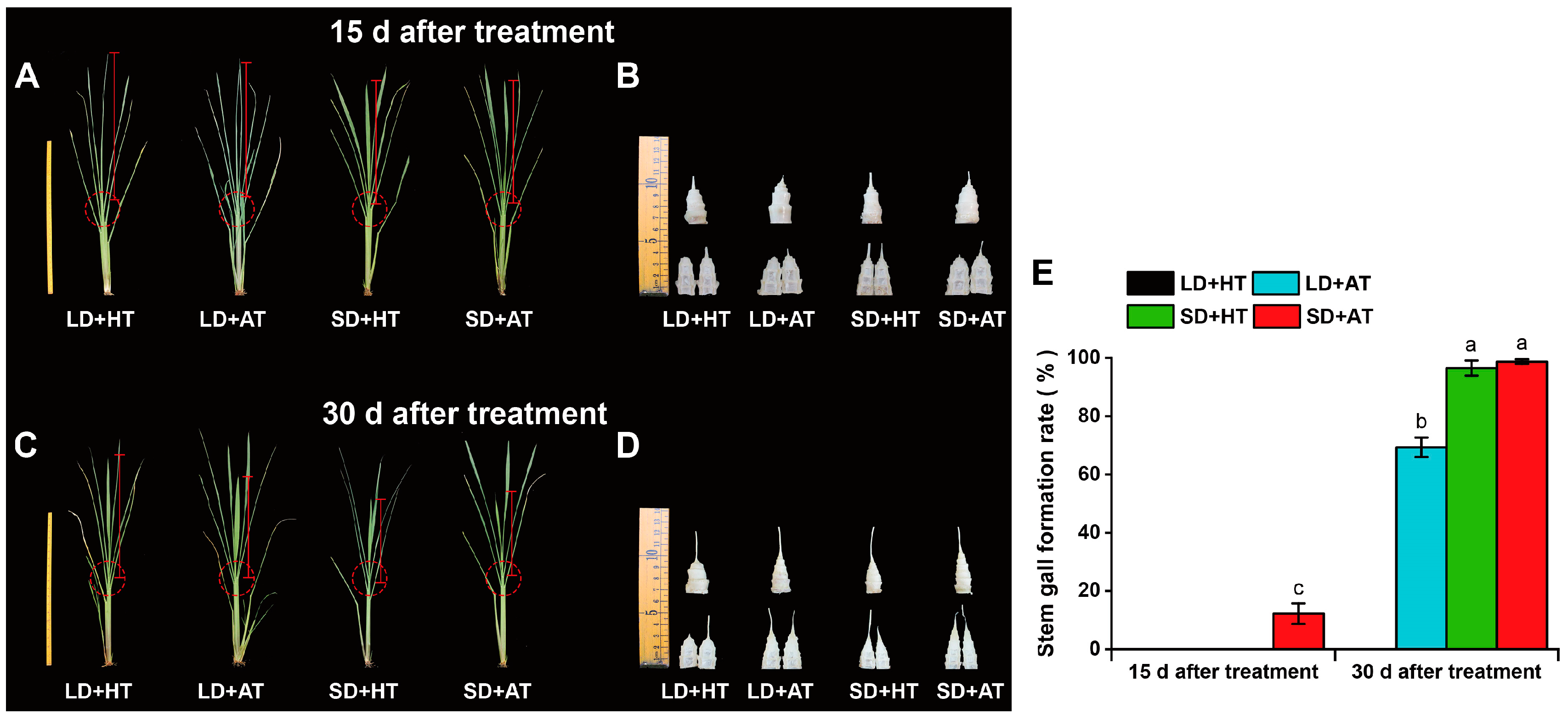
3.6. Short Days Promote the Formation of SG in Z. latifolia
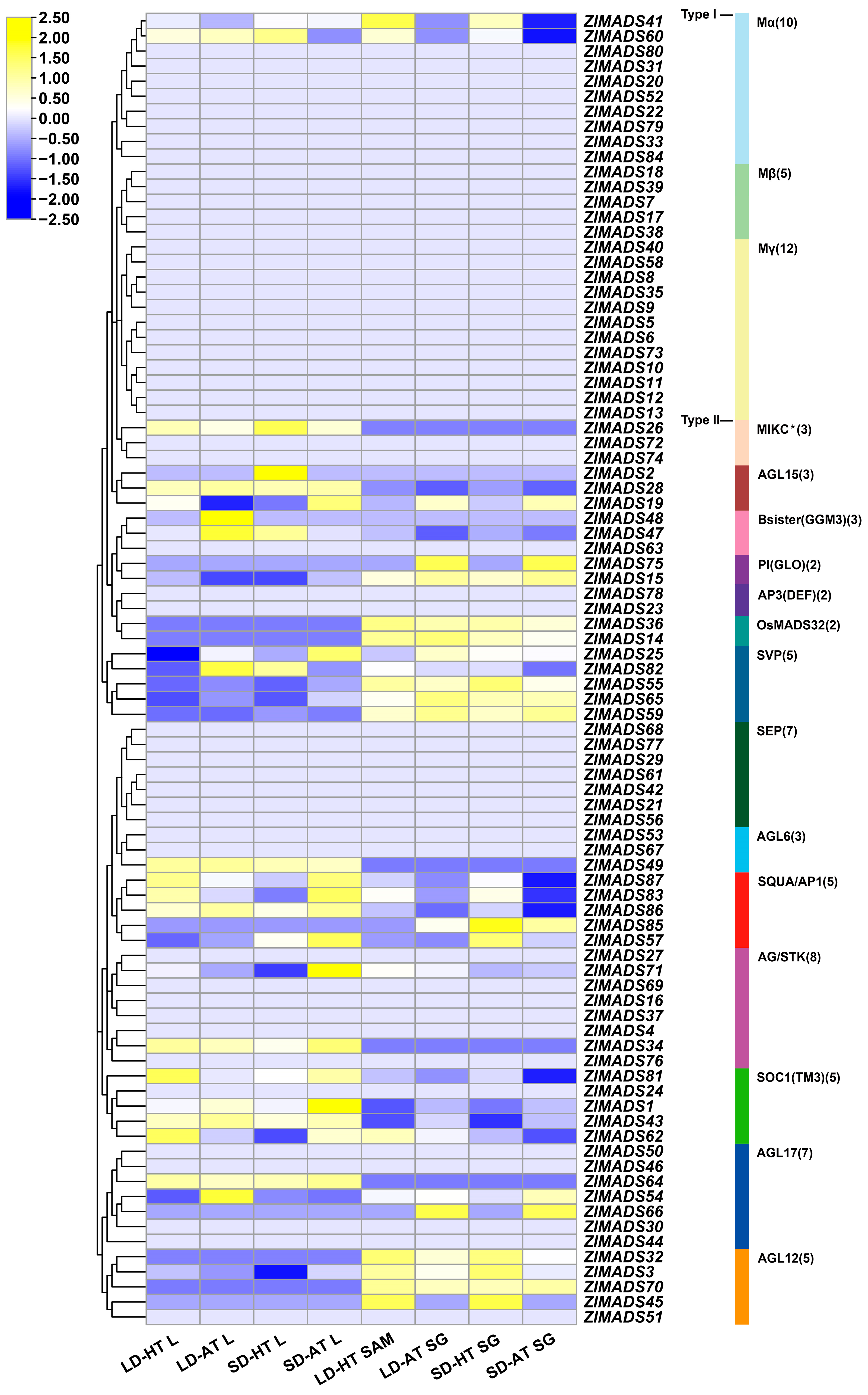
3.7. Expression Analysis of ZlMADS-Box Genes under Different Photoperiods and Temperature Conditions
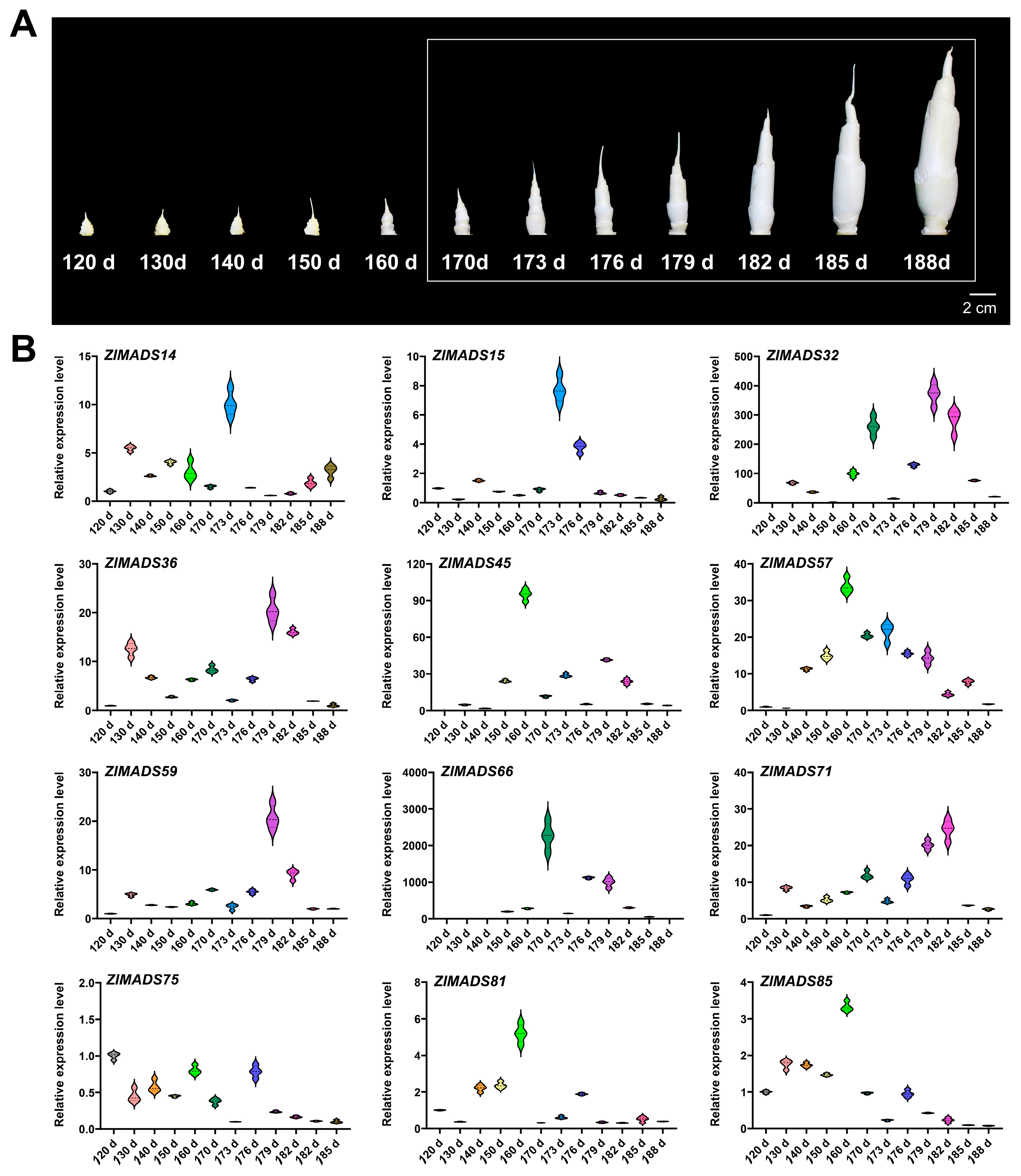
3.8. Expression Analysis of MADS-Box Genes during the Developmental Stage of Z. latifolia
3.9. miRNA Targets and Tertiary Structure of MADS-Box Gene Family in Z. latifolia
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Messenguy, F.; Dubois, E. Role of MADS box proteins and their cofactors in combinatorial control of gene expression and cell development. Gene 2003, 316, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Passmore, S.; Maine, G.T.; Elble, R.; Christ, C.; Tye, B.K. Saccharomyces cerevisiae protein involved in plasmid maintenance is necessary for mating of MATα cells. J. Mol. Biol. 1988, 204, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Yanofsky, M.F.; Ma, H.; Bowman, J.L.; Drews, G.N.; Feldmann, K.A.; Meyerowitz, E.M. The protein encoded by the Arabidopsis homeotic gene agamous resembles transcription factors. Nature 1990, 346, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Sommer, H.; Beltrán, J.P.; Huijser, P.; Pape, H.; Lönnig, W.E.; Saedler, H.; Schwarz-Sommer, Z. Deficiens, a homeotic gene involved in the control of flower orphogenesis in Antirrhinum majus: The protein shows homology to transcription factors. EMBO J. 1990, 9, 605–613. [Google Scholar] [CrossRef]
- Norman, C.; Runswick, M.; Pollock, R.; Treisman, R. Isolation and properties of cDNA clones encoding SRF, a transcription factor that binds to the c-fos serum response element. Cell 1988, 55, 989–1003. [Google Scholar] [CrossRef]
- West, A.G.; Shore, P.; Sharrocks, A.D. DNA binding by MADS-box transcription factors: A molecular mechanism for differential DNA bending. Mol. Cell. Biol. 1997, 17, 2876–2887. [Google Scholar] [CrossRef] [Green Version]
- Gramzow, L.; Ritz, M.S.; Theißen, G. On the origin of MADS-domain transcription factors. Trends Genet. 2010, 26, 149–153. [Google Scholar] [CrossRef]
- Henschel, K.; KOFUJI, R.; Hasebe, M.; Saedler, H.; Münster, T.; Theißen, G. Two ancient classes of MIKC-type MADS-box genes are present in the moss Physcomitrella patens. Mol. Biol. Evol. 2002, 19, 801–814. [Google Scholar] [CrossRef]
- Bodt, S.D.; Raes, J.; Florquin, K.; Rombauts, S.; Rouz, P.; Theien, G.; Peer, Y.V.D. Genomewide Structural Annotation and Evolutionary Analysis of the Type I MADS-Box Genes in Plants. J. Mol. Evol. 2003, 56, 573–586. [Google Scholar] [CrossRef]
- Becker, A.; Theißen, G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenetics Evol. 2003, 29, 464–489. [Google Scholar] [CrossRef]
- Gramzow, L.; Theißen, G. Phylogenomics reveals surprising sets of essential and dispensable clades of MIKCc-group MADS-box genes in flowering plants. J. Exp. Zool. B Mol. Dev. Evol. 2015, 324, 353–362. [Google Scholar] [CrossRef]
- Alvarez-Buylla, E.R.; Liljegren, S.J.; Pelaz, S.; Gold, S.E.; Burgeff, C.; Ditta, G.S.; Vergar-Silva, F.; Yanofsky, M.F. MADS-box gene evolution beyond flowers: Expression in pollen, endosperm, guard cells, roots and trichomes. Plant J. 2000, 24, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Vrebalov, J.; Ruezinsky, D.; Padmanabhan, V.; White, R.; Medrano, D.; Drake, R.; Schuch, W.; Giovannoni, J. A MADS-box gene necessary for fruit ripening at the tomato ripening-inhibitor(rin) locus. Science 2002, 296, 343–346. [Google Scholar] [CrossRef]
- Arora, R.; Agarwal, P.; Ray, S.; Singh, A.K.; Singh, V.P.; Tyagi, A.K.; Kapoor, S. MADS-box gene family in rice: Genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genom. 2007, 8, 242. [Google Scholar] [CrossRef] [Green Version]
- Shao, S.Q.; Li, B.Y.; Zhang, Z.T.; Zhou, Y.; Jiang, J.; Li, X.B. Expression of a cotton MADS-box gene is regulated inanther development and in response to phytohormone signaling. J Genet. Genom. 2010, 37, 805–816. [Google Scholar] [CrossRef]
- Guo, X.H.; Chen, G.P.; Naeem, M.; Yu, X.H.; Tang, B.Y.; Li, A.Z.; Hu, Z.L. The MADS-box gene SlMBP11 regulates plant architecture and affects reproductive development in tomato plants. Plant Sci. 2017, 258, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.H.; Wang, Z.M.; Li, S.L.; Hou, M.L.; Zhou, Y.; Zhao, Y.Q.; Li, G.J.; Zhao, H.; Ma, H.L. Genome-wide survey of potato MADS-box genes reveals that StMADS1 and StMADS13 are putative downstream targets of tuberigen StSP6A. BMC Genom. 2018, 19, 726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.M.; Si, C.; Silva, J.A.T.D.; Li, M.; Duan, J. Genome-wide identification and classification of MIKC-type MADS-box genes in Streptophyte lineages and expression analyses to reveal their role in seed germination of orchid. BMC Plant Biol. 2019, 19, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schilling, S.; Kennedy, A.; Pan, S.; Jermiin, L.S.; Melzer, R. Genome-wide analysis of MIKC-type MADS-box genes in wheat: Pervasive duplications, functional conservation and putative neofunctionalization. New Phytol. 2020, 225, 511–529. [Google Scholar] [CrossRef] [Green Version]
- Shao, Z.W.; He, M.H.; Zeng, Z.P.; Chen, Y.Z.; Hanna, A.D.; Zhu, H.B. Genome-wide identification and expression analysis of the MADS-Box gene family in sweet potato [Ipomoea batatas (L.) Lam]. Front. Genet. 2021, 12, 750137. [Google Scholar] [CrossRef]
- Xue, Y.M.; Ma, L.; Wang, H.T.; Hao, P.B.; Cheng, S.C.; Su, Z.Z.; Li, L.; Yu, S.X.; Wei, H.L. The MADS Transcription factor GhFYF is involved in abiotic stress responses in upland cotton (Gossypium hirsutum L.). Gene 2022, 815, 146138. [Google Scholar] [CrossRef]
- Parenicová, L.; Folter, S.D.; Kieffer, M.; Horner, D.S.; Favalli, C.; Busscher, J.; Cook, H.E.; Ingram, R.M.; Kater, M.M.; Davies, B.; et al. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: New openings to the MADS world. Plant Cell 2003, 15, 1538–1551. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.B.; Li, S.M.; Peng, J.; Ke, W.D. Zizania latifolia turcz. cultivated in china. Genet. Resour. Crop Evol. 2007, 54, 1211–1217. [Google Scholar] [CrossRef]
- Yan, N.; Yang, T.; Yu, X.T.; Shang, L.G.; Guo, D.P.; Zhang, Y.; Meng, L.; Qi, Q.Q.; Li, Y.L.; Du, Y.M.; et al. Chromosome-level genome assembly of Zizania latifolia provides insights into its seed shattering and phytocassane biosynthesis. Commun. Biol. 2022, 5, 36. [Google Scholar] [CrossRef]
- An, M.; Kim, H.; Moon, J.M.; Ko, H.S.; Clayton, P.; Lim, Y.H. Enzyme-treated Zizania latifolia ethanol extract protects from UVA irradiation-induced wrinkle formation via inhibition of lysosome exocytosis and reactive oxygen species generation. Antioxidants 2020, 9, 912. [Google Scholar] [CrossRef]
- Zhang, J.Z.; Chu, F.Q.; Guo, D.P.; Hyde, K.D.; Xie, G.L. Cytology and ultrastructure of interactions between Ustilago esculenta and Zizania latifolia. Mycol. Prog. 2012, 11, 499–508. [Google Scholar] [CrossRef]
- Liang, S.W.; Huang, Y.H.; Chiu, J.Y.; Tseng, H.W.; Haung, J.H.; Shen, W.C. The smut fungus Ustilago esculenta has a bipolar mating system with three idiomorphs larger than 500 kb. Fungal Genet. Biol. 2019, 126, 61–74. [Google Scholar] [CrossRef]
- Wang, Y.M.; Dong, Z.Y.; Zhang, Z.J.; Lin, X.Y.; Shen, Y.; Zhou, D.W.; Liu, B. Extensive de Novo genomic variation in rice induced by introgression from wild rice (Zizania latifolia Griseb.). Genetics 2005, 170, 1945–1956. [Google Scholar] [CrossRef] [Green Version]
- Li, C.X.; Dubcovsky, J. Wheat FT protein regulates VRN1 transcription through interactions with FDL2. Plant J. 2008, 55, 543–554. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Zhang, Z.P.; Song, S.X.; Liu, Y.C.; Zhu, X.R.; Jiang, Y.F.; Shi, L.T.; Jiang, J.Z.; Miao, M.M. Mixed transcriptome analysis revealed the possible tnteraction mechanisms between Zizania latifolia and Ustilago esculenta inducing Jiaobai stem-gall formation. Int. J. Mol. Sci. 2021, 22, 12258. [Google Scholar] [CrossRef]
- Wang, Z.D.; Yan, N.; Wang, Z.H.; Zhang, X.H.; Zhang, J.Z.; Xue, H.M.; Wang, L.X.; Zhan, Q.; Xu, Y.P.; Guo, D.P. RNA-seq analysis provides insight into reprogramming of culm development in Zizania latifolia induced by Ustilago esculenta. Plant Mol. Biol. 2017, 95, 533–547. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Gramzow, L.; Theißen, G. Phylogenomics of MADS-Box genes in plants-two opposing life styles in one gene family. Biology 2013, 2, 1150–1164. [Google Scholar] [CrossRef]
- Duan, W.K.; Song, X.M.; Liu, T.K.; Huang, Z.N.; Ren, J.; Hou, X.L.; Li, Y. Genome-wide analysis of the MADS-box gene family in Brassica rapa (Chinese cabbage). Mol. Genet. Genom. 2015, 290, 239–255. [Google Scholar] [CrossRef]
- Wu, Y.W.; Ke, Y.Z.; Wen, J.; Guo, P.C.; Ran, F.; Wang, M.M.; Liu, M.M.; Li, P.F.; Li, J.N.; Du, H. Evolution and expression analyses of the MADS-box gene family in Brassica napus. PLoS ONE 2018, 13, e200762. [Google Scholar] [CrossRef] [Green Version]
- Airoldi, C.A.; Davies, B. Gene duplication and the evolution of plant MADS-box transcription factors. J. Genet. Genom. 2012, 39, 157–165. [Google Scholar] [CrossRef]
- Edger, P.P.; Pires, J.C. Gene and genome duplications: The impact of dosage-sensitivity on the fate of nuclear genes. Chromosome Res. 2009, 17, 699–717. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Dong, Q.L.; Ji, Z.R.; Chi, F.M.; Cong, P.H.; Zhou, Z.S. Genome-wide identification and analysis of the MADS-box gene family in apple. Gene 2015, 555, 277–290. [Google Scholar] [CrossRef]
- Pérez-Pérez, J.M.; Esteve-Bruna, D.; González-Bayón, R.; Kangasjärvi, S.; Caldana, C.; Hannah, M.A.; Willmitzer, L.; Ponce, M.R.; Micol, J.L. Functional redundancy and divergence within the Arabidopsis RETICULATA-RELATED gene family. Plant Physiol. 2013, 162, 589–603. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Kim, J.; Han, J.J.; Han, M.J.; An, G.H. Functional analyses of the flowering time gene OsMADS50, the putative SUPPRESSOR OF OVEREXPRESSION OF CO 1/AGAMOUS-LIKE 20 (SOC1/AGL20) ortholog in rice. Plant J. 2004, 38, 754–764. [Google Scholar] [CrossRef]
- Ciaffi, M.; Paolacci, A.R.; Tanzarella, O.A.; Porceddu, E. Molecular Aspects of Flower Development in Grasses. Sex. Plant Reprod. 2011, 24, 247–282. [Google Scholar] [CrossRef]
- Shima, Y.; Kitagawa, M.; Fujisawa, M.; Nakano, T.; Kato, H.; Kimbara, J.; Kasumi, T.; Ito, Y. Tomato FRUITFULL homologues act in fruit ripening via forming MADS-box transcription factor complexes with RIN. Plant Mol. Biol. 2013, 82, 427–438. [Google Scholar] [CrossRef]
- Wang, Y.S.; Guo, P.Y.; Zhang, J.L.; Xie, Q.L.; Shen, H.; Hu, Z.L.; Chen, G.P. Overexpression of the MADS-box gene SIMBP21 alters leaf morphology and affects reproductive development in tomato. J. Integr. Agric. 2021, 20, 3170–3185. [Google Scholar] [CrossRef]
- Gu, Q.; Ferrándiz, C.; Yanofsky, M.F.; Martienssen, R. The FRUITFULL MADS-box gene mediates cell differentiation during Arabidopsis fruit development. Development 1998, 125, 1509–1517. [Google Scholar] [CrossRef]
- Burko, Y.; Shleizer-Burko, S.; Yanai, O.; Shwartz, I.; Zelnik, I.D.; Jacob-Hirsch, J.; Kela, I.; Eshed-Williams, L.; Ori, N. A role for APETALA1/fruitfull transcription factors in tomato leaf development. Plant Cell 2013, 25, 2070–2083. [Google Scholar] [CrossRef] [Green Version]
- He, S.C.; Ma, R.; Liu, Z.J.; Zhang, D.; Wang, S.X.; Guo, Y.; Chen, M.X. Overexpression of BnaAGL11, a MADS-Box transcription factor, regulates leaf morphogenesis and senescence in Brassica napus. J. Agric. Food Chem. 2022, 70, 3420–3434. [Google Scholar] [CrossRef]
- Kim, S.H.; Mizuno, K.; Fujimura, T. Isolation of MADS-box genes from sweet potato (Ipomoea batatas L. Lam.) expressed specifically in vegetative tissues. Plant Cell Physiol. 2002, 43, 314–322. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Hamada, T.; Otani, M.; Shimada, T. Isolation and characterization of MADS box genes possibly related to root development in sweet potato (Ipomoea batatas L. Lam.). J. Plant Biol. 2005, 48, 387–393. [Google Scholar] [CrossRef]
- Taoka, K.I.; Ohki, I.; Tsuji, H.; Furuita, K.; Hayashi, K.; Yanase, T.; Yamaguchi, M.; Nakashima, C.; Purwestri, Y.A.; Tamaki, S.; et al. 14-3-3 proteins act as intracellular receptors for rice Hd3a florigen. Nature 2011, 476, 332–335. [Google Scholar] [CrossRef]
- Li, D.; Zhang, H.Y.; Mou, M.H.; Chen, Y.L.; Xiang, S.Y.; Chen, L.G.; Yu, D.Q. Arabidopsis class II TCP transcription factors integrate with the FT–FD module to control flowering. Plant Physiol. 2019, 181, 97–111. [Google Scholar] [CrossRef]
- Hemming, M.N.; Trevaskis, B. Make hay when the sun shines: The role of MADS-box genes in temperature-dependant seasonal flowering responses. Plant Sci. 2011, 180, 447–453. [Google Scholar] [CrossRef]
- Yang, Q.S.; Yang, B.; Li, Y.Z.; Wang, Y.; Tao, R.Y.; Yang, F.; Wu, X.Y.; Yan, X.H.; Ahmad, M.; Shen, J.Q.; et al. ABA-responsive ABRE-BINDING FACTOR3 activates DAM3 expression to promote bud dormancy in Asian pear. Plant Cell Environ. 2020, 43, 1360–1375. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Xiao, M.; Song, S.; Jiang, Y.; Zhu, X.; Shi, L.; Zheng, X.; Jiang, J.; Miao, M. Genome-Wide Identification, Classification and Expression Analyses of MADS-Box Genes Reveal Their Role in Stem Gall Formation and Expansion of Zizania latifolia. Agronomy 2023, 13, 1758. https://doi.org/10.3390/agronomy13071758
Zhang Z, Xiao M, Song S, Jiang Y, Zhu X, Shi L, Zheng X, Jiang J, Miao M. Genome-Wide Identification, Classification and Expression Analyses of MADS-Box Genes Reveal Their Role in Stem Gall Formation and Expansion of Zizania latifolia. Agronomy. 2023; 13(7):1758. https://doi.org/10.3390/agronomy13071758
Chicago/Turabian StyleZhang, Zhiping, Meng Xiao, Sixiao Song, Yifeng Jiang, Xinrui Zhu, Lingtong Shi, Xiaomeng Zheng, Jiezeng Jiang, and Minmin Miao. 2023. "Genome-Wide Identification, Classification and Expression Analyses of MADS-Box Genes Reveal Their Role in Stem Gall Formation and Expansion of Zizania latifolia" Agronomy 13, no. 7: 1758. https://doi.org/10.3390/agronomy13071758