
Identification of Candidate Genes for Soybean Storability via GWAS and WGCNA Approaches

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Evaluation of Germination Test
2.2. SNP Genotyping Data Collection
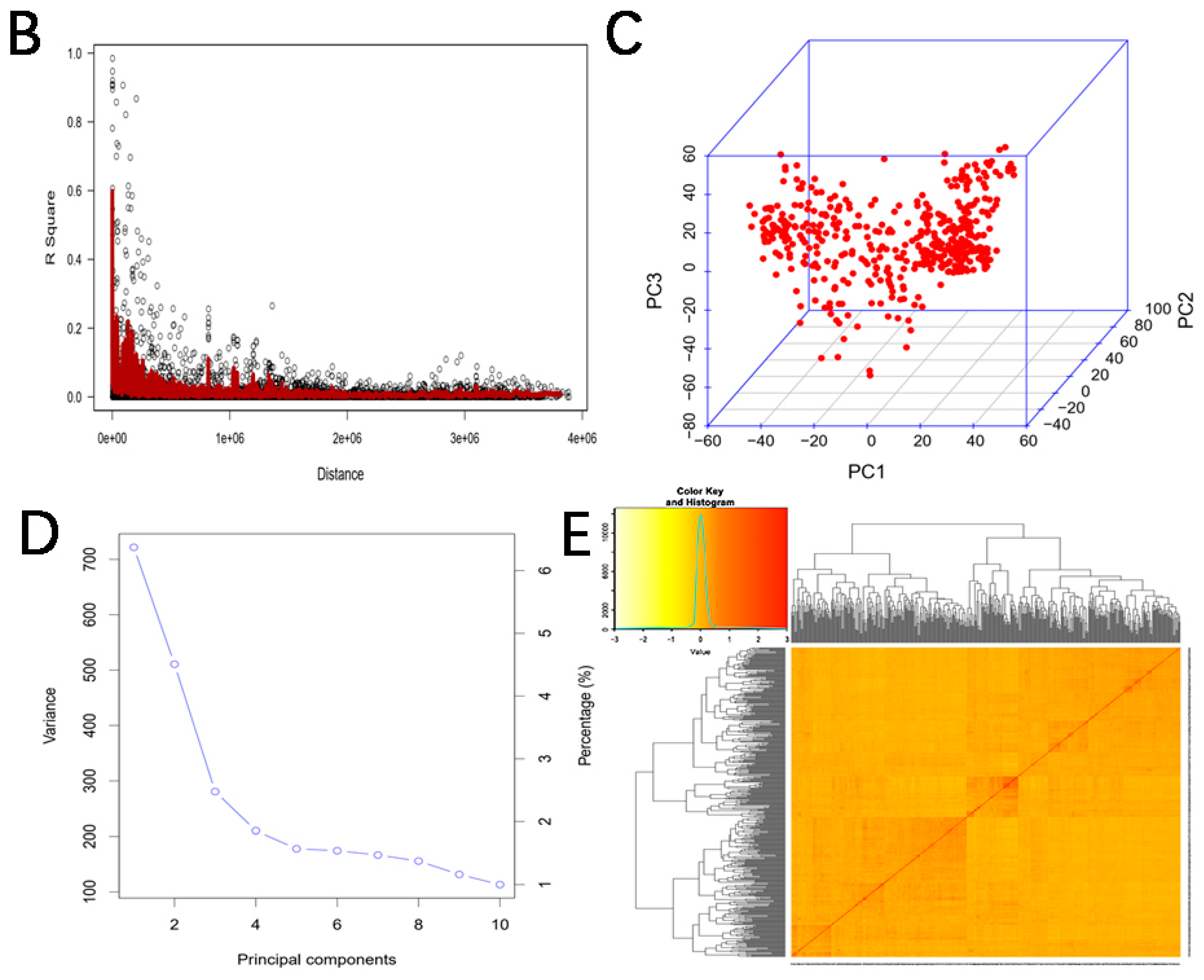
2.3. Population Structure Evaluation and Linkage Disequilibrium (LD) Analysis
2.4. Association Analysis and Candidate Gene Prediction and Annotation
2.5. Metabolomic and Transcriptomics Data Processing and Analysis
2.6. Weighted Gene Co-Expression Network Analysis
3. Results
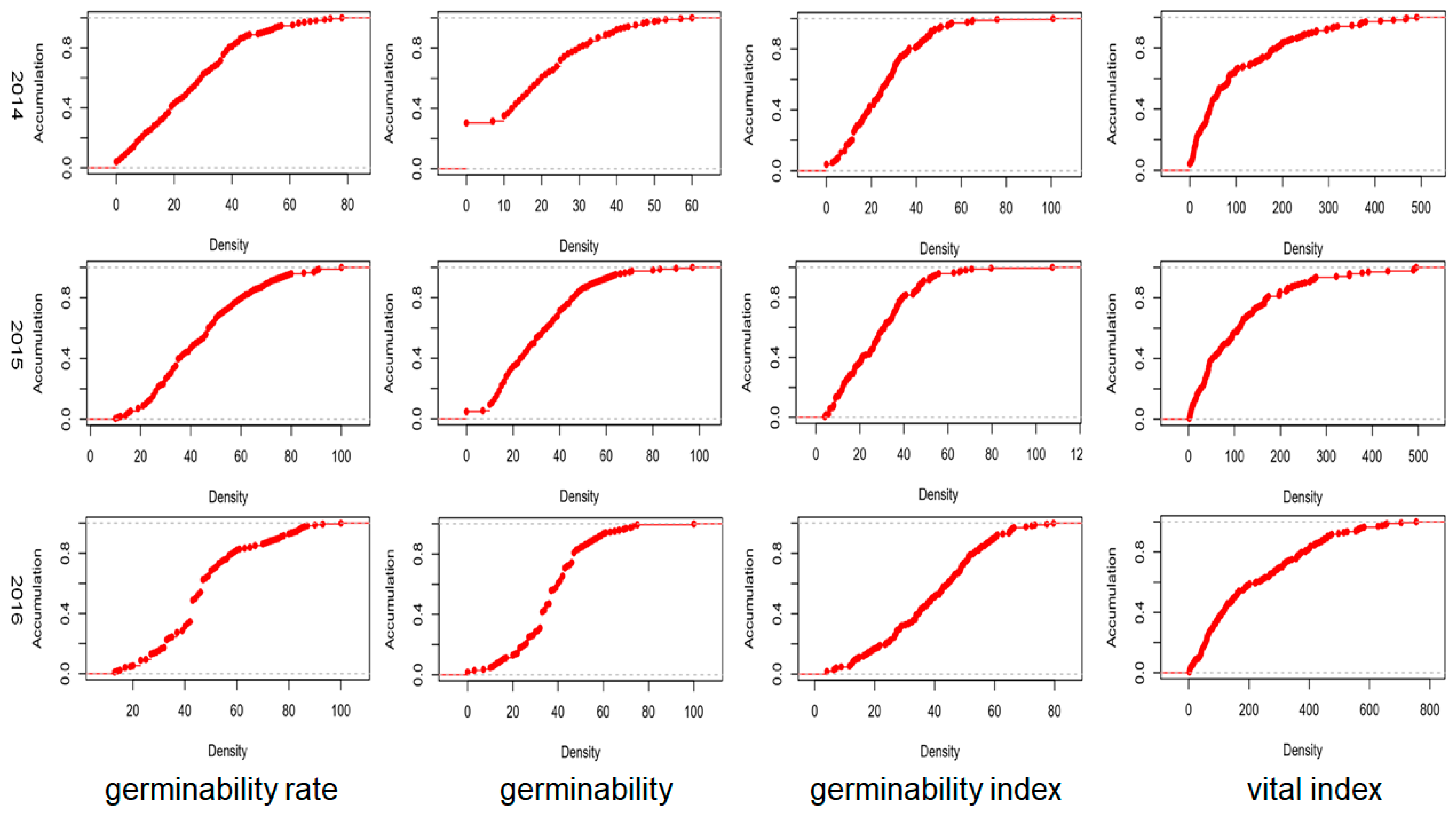
3.1. Relates Traits of Seed Vigor in Soybean
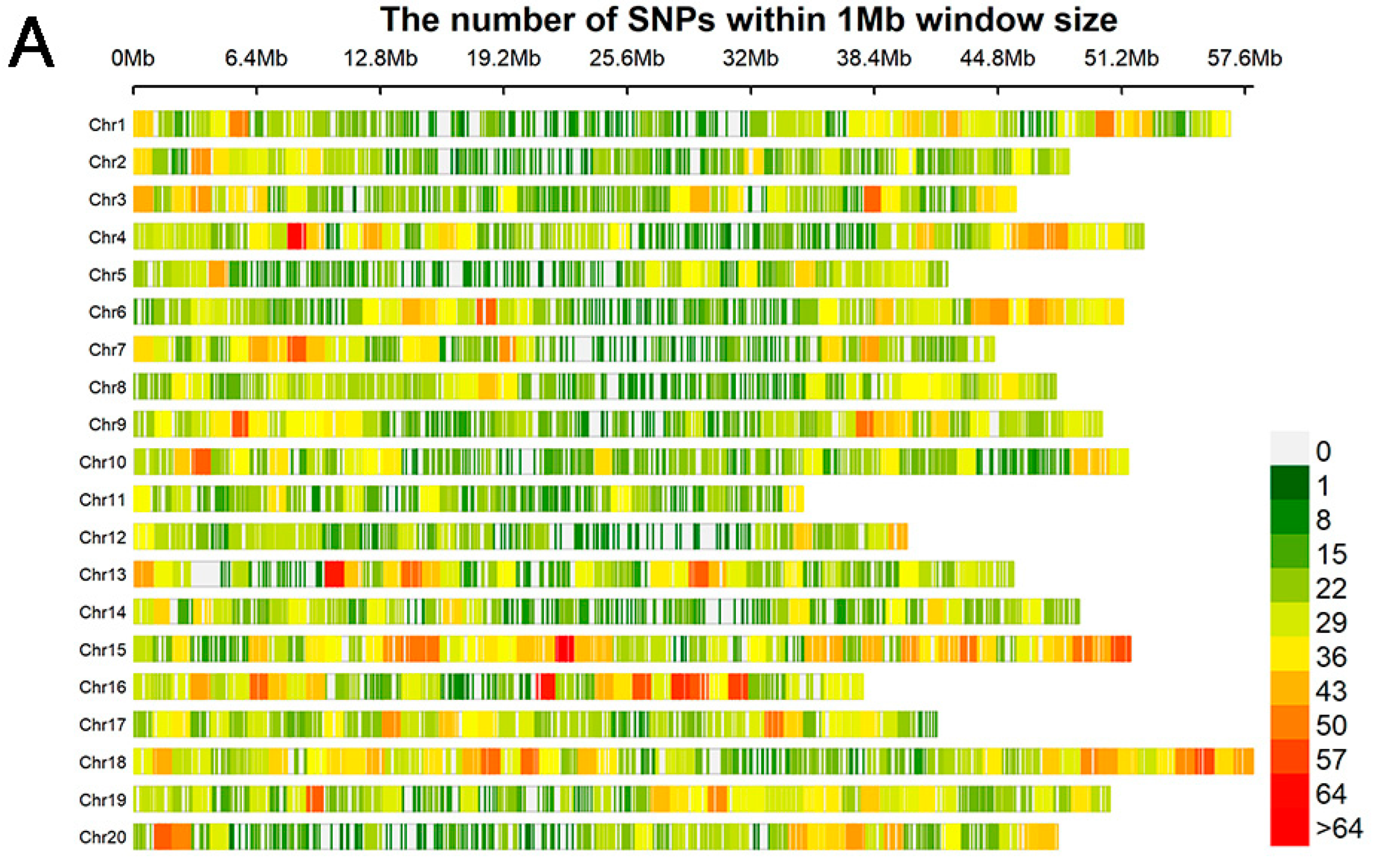
3.2. Distribution of Markers and Analysis of Mapping Population
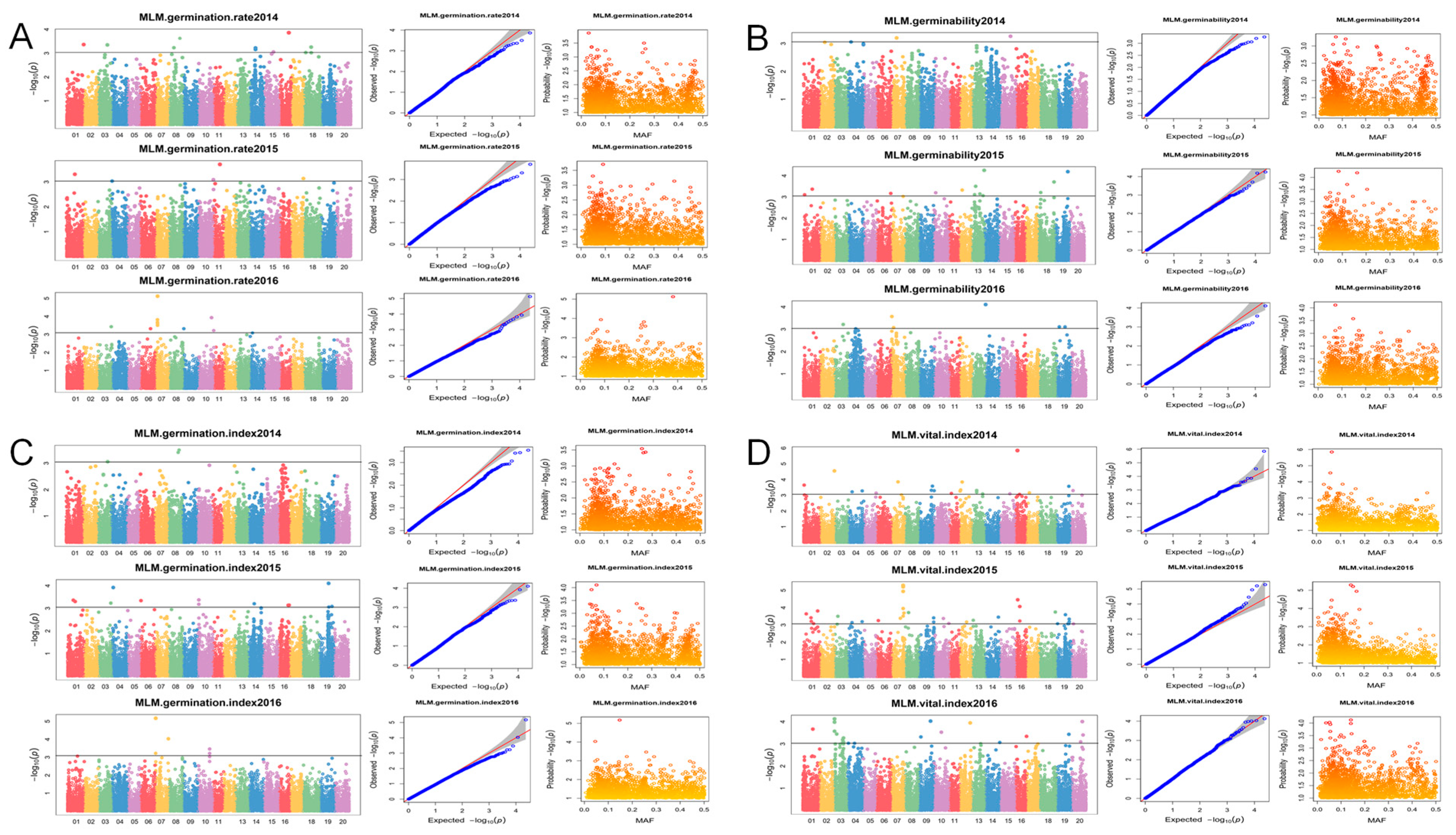
3.3. Quantitative Trait Nucleotide (QTN) Associated with Soybean Seed-Vigor-Related Traits by GWAS
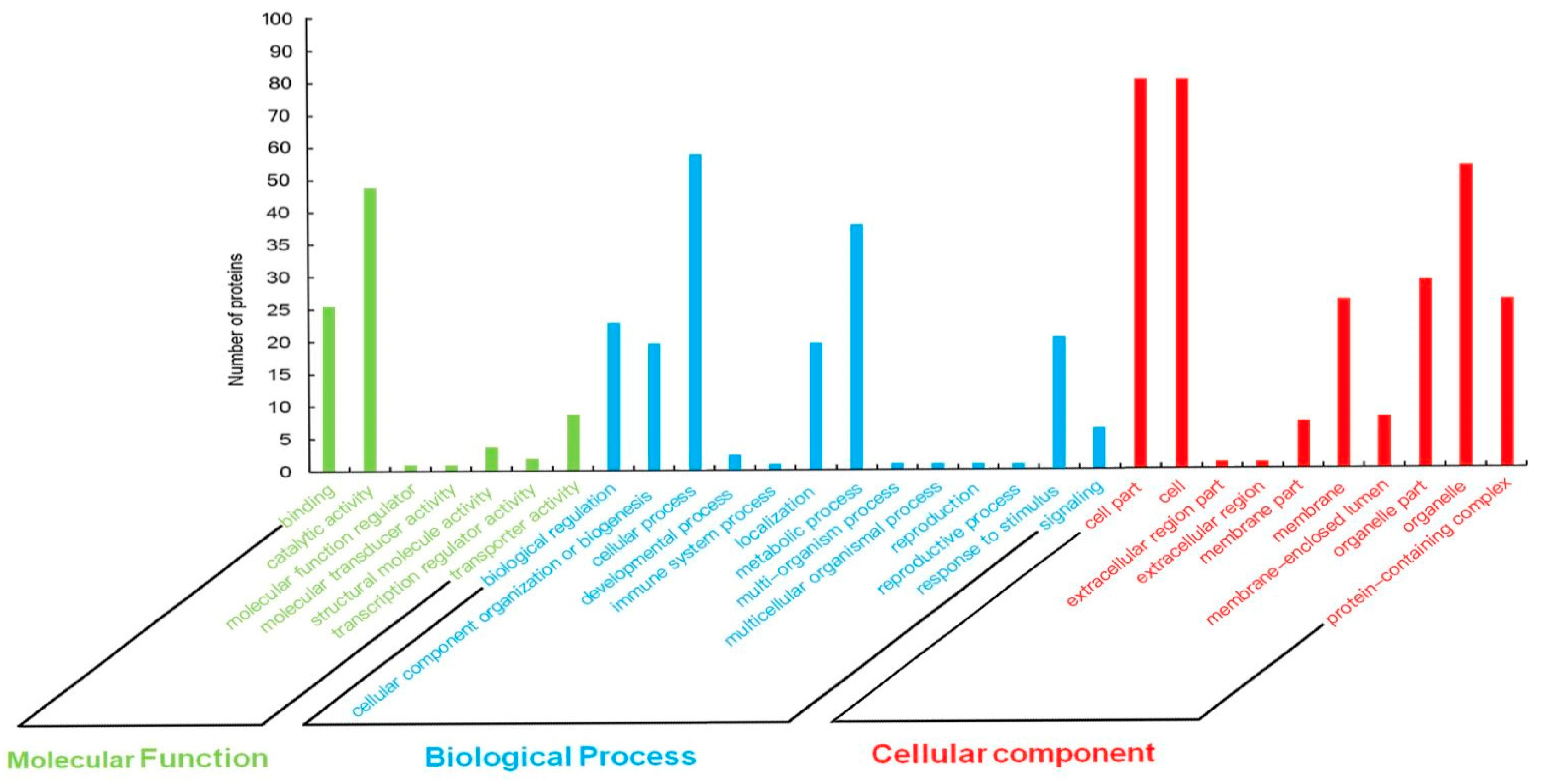
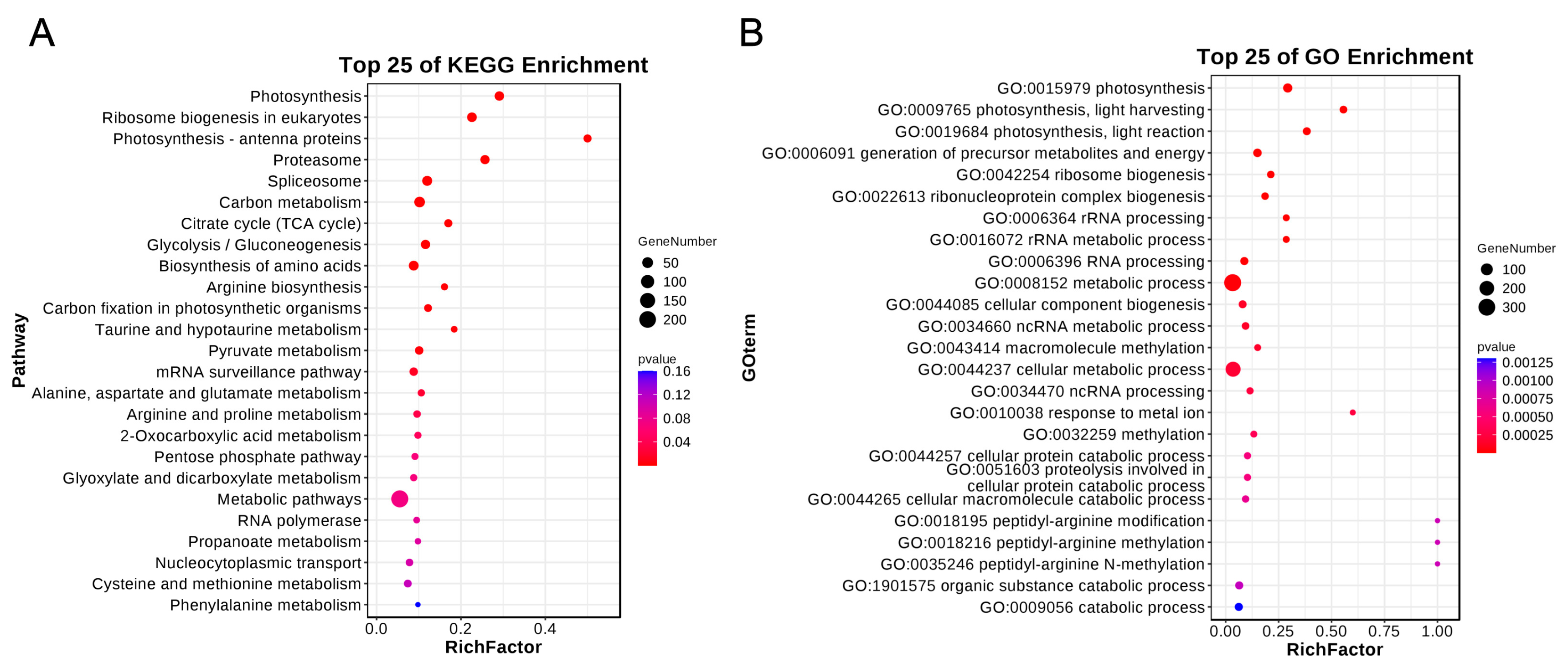
3.4. Gene Enrichment Analysis of Candidate Genes
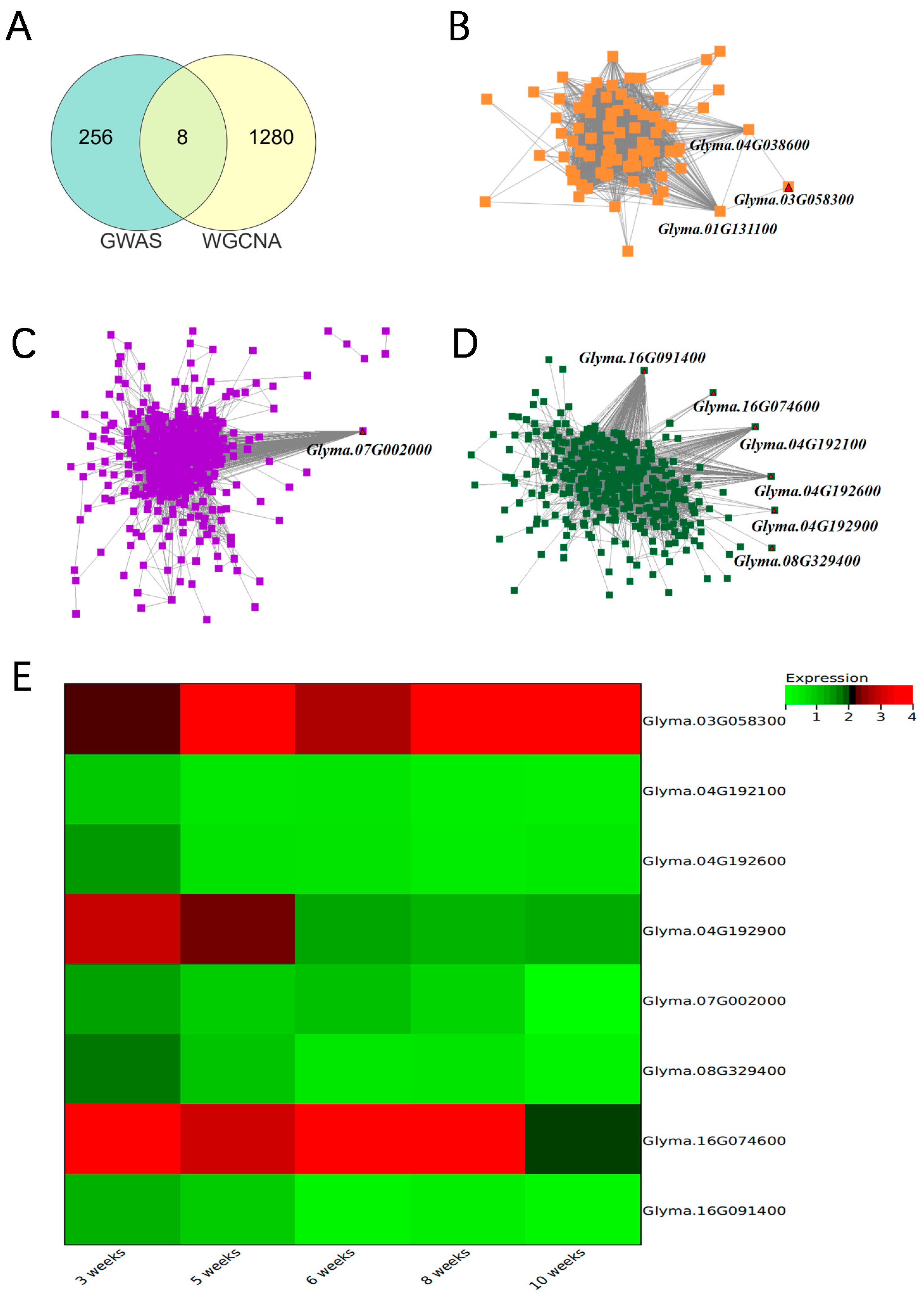
3.5. Identification of Key Modules Possessing Candidate Genes via WGCNA
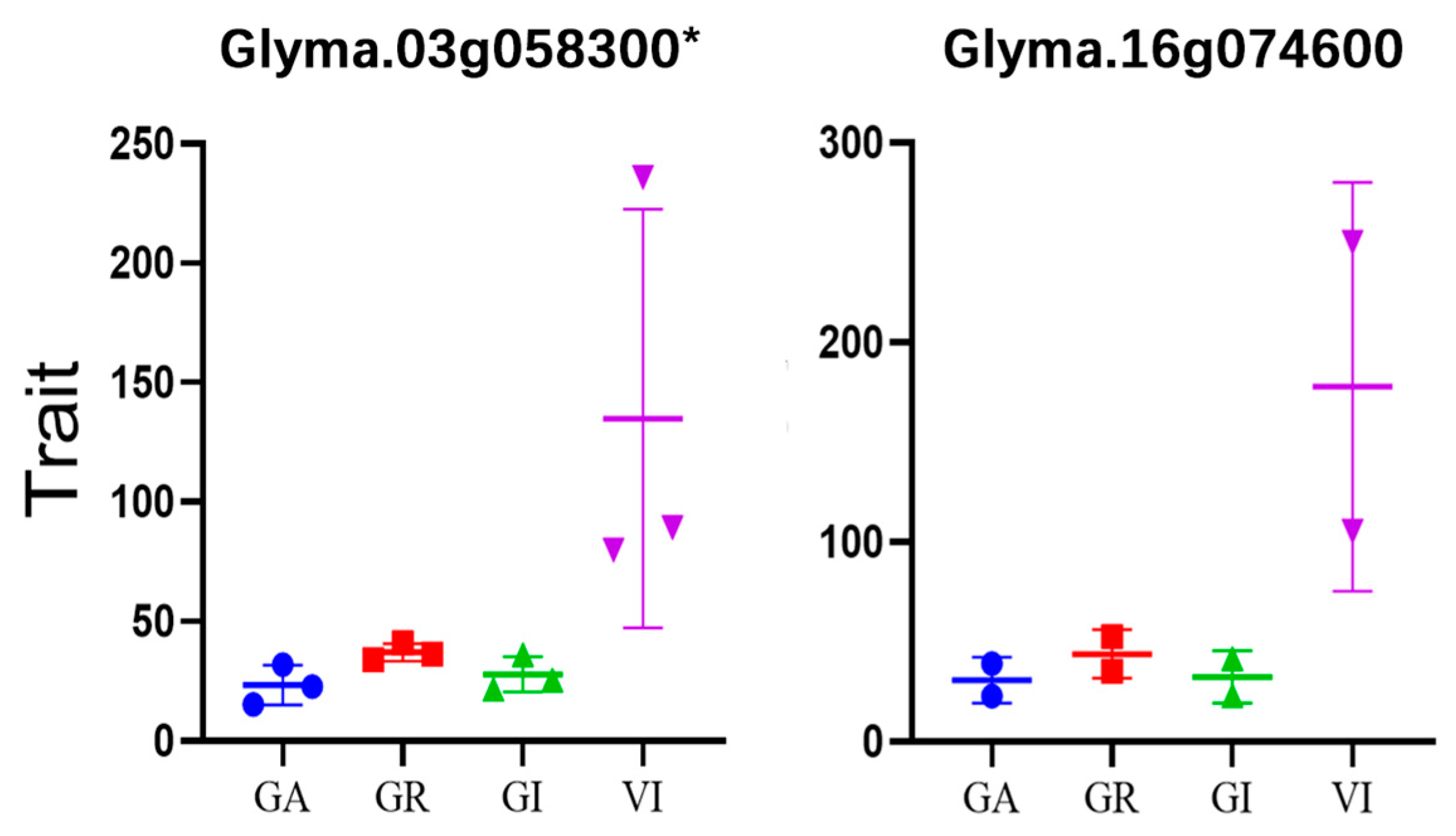
3.6. Prediction of Candidate Genes for Storage Tolerant Traits
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Pirredda, M.; Fañanás-Pueyo, I.; Oñate-Sánchez, L.; Mira, S. Seed Longevity and Ageing: A Review on Physiological and Genetic Factors with an Emphasis on Hormonal Regulation. Plants 2024, 13, 41. [Google Scholar] [CrossRef] [PubMed]
- Souza, A.D.V.; Santos, D.; Rodrigues, A.A.; Zuchi, J.; Vieira, M.C.; Sales, J.F. Physical and Physiological Soybean Seed Qualities Stored under Different Environmental Conditions and Storage Bag Depths. Braz. J. Biol. 2023, 83, e277916. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Zhou, Y.; Mu, K.; Hu, H.; Chen, M.; He, Q.; Huang, S.; Ma, H.; Yu, X. A Transcriptomic Analysis Reveals Soybean Seed Pre-Harvest Deterioration Resistance Pathways under High Temperature and Humidity Stress. Genome 2020, 63, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Chen, F.; Luo, X.; Dai, Y.; Yang, Y.; Zheng, C.; Yang, W.; Shu, K. A Matter of Life and Death: Molecular, Physiological, and Environmental Regulation of Seed Longevity. Plant Cell Environ. 2020, 43, 293–302. [Google Scholar] [CrossRef]
- Nagel, M.; Kranner, I.; Neumann, K.; Rolletschek, H.; Seal, C.E.; Colville, L.; Fernández-Marín, B.; Börner, A. Genome-Wide Association Mapping and Biochemical Markers Reveal That Seed Ageing and Longevity Are Intricately Affected by Genetic Background and Developmental and Environmental Conditions in Barley. Plant Cell Environ. 2015, 38, 1011–1022. [Google Scholar] [CrossRef]
- Zheng, Q.; Teng, Z.; Zhang, J.; Ye, N. ABA Inhibits Rice Seed Aging by Reducing H2O2 Accumulation in the Radicle of Seeds. Plants 2024, 13, 809. [Google Scholar] [CrossRef]
- Dargahi, H.; Tanya, P.; Srinives, P. Mapping of the Genomic Regions Controlling Seed Storability in Soybean (Glycine max L.). J. Genet. 2014, 93, 365–370. [Google Scholar] [CrossRef]
- Bentsink, L.; Alonso-Blanco, C.; Vreugdenhil, D.; Tesnier, K.; Groot, S.P.; Koornneef, M. Genetic Analysis of Seed-Soluble Oligosaccharides in Relation to Seed Storability of Arabidopsis. Plant Physiol. 2000, 124, 1595–1604. [Google Scholar] [CrossRef]
- Ramtekey, V.; Cherukuri, S.; Kumar, S.; Kudekallu V., S.; Sheoran, S.; Bhaskar K., U.; Naik K., B.; Kumar, S.; Singh, A.N.; Singh, H.V. Seed Longevity in Legumes: Deeper Insights Into Mechanisms and Molecular Perspectives. Front. Plant Sci. 2022, 13, 918206. [Google Scholar] [CrossRef]
- Lee, J.; Welti, R.; Roth, M.; Schapaugh, W.T.; Li, J.; Trick, H.N. Enhanced Seed Viability and Lipid Compositional Changes during Natural Ageing by Suppressing Phospholipase Dα in Soybean Seed. Plant Biotechnol. J. 2012, 10, 164–173. [Google Scholar] [CrossRef]
- Jong, C.; Yu, Z.; Zhang, Y.; Choe, K.; Uh, S.; Kim, K.; Jong, C.; Cha, J.; Kim, M.; Kim, Y.; et al. Multi-Omics Analysis of a Chromosome Segment Substitution Line Reveals a New Regulation Network for Soybean Seed Storage Profile. Int. J. Mol. Sci. 2024, 25, 5614. [Google Scholar] [CrossRef]
- Nakajima, S.; Ito, H.; Tanaka, R.; Tanaka, A. Chlorophyll b Reductase Plays an Essential Role in Maturation and Storability of Arabidopsis Seeds. Plant Physiol. 2012, 160, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Rissel, D.; Losch, J.; Peiter, E. The Nuclear Protein Poly(ADP-Ribose) Polymerase 3 (AtPARP3) Is Required for Seed Storability in Arabidopsis Thaliana. Plant Biol. 2014, 16, 1058–1064. [Google Scholar] [CrossRef]
- Yan, S.; Huang, W.; Gao, J.; Fu, H.; Liu, J. Comparative Metabolomic Analysis of Seed Metabolites Associated with Seed Storability in Rice (Oryza sativa L.) during Natural Aging. Plant Physiol. Biochem. 2018, 127, 590–598. [Google Scholar] [CrossRef]
- Yuan, Z.; Fan, K.; Xia, L.; Ding, X.; Tian, L.; Sun, W.; He, H.; Yu, S. Genetic Dissection of Seed Storability and Validation of Candidate Gene Associated with Antioxidant Capability in Rice (Oryza sativa L.). Int. J. Mol. Sci. 2019, 20, 4442. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Ding, Y.; Huang, S.; Zhu, C. Identification of miRNAs and Their Target Genes Associated with Sweet Corn Seed Vigor by Combined Small RNA and Degradome Sequencing. J. Agric. Food Chem. 2015, 63, 5485–5491. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Lv, Z.; Wang, Y.; Li, C.; Jia, Y.; Zhu, Y.; Cao, M.; Zhou, Y.; Zeng, X.; Wang, Z.; et al. Identification of miRNAs Mediating Seed Storability of Maize during Germination Stage by High-Throughput Sequencing, Transcriptome and Degradome Sequencing. Int. J. Mol. Sci. 2022, 23, 12339. [Google Scholar] [CrossRef] [PubMed]
- Groot, S.P.C.; Surki, A.A.; de Vos, R.C.H.; Kodde, J. Seed Storage at Elevated Partial Pressure of Oxygen, a Fast Method for Analysing Seed Ageing under Dry Conditions. Ann. Bot. 2012, 110, 1149–1159. [Google Scholar] [CrossRef]
- Singh, R.K.; Raipuria, R.K.; Bhatia, V.S.; Rani, A.; Pushpendra; Husain, S.M.; Chauhan, D.; Chauhan, G.S.; Mohapatra, T. SSR Markers Associated with Seed Longevity in Soybean. Seed Sci. Technol. 2008, 36, 162–167. [Google Scholar] [CrossRef]
- Li, L.; Lin, Q.; Liu, S.; Liu, X.; Wang, W.; Hang, N.T.; Liu, F.; Zhao, Z.; Jiang, L.; Wan, J. Identification of Quantitative Trait Loci for Seed Storability in Rice (Oryza sativa L.). Plant Breed. 2012, 131, 739–743. [Google Scholar] [CrossRef]
- Zhang, X.; Hina, A.; Song, S.; Kong, J.; Bhat, J.A.; Zhao, T. Whole-Genome Mapping Identified Novel “QTL Hotspots Regions” for Seed Storability in Soybean (Glycine max L.). BMC Genom. 2019, 20, 499. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Jiang, Y.; Wang, Z.; Gou, Z.; Lyu, J.; Li, W.; Yu, Y.; Shu, L.; Zhao, Y.; Ma, Y.; et al. Resequencing 302 Wild and Cultivated Accessions Identifies Genes Related to Domestication and Improvement in Soybean. Nat. Biotechnol. 2015, 33, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wu, F.; Xie, X.; Yang, C. Quantitative Trait Locus Mapping of Seed Vigor in Soybean under −20 °C Storage and Accelerated Aging Conditions via RAD Sequencing. Curr. Issues Mol. Biol. 2021, 43, 1977–1996. [Google Scholar] [CrossRef]
- Hayes, B. Overview of Statistical Methods for Genome-Wide Association Studies (GWAS). Methods Mol. Biol. 2013, 1019, 149–169. [Google Scholar] [CrossRef]
- He, J.; Gai, J. Genome-Wide Association Studies (GWAS). Methods Mol. Biol. 2023, 2638, 123–146. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Liu, X.; Zhou, J.; Deng, H.; Zhang, G.; Xiao, Y.; Tang, W. WGCNA Analysis Identifies the Hub Genes Related to Heat Stress in Seedling of Rice (Oryza sativa L.). Genes 2022, 13, 1020. [Google Scholar] [CrossRef]
- Zong, J.; Chen, P.; Luo, Q.; Gao, J.; Qin, R.; Wu, C.; Lv, Q.; Zhao, T.; Fu, Y. Transcriptome-Based WGCNA Analysis Reveals the Mechanism of Drought Resistance Differences in Sweetpotato (Ipomoea batatas (L.) Lam.). Int. J. Mol. Sci. 2023, 24, 14398. [Google Scholar] [CrossRef]
- Azam, M.; Zhang, S.; Li, J.; Ahsan, M.; Agyenim-Boateng, K.G.; Qi, J.; Feng, Y.; Liu, Y.; Li, B.; Qiu, L.; et al. Identification of Hub Genes Regulating Isoflavone Accumulation in Soybean Seeds via GWAS and WGCNA Approaches. Front. Plant Sci. 2023, 14, 1120498. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, Y.; Wang, J.; Zhao, X.; Li, Y.; Teng, W.; Han, Y.; Zhan, Y. GWAS and WGCNA Analysis Uncover Candidate Genes Associated with Oil Content in Soybean. Plants 2024, 13, 1351. [Google Scholar] [CrossRef]
- Zhang, Z.-H.; Yu, S.-B.; Yu, T.; Huang, Z.; Zhu, Y.-G. Mapping Quantitative Trait Loci (QTLs) for Seedling-Vigor Using Recombinant Inbred Lines of Rice (Oryza sativa L.). Field Crops Res. 2005, 91, 161–170. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, M.; Hina, A.; Kong, J.; Gai, J.; He, X.; Zhao, T. Seed Storability of Summer-Planting Soybeans under Natural and Artificial Aging Conditions. Legume Res. 2019, 42, 250–259. [Google Scholar] [CrossRef]
- Xia, Y.; Chen, F.; Du, Y.; Liu, C.; Bu, G.; Xin, Y.; Liu, B. A Modified SDS-Based DNA Extraction Method from Raw Soybean. Biosci. Rep. 2019, 39, BSR20182271. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47, 11.12.1–11.12.34. [Google Scholar] [CrossRef]
- Ma, L.; Qing, C.; Zhang, M.; Zou, C.; Pan, G.; Shen, Y. GWAS with a PCA Uncovers Candidate Genes for Accumulations of Microelements in Maize Seedlings. Physiol. Plant 2021, 172, 2170–2180. [Google Scholar] [CrossRef]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome Association and Prediction Integrated Tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, X.; Wang, W.; Qu, Y.; Teng, W.; Qiu, L.; Zheng, H.; Han, Y.; Li, W. Genome-Wide Association Study of Inflorescence Length of Cultivated Soybean Based on the High-throughout Single-Nucleotide Markers. Mol. Genet. Genom. 2019, 294, 607–620. [Google Scholar] [CrossRef]
- Sedgwick, P. Multiple Hypothesis Testing and Bonferroni’s Correction. BMJ 2014, 349, g6284. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for Taxonomy-Based Analysis of Pathways and Genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef]
- Rio, D.C.; Ares, M.; Hannon, G.J.; Nilsen, T.W. Purification of RNA Using TRIzol (TRI Reagent). Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5439. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.A.; Park, J.L.; Kim, J.-H.; Kim, J.H.; Kim, Y.S.; Kim, J.C.; Kim, S.-Y. Comparison of the MGISEQ-2000 and Illumina HiSeq 4000 Sequencing Platforms for RNA Sequencing. Genom. Inf. 2019, 17, e32. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wang, Y.; Yao, W.; Ge, W.; Jiang, T.; Zhou, B. Transcriptome Sequencing and WGCNA Reveal Key Genes in Response to Leaf Blight in Poplar. Int. J. Mol. Sci. 2023, 24, 10047. [Google Scholar] [CrossRef]
- DiLeo, M.V.; Strahan, G.D.; den Bakker, M.; Hoekenga, O.A. Weighted Correlation Network Analysis (WGCNA) Applied to the Tomato Fruit Metabolome. PLoS ONE 2011, 6, e26683. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Singh, R.K.; Raipuria, R.K.; Bhatia, V.S.; Rani, A.; Pushpendra, null; Husain, S.M.; Tara Satyavathi, C.; Chauhan, G.S.; Mohapatra, T. Identification of SSR Markers Associated with Seed Coat Permeability and Electrolyte Leaching in Soybean. Physiol. Mol. Biol. Plants 2008, 14, 173–177. [Google Scholar] [CrossRef]
- Müller, M.; Munné-Bosch, S. Ethylene Response Factors: A Key Regulatory Hub in Hormone and Stress Signaling. Plant Physiol. 2015, 169, 32–41. [Google Scholar] [CrossRef]
- Jia, D.; Gao, H.; He, Y.; Liao, G.; Lin, L.; Huang, C.; Xu, X. Kiwifruit Monodehydroascorbate Reductase 3 Gene Negatively Regulates the Accumulation of Ascorbic Acid in Fruit of Transgenic Tomato Plants. Int. J. Mol. Sci. 2023, 24, 17182. [Google Scholar] [CrossRef]
- Murthy, S.S.; Zilinskas, B.A. Molecular Cloning and Characterization of a cDNA Encoding Pea Monodehydroascorbate Reductase. J. Biol. Chem. 1994, 269, 31129–31133. [Google Scholar] [CrossRef]
- Shao, Q.; Liu, X.; Su, T.; Ma, C.; Wang, P. New Insights Into the Role of Seed Oil Body Proteins in Metabolism and Plant Development. Front. Plant Sci. 2019, 10, 1568. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Wang, Q.; Zhang, W.; Zhang, H.; Liu, X.; Song, Q.; Zhu, Y.; Cui, X.; Chen, X.; Chen, H. Using Transcriptomic and Metabolomic Data to Investigate the Molecular Mechanisms That Determine Protein and Oil Contents during Seed Development in Soybean. Front. Plant Sci. 2022, 13, 1012394. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, V.; Berghetti, M.R.P.; Ribeiro, S.R.; Rossato, F.P.; Wendt, L.M.; Thewes, F.R.; Thewes, F.R.; Brackmann, A.; Both, V.; Wagner, R. The Effects of Soybean Storage under Controlled Atmosphere at Different Temperatures on Lipid Oxidation and Volatile Compounds Profile. Food Res. Int. 2021, 147, 110483. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.T.; Choi, U.-K.; Ryu, H.S.; Lee, S.J.; Kwon, O.-S. Mobilization of Storage Proteins in Soybean Seed (Glycine max L.) during Germination and Seedling Growth. Biochim. Biophys. Acta 2011, 1814, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Fleming, M.B.; Hill, L.M.; Walters, C. The Kinetics of Ageing in Dry-Stored Seeds: A Comparison of Viability Loss and RNA Degradation in Unique Legacy Seed Collections. Ann. Bot. 2019, 123, 1133–1146. [Google Scholar] [CrossRef]
- Wu, F.; Luo, X.; Wang, L.; Wei, Y.; Li, J.; Xie, H.; Zhang, J.; Xie, G. Genome-Wide Association Study Reveals the QTLs for Seed Storability in World Rice Core Collections. Plants 2021, 10, 812. [Google Scholar] [CrossRef]
- Hartanto, M.; Joosen, R.V.L.; Snoek, B.L.; Willems, L.A.J.; Sterken, M.G.; de Ridder, D.; Hilhorst, H.W.M.; Ligterink, W.; Nijveen, H. Network Analysis Prioritizes DEWAX and ICE1 as the Candidate Genes for Major eQTL Hotspots in Seed Germination of Arabidopsis Thaliana. G3 2020, 10, 4215–4226. [Google Scholar] [CrossRef]
- Liu, F.; Li, N.; Yu, Y.; Chen, W.; Yu, S.; He, H. Insights into the Regulation of Rice Seed Storability by Seed Tissue-Specific Transcriptomic and Metabolic Profiling. Plants 2022, 11, 1570. [Google Scholar] [CrossRef]
- Li, L.; Wang, F.; Li, X.; Peng, Y.; Zhang, H.; Hey, S.; Wang, G.; Wang, J.; Gu, R. Comparative Analysis of the Accelerated Aged Seed Transcriptome Profiles of Two Maize Chromosome Segment Substitution Lines. PLoS ONE 2019, 14, e0216977. [Google Scholar] [CrossRef]
- Shvachko, N.A.; Khlestkina, E.K. Molecular Genetic Bases of Seed Resistance to Oxidative Stress during Storage. Vavilovskii Zhurnal Genet. Sel. 2020, 24, 451–458. [Google Scholar] [CrossRef]
- Shi, H.; Guan, W.; Shi, Y.; Wang, S.; Fan, H.; Yang, J.; Chen, W.; Zhang, W.; Sun, D.; Jing, R. QTL Mapping and Candidate Gene Analysis of Seed Vigor-Related Traits during Artificial Aging in Wheat (Triticum aestivum). Sci. Rep. 2020, 10, 22060. [Google Scholar] [CrossRef] [PubMed]
- Bizouerne, E.; Ly Vu, J.; Ly Vu, B.; Diouf, I.; Bitton, F.; Causse, M.; Verdier, J.; Buitink, J.; Leprince, O. Genetic Variability in Seed Longevity and Germination Traits in a Tomato MAGIC Population in Contrasting Environments. Plants 2023, 12, 3632. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Kong, Y.; Shao, Z.; Zhang, H.; Li, X.; Zhang, C. Discovery of Genetic Loci and Causal Genes for Seed Germination via Deep Re-Sequencing in Soybean. Mol. Breed. 2022, 42, 45. [Google Scholar] [CrossRef]
- Cao, Y.; Zhang, X.; Jia, S.; Karikari, B.; Zhang, M.; Xia, Z.; Zhao, T.; Liang, F. Genome-Wide Association among Soybean Accessions for the Genetic Basis of Salinity-Alkalinity Tolerance during Germination. Crop Pasture Sci. 2021, 72, 255–267. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, W.; Zhang, H.; Liu, X.; Cui, X.; Li, S.; Song, L.; Zhu, Y.; Chen, X.; Chen, H. Comparative Selective Signature Analysis and High-Resolution GWAS Reveal a New Candidate Gene Controlling Seed Weight in Soybean. Theor. Appl. Genet. 2021, 134, 1329–1341. [Google Scholar] [CrossRef]
- Liu, Z.; Li, H.; Gou, Z.; Zhang, Y.; Wang, X.; Ren, H.; Wen, Z.; Kang, B.-K.; Li, Y.; Yu, L.; et al. Genome-Wide Association Study of Soybean Seed Germination under Drought Stress. Mol. Genet. Genom. 2020, 295, 661–673. [Google Scholar] [CrossRef]
- Nissan, N.; Hooker, J.; Pattang, A.; Charette, M.; Morrison, M.; Yu, K.; Hou, A.; Golshani, A.; Molnar, S.J.; Cober, E.R.; et al. Novel QTL for Low Seed Cadmium Accumulation in Soybean. Plants 2022, 11, 1146. [Google Scholar] [CrossRef]
- Hu, Z.; Zhang, D.; Zhang, G.; Kan, G.; Hong, D.; Yu, D. Association Mapping of Yield-Related Traits and SSR Markers in Wild Soybean (Glycine soja Sieb. and Zucc.). Breed. Sci. 2014, 63, 441–449. [Google Scholar] [CrossRef]
- Alam, M.; Wang, Y.; Chen, J.; Lou, G.; Yang, H.; Zhou, Y.; Luitel, S.; Jiang, G.; He, Y. QTL Detection for Rice Grain Storage Protein Content and Genetic Effect Verifications. Mol. Breed. 2023, 43, 89. [Google Scholar] [CrossRef]
- Scheben, A.; Batley, J.; Edwards, D. Genotyping-by-Sequencing Approaches to Characterize Crop Genomes: Choosing the Right Tool for the Right Application. Plant Biotechnol. J. 2017, 15, 149–161. [Google Scholar] [CrossRef]
- Naflath, T.V.; Rajendraprasad, S.; Ravikumar, R.L. Evaluation of Diverse Soybean Genotypes for Seed Longevity and Its Association with Seed Coat Colour. Sci. Rep. 2023, 13, 4313. [Google Scholar] [CrossRef]
- Yu, J.-Y.; Zhang, Z.-G.; Huang, S.-Y.; Han, X.; Wang, X.-Y.; Pan, W.-J.; Qin, H.-T.; Qi, H.-D.; Yin, Z.-G.; Qu, K.-X.; et al. Analysis of miRNAs Targeted Storage Regulatory Genes during Soybean Seed Development Based on Transcriptome Sequencing. Genes 2019, 10, 408. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Ahn, J.K.; Kim, S.H.; Kim, J.T.; Han, S.J.; Jung, M.Y.; Chung, I.M. Variation in Isoflavone of Soybean Cultivars with Location and Storage Duration. J. Agric. Food Chem. 2003, 51, 3382–3389. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C. The Signalling Role of ROS in the Regulation of Seed Germination and Dormancy. Biochem. J. 2019, 476, 3019–3032. [Google Scholar] [CrossRef]
- Waterworth, W.; Balobaid, A.; West, C. Seed Longevity and Genome Damage. Biosci. Rep. 2024, 44, BSR20230809. [Google Scholar] [CrossRef]
- Pagano, A.; Araújo, S.D.S.; Macovei, A.; Leonetti, P.; Balestrazzi, A. The Seed Repair Response during Germination: Disclosing Correlations between DNA Repair, Antioxidant Response, and Chromatin Remodeling in Medicago Truncatula. Front. Plant Sci. 2017, 8, 1972. [Google Scholar] [CrossRef]
- Min, C.W.; Lee, S.H.; Cheon, Y.E.; Han, W.Y.; Ko, J.M.; Kang, H.W.; Kim, Y.C.; Agrawal, G.K.; Rakwal, R.; Gupta, R.; et al. In-Depth Proteomic Analysis of Glycine max Seeds during Controlled Deterioration Treatment Reveals a Shift in Seed Metabolism. J. Proteom. 2017, 169, 125–135. [Google Scholar] [CrossRef]
- Vijayakumar, H.P.; Vijayakumar, A.; Srimathi, P.; Somasundaram, G.; Prasad, S.R.; Natarajan, S.; Dhandapani, R.; Boraiah, K.M.; Vishwanath, K. Biochemical Changes in Naturally Aged Seeds of Soybean Genotypes with Good and Poor Storability. Legume Res. 2019, 42, 782–788. [Google Scholar] [CrossRef]
- Manohar, M.; Shigaki, T.; Mei, H.; Park, S.; Marshall, J.; Aguilar, J.; Hirschi, K.D. Characterization of Arabidopsis Ca2+/H+ Exchanger CAX3. Biochemistry 2011, 50, 6189–6195. [Google Scholar] [CrossRef]
- Cheng, N.-H.; Pittman, J.K.; Shigaki, T.; Lachmansingh, J.; LeClere, S.; Lahner, B.; Salt, D.E.; Hirschi, K.D. Functional Association of Arabidopsis CAX1 and CAX3 Is Required for Normal Growth and Ion Homeostasis. Plant Physiol. 2005, 138, 2048–2060. [Google Scholar] [CrossRef]
- Park, S.; Cheng, N.H.; Pittman, J.K.; Yoo, K.S.; Park, J.; Smith, R.H.; Hirschi, K.D. Increased Calcium Levels and Prolonged Shelf Life in Tomatoes Expressing Arabidopsis H+/Ca2+ Transporters. Plant Physiol. 2005, 139, 1194–1206. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Year | Min a | Max a | Mean a | SD b | CV (%) c | Skewness | Kurtosis |
|---|---|---|---|---|---|---|---|---|
| Germinability | 2014 | 0.00 | 60.00 | 17.39 | 15.18 | 87.26 | 0.52 | −0.48 |
| 2015 | 0.00 | 97.00 | 31.35 | 18.69 | 59.64 | 0.74 | 0.64 | |
| 2016 | 0.00 | 100.00 | 37.21 | 15.86 | 42.62 | 0.32 | 1.25 | |
| Average | 0.00 | 85.67 | 28.65 | 17.70 | 63.17 | 0.53 | 0.47 | |
| Germinability rate | 2014 | 0.00 | 78.00 | 26.19 | 19.30 | 67.58 | 0.59 | −0.06 |
| 2015 | 10.00 | 100.00 | 44.58 | 17.90 | 43.30 | 0.60 | −0.05 | |
| 2016 | 13.00 | 100.00 | 47.33 | 16.40 | 37.83 | 0.62 | 0.23 | |
| Average | 7.67 | 92.67 | 39.37 | 16.82 | 49.57 | 0.60 | 0.04 | |
| Germinability index | 2014 | 0.00 | 100.75 | 25.18 | 16.91 | 65.13 | 1.01 | 2.09 |
| 2015 | 4.06 | 107.43 | 28.27 | 112.37 | 59.51 | 1.06 | 2.28 | |
| 2016 | 4.06 | 79.61 | 38.66 | 108.51 | 43.74 | −0.05 | −0.59 | |
| Average | 2.71 | 95.93 | 30.70 | 178.79 | 56.13 | 0.67 | 1.26 | |
| Vitality index | 2014 | 0.00 | 490.27 | 105.33 | 15.18 | 106.69 | 1.45 | 1.61 |
| 2015 | 2.01 | 495.74 | 112.57 | 18.69 | 96.40 | 1.60 | 2.65 | |
| 2016 | 2.49 | 754.77 | 215.11 | 15.86 | 83.11 | 0.87 | −0.11 | |
| Average | 1.50 | 580.26 | 144.34 | 17.70 | 95.40 | 1.31 | 1.38 |
| SNP | Chr. | Position | Trait | Year | −Log10(P) | MAF | R2 | Allele 1 | Allele 2 | Allele Effect | References |
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs25887810 | 1 | 25887810 | germination index | 2015 | 3.30 | 0.066 | 0.100 | C | T | 9.354 | |
| vitality index | 3.42 | 0.109 | 60.898 | ||||||||
| rs27941858 | 1 | 27941858 | germination rate | 2015 | 3.31 | 0.048 | 0.131 | G | A | −12.509 | |
| germination | 3.37 | 0.128 | −12.301 | ||||||||
| rs33981296 | 1 | 33981296 | germination index | 2016 | 3.07 | 0.054 | 0.104 | G | A | −9.849 | |
| vitality index | 3.67 | 0.118 | −116.141 | ||||||||
| rs8468280 | 3 | 8468280 | vitality index | 2015 | 3.04 | 0.093 | 0.098 | G | A | −50.777 | |
| 2016 | 3.45 | 0.112 | −90.582 | ||||||||
| rs12451980 | 4 | 12451980 | vitality index | 2014 | 3.22 | 0.111 | 0.123 | C | A | −51.458 | |
| 2015 | 3.14 | 0.101 | −49.905 | ||||||||
| rs46324094 | 4 | 46324094 | vitality index | 2014 | 3.28 | 0.144 | 0.124 | T | C | −45.041 | |
| 2015 | 3.19 | 0.102 | −44.039 | ||||||||
| rs249786 | 7 | 249786 | germination | 2016 | 3.57 | 0.147 | 0.113 | A | T | −6.939 | |
| germination index | 2016 | 5.17 | 0.164 | −9.130 | |||||||
| rs5985722 | 7 | 5985722 | germination | 2016 | 3.08 | 0.380 | 0.100 | G | T | 4.471 | |
| germination rate | 2016 | 5.12 | 0.134 | 6.886 | |||||||
| rs44713950 | 8 | 44713950 | germination rate | 2014 | 3.41 | 0.263 | 0.115 | C | T | −5.464 | |
| germination index | 2014 | 3.29 | 0.112 | −5.141 | |||||||
| rs46769428 | 8 | 46769428 | germination rate | 2014 | 3.50 | 0.257 | 0.118 | C | A | −6.006 | Singh R.K., et al. [47] |
| germination index | 2014 | 3.54 | 0.119 | 1.877 | |||||||
| rs18610980 | 14 | 18610980 | germination rate | 2014 | 3.16 | 0.251 | 0.108 | T | C | −5.662 | |
| germination index | 2015 | 3.21 | 0.097 | −5.806 | |||||||
| rs14369289 | 16 | 14369289 | vitality index | 2014 | 3.07 | 0.084 | 0.118 | G | T | 60.125 | |
| 2015 | 3.23 | 0.103 | 61.370 | ||||||||
| rs7372359 | 16 | 7372359 | vitality index | 2014 | 5.85 | 0.063 | 0.197 | A | T | 93.122 | |
| 2015 | 4.46 | 0.138 | 78.454 | ||||||||
| rs42627530 | 19 | 42627530 | germination index | 2015 | 3.09 | 0.162 | 0.094 | G | T | 6.383 | |
| vitality index | 2015 | 3.34 | 0.106 | 45.477 | |||||||
| 2014 | 3.57 | 0.132 | 43.023 | ||||||||
| germination | 2015 | 4.19 | 0.151 | 8.425 |
| Gene ID | Chromosome | Physical Position (bp) | Region | Trait | Alleles | −Log10(P) | R2 (%) | Functional Annotation |
|---|---|---|---|---|---|---|---|---|
| Glyma.03G058300 | 3 | 8286280 | intronic | VI15 | C/A | 2.581699 | 0.455 | cation exchanger 3 |
| 8287385 | intronic | VI16 | G/T | 2.853872 | 0.422 | |||
| 8287566 | intronic | VI14 | G/A | 2.636388 | 0.397 | |||
| 8287572 | intronic | VI15 | G/A | 5.137404 | 0.627 | |||
| 8287590 | intronic | VI14 | A/G | 3.044288 | 0.362 | |||
| 8287592 | intronic | VI15 | A/C | 5.137404 | 0.627 | |||
| 8287668 | intronic | VI14 | G/A | 3.044288 | 0.362 | |||
| 8287672 | intronic | VI14 | A/G | 3.044288 | 0.362 | |||
| 8287673 | intronic | VI14 | T/C | 3.044288 | 0.362 | |||
| 8287675 | intronic | VI14 | A/G | 3.044288 | 0.362 | |||
| 8287680 | intronic | VI15 | C/A | 2.747147 | 0.41 | |||
| 8287786 | intronic | VI15 | G/A | 3.31502 | 0.471 | |||
| 8280562 | upstream | GR15 | C/T | 2.928118 | 0.349 | |||
| 8288104 | UTR3 | VI15 | G/T | 2.576754 | 0.39 | |||
| Glyma.16G074600 | 16 | 7490751 | intronic | VI15 | G/A | 3.035858 | 0.442 | breast basic conserved 1 |
| 7490754 | intronic | VI15 | G/A | 3.035858 | 0.442 | |||
| 7492051 | intronic | GR14 | T/A | 2.530178 | 0.385 | |||
| 7492068 | intronic | VI15 | A/G | 2.59176 | 0.456 | |||
| 7492080 | intronic | VI15 | G/A | 2.537602 | 0.449 | |||
| 7492199 | downstream | VI15 | T/A | 2.66354 | 0.464 | |||
| 7492973 | downstream | GR14 | A/C | 3.672518 | 0.428 | |||
| 7493438 | downstream | GR14 | G/A | 2.527244 | 0.302 | |||
| 7493443 | synonymous | VI15 | G/A | 2.50307 | 0.299 | |||
| 7493464 | synonymous | GR14 | T/C | 3.335631 | 0.473 | |||
| 7493761 | synonymous | VI15 | T/A | 2.832683 | 0.419 | |||
| 7494765 | UTR5 | VI15 | A/T | 2.982967 | 0.498 | |||
| 7494812 | UTR5 | VI15 | C/T | 3.468981 | 0.486 | |||
| 7495467 | UTR5 | VI15 | A/T | 3.19135 | 0.458 | |||
| 7495873 | UTR5 | VI15 | A/T | 3.525653 | 0.492 | |||
| 7495915 | UTR5 | VI15 | T/A | 2.653647 | 0.463 | |||
| 7496419 | upstream | VI15 | C/T | 2.546682 | 0.45 | |||
| 7496426 | upstream | VI15 | A/G | 2.821023 | 0.418 | |||
| 7496457 | upstream | VI15 | T/C | 2.860121 | 0.423 | |||
| 7496477 | upstream | GR15 | T/A | 2.605548 | 0.393 | |||
| 7496478 | upstream | GR15 | G/A | 2.605548 | 0.393 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Wang, Y.; Xie, J.; Yang, Z.; Li, H.; Li, Y.; Teng, W.; Zhao, X.; Zhan, Y.; Han, Y. Identification of Candidate Genes for Soybean Storability via GWAS and WGCNA Approaches. Agronomy 2024, 14, 2457. https://doi.org/10.3390/agronomy14112457
Wu X, Wang Y, Xie J, Yang Z, Li H, Li Y, Teng W, Zhao X, Zhan Y, Han Y. Identification of Candidate Genes for Soybean Storability via GWAS and WGCNA Approaches. Agronomy. 2024; 14(11):2457. https://doi.org/10.3390/agronomy14112457
Chicago/Turabian StyleWu, Xu, Yuhe Wang, Jiapei Xie, Zhenhong Yang, Haiyan Li, Yongguang Li, Weili Teng, Xue Zhao, Yuhang Zhan, and Yingpeng Han. 2024. "Identification of Candidate Genes for Soybean Storability via GWAS and WGCNA Approaches" Agronomy 14, no. 11: 2457. https://doi.org/10.3390/agronomy14112457
APA StyleWu, X., Wang, Y., Xie, J., Yang, Z., Li, H., Li, Y., Teng, W., Zhao, X., Zhan, Y., & Han, Y. (2024). Identification of Candidate Genes for Soybean Storability via GWAS and WGCNA Approaches. Agronomy, 14(11), 2457. https://doi.org/10.3390/agronomy14112457