
Genome-Wide Association Analysis Identifies Loci and Candidate Genes for 100-Kernel Weight in Maize

Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Materials and Field Design
2.2. Phenotypic Data Collection and Statistical Analysis
2.3. Genotyping and Quality Control
2.4. Genome-Wide Association Analysis
2.5. Candidate Gene Identification
3. Results
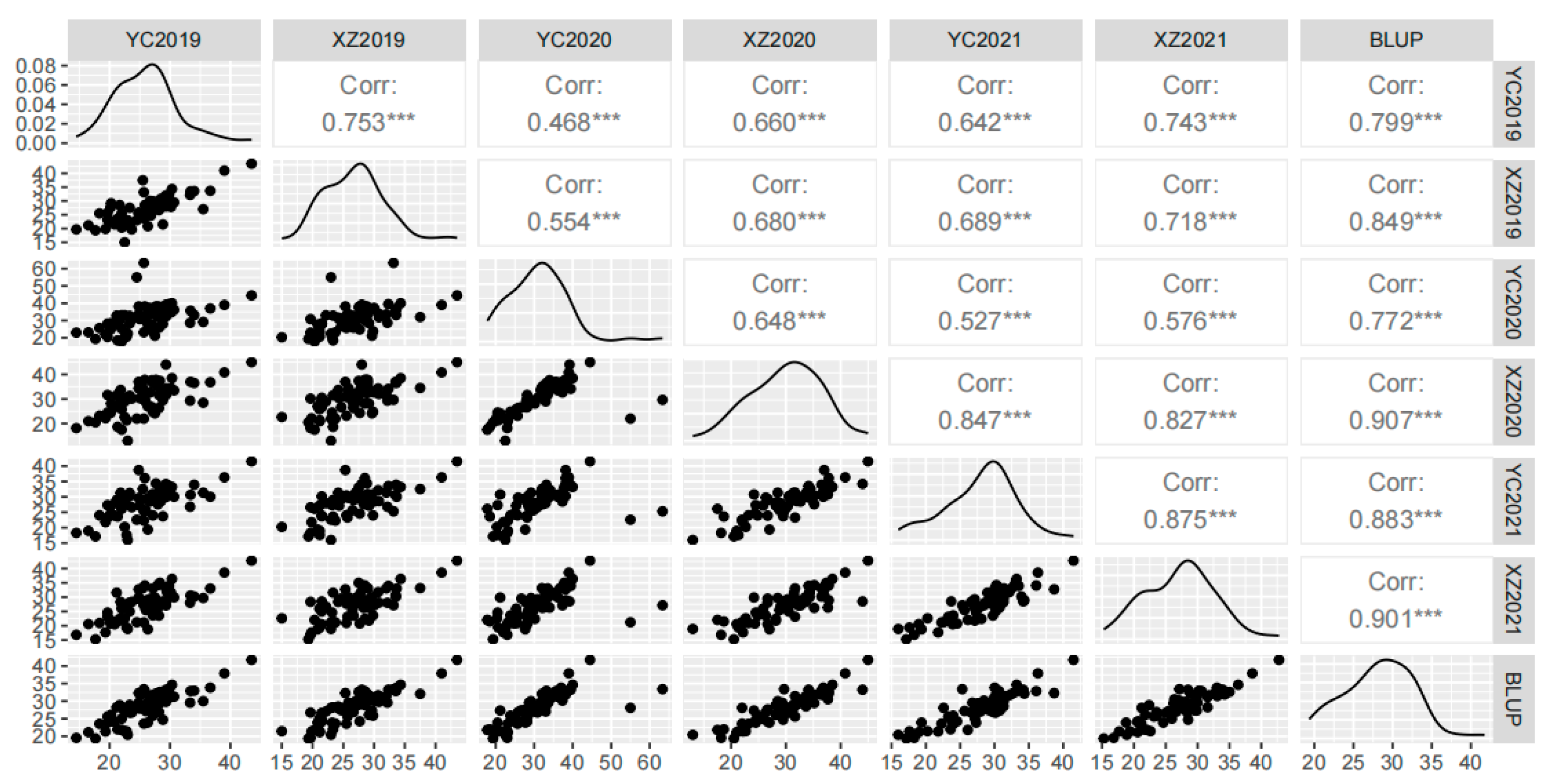
3.1. Phenotypic Analysis of 100-Kernel Weight
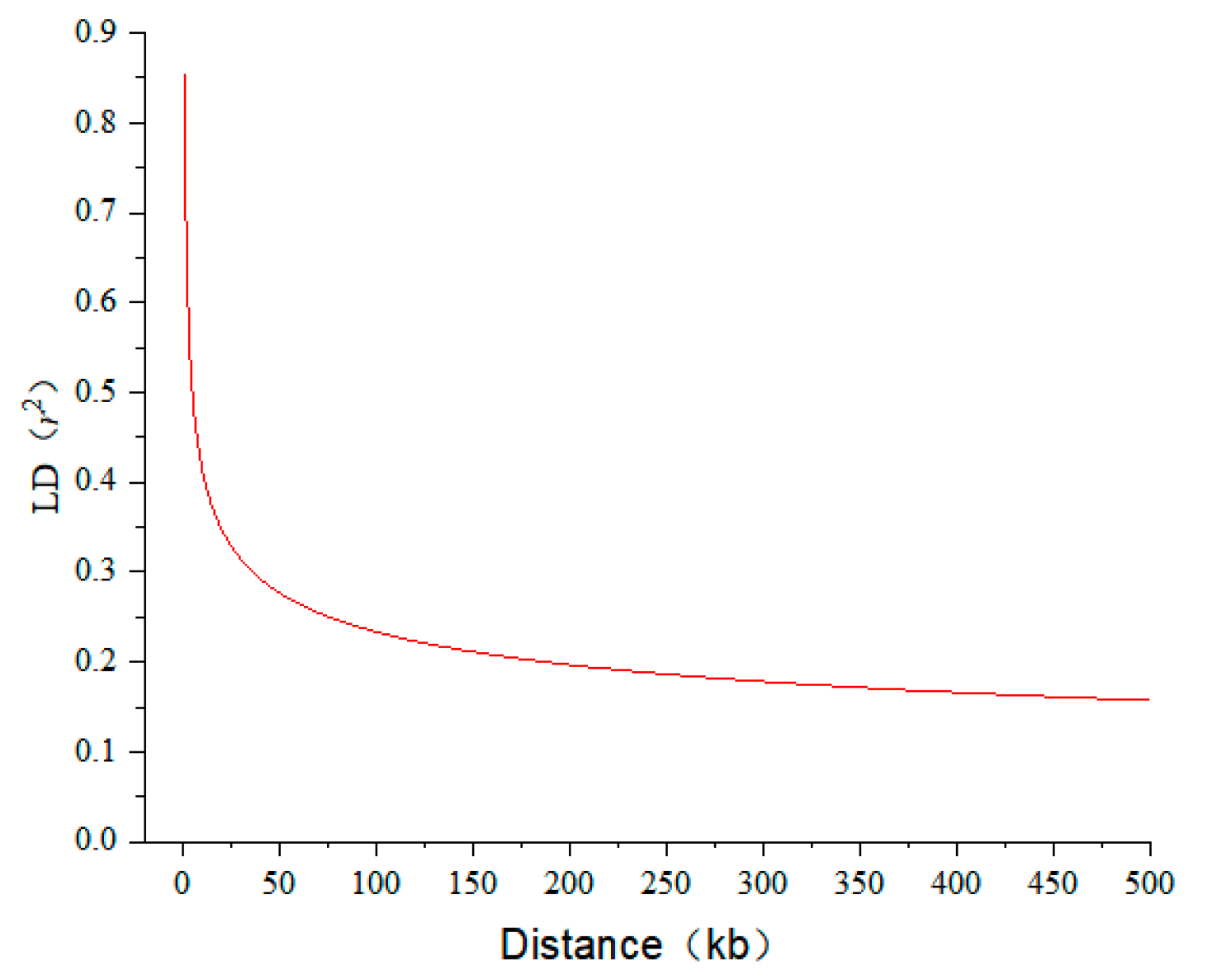
3.2. Genome-Wide Association Analysis of 100-Kernel Weight
3.3. HKW Candidate Gene Analysis
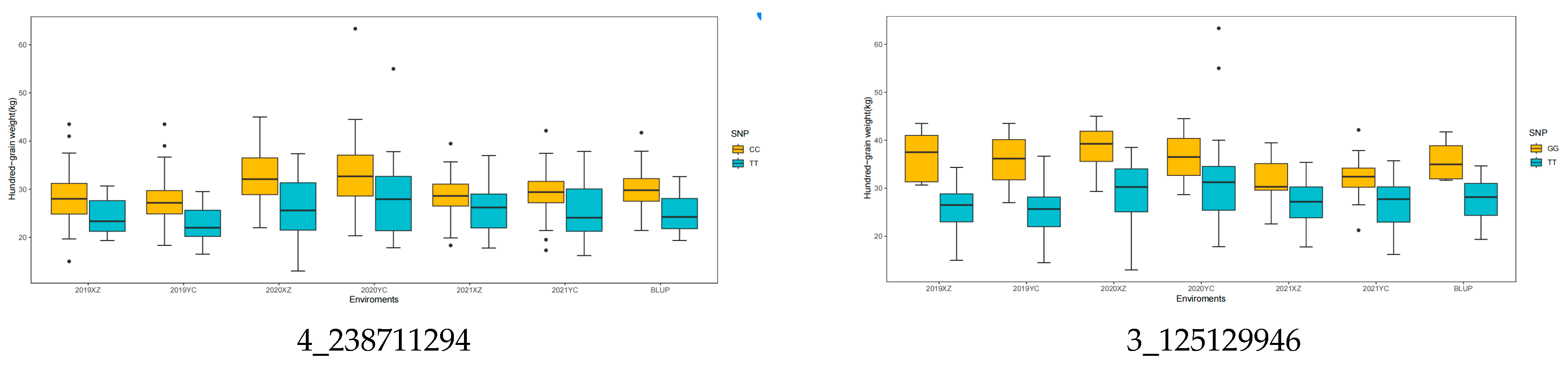
3.4. Allelic Variation Effects
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shen, T.L. Development of corn industry and food security in China. Agr. Econ. 2021, 5, 44–49. [Google Scholar]
- Alvarez Prado, S.; Sadras, V.O.; Borrás, L. Independent genetic control of maize (Zea mays L.) kernel weight determination and its phenotypic plasticity. J. Exp. Bot. 2014, 65, 4479–4487. [Google Scholar] [CrossRef] [PubMed]
- Sadras, V.O. Evolutionary aspects of the trade-off between seed size and number in crops. Field Crop Res. 2007, 100, 125–138. [Google Scholar] [CrossRef]
- Han, Y.; Li, D.; Zhu, D.; Li, H.; Li, X.; Teng, W.; Li, W. QTL analysis of soybean seed weight across multi-genetic backgrounds and environments. Theor. Appl. Genet. 2012, 125, 671–683. [Google Scholar] [CrossRef]
- Kang, Y.J.; Shim, K.C.; Lee, H.S.; Jeon, Y.A.; Kim, S.H.; Kang, J.W.; Yun, Y.T.; Park, I.K.; Ahn, S.N. Fine mapping and candidate gene analysis of the quantitative trait locus gw8.1 associated with grain length in rice. Genes Genom. 2018, 40, 389–397. [Google Scholar] [CrossRef]
- Li, Y.; Fan, C.; Xing, Y.; Jiang, Y.; Luo, L.; Sun, L.; Shao, D.; Xu, C.; Li, X.; Xiao, J.; et al. Natural variation in GS5 plays an important role in regulating grain size and yield in rice. Nat. Genet. 2011, 43, 1266. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, J.; Huang, J.; Lan, H.; Wang, C.; Yin, C.; Wu, Y.; Tang, H.; Qian, Q.; Li, J. Rare allele of OsPPKL1 associated with grain length causes extra-large grain and a significant yield increase in rice. Proc. Natl. Acad. Sci. USA 2012, 109, 21534–21539. [Google Scholar] [CrossRef]
- Song, X.-J.; Huang, W.; Shi, M.; Zhu, M.-Z.; Lin, H.-X. A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat. Genet. 2007, 39, 623. [Google Scholar] [CrossRef]
- Raihan, M.S.; Liu, J.; Huang, J.; Guo, H.; Pan, Q.; Yan, J. Multi-environment QTL analysis of grain morphology traits and fine mapping of a kernel-width QTL in Zheng58 x SK maize population. Theor. Appl. Genet. 2016, 129, 1465–1477. [Google Scholar] [CrossRef]
- Liu, J.; Huang, J.; Guo, H.; Lan, L.; Wang, H.; Xu, Y.; Yang, X.; Li, W.; Tong, H.; Xiao, Y.; et al. The Conserved and Unique Genetic Architecture of Kernel Size and Weight in Maize and Rice. Plant Physiol. 2017, 175, 774–785. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, J.; Fang, P.; Peng, Y. Comparative QTL analysis for yield components and morphological traits in maize (Zea mays L.)under water-stressed and well-watered conditions. Breed Sci. 2019, 69, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Wang, W.; Gong, S.; Zuo, J.; Li, S.; Xu, S. High Density Linkage Map Construction and Mapping of Yield Trait QTLs in Maize (Zea mays L.) Using the Genotyping-by-Sequencing (GBS) Technology. Front. Plant Sci. 2017, 8, 706. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Lu, G.; Liu, L.; Raihan, M.S.; Xu, J.; Jian, L.; Zhao, L.; Tran, T.M.; Zhang, Q.; Liu, J.; et al. The kernel size-related quantitative trait locus qKW9 encodes a pentatricopeptide repeat protein that affects photosynthesis and grain flling. Plant Physiol. 2020, 183, 1696–1709. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, Y.X.; Li, C.; Shi, Y.; Song, Y.; Zhang, D.; Wang, H.; Li, Y.; Wang, T. The retromer protein ZmVPS29 regulates maize kernel morphology likely through an auxin-dependent process(es). Plant Biotechnol. J. 2020, 18, 1004–1014. [Google Scholar] [CrossRef]
- Pasam, R.K.; Sharma, R.; Malosetti, M.; van Eeuwijk, F.A.; Haseneyer, G.; Kilian, B.; Graner, A. Genome-wide association studies for agronomical traits in a world wide spring barley collection. BMC Plant Biol. 2012, 12, 16. [Google Scholar] [CrossRef]
- Thornsberry, J.M.; Goodman, M.M.; Doebley, J.; Kresovich, S.; Nielsen, D.; Buckler, E.S. Dwarf8 polymorphisms associate with variation in flowering time. Nat. Genet. 2001, 28, 286–289. [Google Scholar] [CrossRef]
- Wilson, L.M.; Whitt, S.R.; Ibáñez, A.M.; Rocheford, T.R.; Goodman, M.M.; Buckler, E.S., IV. Dissection of maize kernel composition and starch production by candidate gene associations. Plant Cell 2004, 16, 2719–2733. [Google Scholar] [CrossRef]
- Godfray, H.C.; Beddington, J.R.; Crute, I.R.; Haddad, L.; Lawrence, D.; Muir, J.F.; Pretty, J.; Robinson, S.; Thomas, S.M.; Toulmin, C. Food security: The challenge of feeding 9 billion people. Science 2010, 327, 812–818. [Google Scholar] [CrossRef]
- Peng, B.; Zhao, X.L.; Wang, W.Y.; Li, C.H.; Li, Y.X.; Zhang, D.F.; Shi, Y.S.; Song, C.Y.; Wang, T.Y.; Li, Y. Genome wide association analysis of leaf orientation values in Maize. Crop J. 2020, 46, 819–831. [Google Scholar]
- Ma, J.; Cao, Y.Y.; Li, H.Y. Genome wide association analysis of corn cob diameter. Crop J. 2021, 47, 1228–1238. [Google Scholar]
- Qu, J.Z.; Feng, W.H.; Zhang, X.H.; Xu, S.T.; Xue, J.Q. Genome wide association analysis of maize grain size. Crop J. 2022, 48, 304–319. [Google Scholar]
- Knapp, S.J.; Stroup, W.W.; Ross, W.M. Exact confidence intervals for heritability on a progeny mean basis. Crop Sci. 1985, 25, 192–194. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Yang, L.; Li, T.; Tian, X.; Yang, B.; Lao, Y.; Wang, Y.; Zhang, X.; Xue, J.; Xu, S. Genome-wide association study (GWAS) reveals genetic basis of ear-related traits in maize. Euphytica 2020, 216, 172. [Google Scholar] [CrossRef]
- Zhu, X.M.; Shao, X.Y.; Pei, Y.H.; Guo, X.M.; Li, J.; Song, X.Y.; Zhao, M.A. Genetic diversity and genome-wide association study of major ear quantitative traits using high-density SNPs in maize. Front. Plant Sci. 2018, 9, 966. [Google Scholar] [CrossRef]
- Xiao, Y.; Tong, H.; Yang, X.; Xu, S.; Pan, Q.; Qiao, F.; Raihan, M.S.; Luo, Y.; Liu, H.; Zhang, X.; et al. Genome-wide dissection of the maize ear genetic architecture using multiple populations. New Phytol. 2016, 210, 1095–1106. [Google Scholar] [CrossRef]
- Xiao, Y.; Liu, H.; Wu, L.; Warburton, M.; Yan, J. Genome-wide Association Studies in maize: Praise and Stargaze. Mol. Plant. 2017, 10, 359–374. [Google Scholar] [CrossRef]
- Melchinger, A.E.; Utz, H.F.; Schön, C.C. Quantitative trait locus (QTL) mapping using different testers and independent population samples in maize reveals low power of QTL detection and large bias in estimates of QTL effects. Genetics 1998, 149, 383–403. [Google Scholar] [CrossRef] [PubMed]
- Ribaut, J.M.; Jiang, C.; Gonzalez-de-Leon, D.; Edmeades, G.O.; Hoisington, D. A Identification of quantitative trait loci under drought conditions in tropical maize. 2. Yield components and marker-assisted selection strategies. Theor. Appl. Genet. 1997, 94, 887–896. [Google Scholar] [CrossRef]
- Peng, B.; Li, Y.; Wang, Y.; Liu, C.; Liu, Z.; Tan, W.; Zhang, Y.; Wang, D.; Shi, Y.; Sun, B.; et al. QTL analysis for yield components and kernel-related traits in maize across multi-environments. Theor. Appl. Genet. 2011, 122, 1305–1320. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Peng, Y.; Zhang, J.; Fang, P.; Wu, B. Identification of QTLs and meta-QTLs for seven agronomic traits in multiple maize populations under well-watered and water-stressed conditions. Crop Sci. 2018, 58, 507–520. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, N.; Ku, L.; Tian, Z.; Shi, Y.; Guo, S.; Su, H.; Zhang, L.; Ren, Z.; Li, G.; et al. Dissection of the genetic architecture for grain quality-related traits in three RIL populations of maize (Zea mays L.). Plant Breed. 2016, 135, 38–46. [Google Scholar] [CrossRef]
- Yang, G.; Dong, Y.; Li, Y.; Wang, Q.; Shi, Q.; Zhou, Q. Verification of QTL for grain starch content and its genetic correlation with oil content using two connected RIL populations in high-oil maize. PLoS ONE 2013, 8, e53770. [Google Scholar] [CrossRef]
- Jiménez-Galindo, J.C.; Ordás, B.; Butrón, A.; Samayoa, L.F.; Malvar, R.A. QTL mapping for yield and resistance against mediterranean corn borer in maize. Front. Plant Sci. 2017, 8, 698. [Google Scholar] [CrossRef]
- Watanabe, C.K.; Hachiya, T.; Takahara, K. Effects of AOX1a deficiency on plant growth, gene expression of respiratory components and metabolic profile under low-nitrogen stress in Arabidopsis thaliana. Plant Cell Physiol. 2010, 51, 810–822. [Google Scholar] [CrossRef]
- Gandin, A.; Denysyuk, M.; Cousins, A.B. Disruption of the mitochondrial alternative oxidase (AOX) and uncoupling protein (UCP) alters rates of foliar nitrate and carbon assimilation in Arabidopsis thaliana. J. Exp. Bot. 2014, 65, 3133–3142. [Google Scholar] [CrossRef]
- Chai, T.T.; Simmonds, D.; Day, D.A.; Colmer, T.D.; Finnegan, P.M. A GmAOX2b antisense gene compromises vegetative growth and seed production in soybean. Planta 2012, 236, 199–207. [Google Scholar] [CrossRef]
- Li, N.; Xu, R.; Li, Y. Molecular networks of seed size control in plants. Annu. Rev. Plant Biol. 2019, 70, 435–463. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yue, J.; Yang, N.; Chuan, Z.; Zheng, C.; Zheng, Y.N.; Wu, X.; Yang, J.; Zhang, H.W.; Liu, L.L.; et al. An ERAD-related ubiquitin-conjugating enzyme boosts broad-spectrum disease resistance and yield in rice. Nat. Food 2023, 4, 774–787. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cao, H.; Chen, F.; Liu, Y. The roles of histone acetylation in seed performance and plant development. Plant Physiol. Biochem. 2014, 84, 125–133. [Google Scholar] [CrossRef]
- Rossi, V.; Locatelli, S.; Varotto, S.; Donn, G.; Pirona, R.; Henderson, D.A.; Hartings, H.; Motto, M. Maize histone deacetylase hda101 is involved in plant development, gene transcription, and sequence-specific modulation of histone modification of genes and repeats. Plant Cell 2007, 19, 1145–1162. [Google Scholar] [CrossRef]
- Huangfu, L.; Chen, R.; Lu, Y.; Zhang, E.; Miao, J.; Zuo, Z.; Zhao, Y.; Zhu, M.; Zhang, Z.; Li, P. OsCOMT, encoding a caffeic acid O-methyltransferase in melatonin biosynthesis, increases rice grain yield through dual regulation of leaf senescence and vascular development. Plant Biotechnol. J. 2022, 20, 1122–1139. [Google Scholar] [CrossRef]
- Malla, K.B.; Thapa, G.; Doohan, F.M. Mitochondrial phosphate transporter and methyltransferase genes contribute to Fusarium head blight Type II disease resistance and grain development in wheat. PLoS ONE 2021, 16, e0258726. [Google Scholar] [CrossRef]
- Qi, X.; Li, S.; Zhu, Y.; Zhao, Q.; Zhu, D.; Yu, J. ZmDof3, a maize endosperm-specific Dof protein gene, regulates starch accumulation and aleurone development in maize endosperm. Plant Mol. Biol. 2017, 93, 7–20. [Google Scholar] [CrossRef]
- Dong, Q.; Xu, Q.; Kong, J.; Peng, X.; Zhou, W.; Chen, L.; Wu, J.; Xiang, Y.; Jiang, H.; Cheng, B. Overexpression of ZmbZIP22 gene alters endosperm starch content and composition in maize and rice. Plant Sci. 2019, 283, 407–415. [Google Scholar] [CrossRef]
- Sun, F.; Ding, L.; Feng, W.Q.; Cao, Y.; Lu, F.Z.; Yang, Q.Q.; Li, W.C.; Lu, Y.L.; Shabek, N.; Fu, F.L.; et al. Maize transcription factor ZmBES1/BZR1-5 positively regulates kernel size. J. Exp. Bot. 2021, 72, 1714–1726. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Location | Mean | Max | Min | KURT | SKEW | SD | CV (%) | H2 (%) |
|---|---|---|---|---|---|---|---|---|---|
| 2019 | YC | 26.01 | 44.52 | 15.81 | 0.79 | 0.58 | 5.30 | 20.35 | 85.64 |
| XZ | 26.97 | 44.58 | 16.72 | 0.77 | 0.48 | 5.22 | 19.35 | ||
| 2020 | YC | 30.79 | 55.00 | 17.83 | 0.44 | 0.38 | 6.92 | 22.49 | |
| XZ | 30.02 | 44.21 | 14.23 | −0.17 | −0.29 | 6.47 | 21.54 | ||
| 2021 | YC | 28.12 | 42.54 | 16.76 | −0.15 | 0.34 | 5.19 | 18.46 | |
| XZ | 29.34 | 41.56 | 16.34 | 0.08 | −0.24 | 5.42 | 19.93 | ||
| BLUP | — | 28.24 | 42.34 | 18.43 | 0.12 | 0.41 | 4.56 | 16.17 |
| Source of Variation | Sum of Squares | DF | Mean Squares | F-Value |
|---|---|---|---|---|
| Location (L) | 6604 | 1 | 3302 | 149.803 *** |
| Year (Y) | 16 | 2 | 16 | 0.710 |
| Genotype (G) | 49,549 | 198 | 250 | 11.352 *** |
| Y × L | 545 | 2 | 273 | 12.368 *** |
| L × G | 11,372 | 330 | 34 | 1.563 *** |
| Y × G | 3119 | 168 | 19.00 | 0.842 |
| Y × L × G | 6867 | 326 | 21 | 0.969 |
| Residuals | 21,691 | 984 | 22 |
| Method | SNPID | Chr. | p-Value | R2 (%) | Envir. | Candidate Genes |
|---|---|---|---|---|---|---|
| FarmCPU | 1_274302441 | 1 | 1.09 × 10−8 | 4.91 | 2021YC | GRMZM2G135283 (Serine hydroxymethyltransferase) |
| FarmCPU | 2_2636802 | 2 | 1.17 × 10−5 | 1.02 | 2020YC | GRMZM2G010555 (alternative oxidase); GRMZM2G102471 (ubiquitin-conjugating enzyme 30) |
| FarmCPU | 3_122617408 | 3 | 1.93 × 10−5 | 1.50 | 2019XZ | -- |
| FarmCPU | 3_122617431 | 3 | 1.93 × 10−5 | 7.01 | 2019XZ | -- |
| FarmCPU | 3_122928920 | 3 | 1.93 × 10−5 | 2.10 | 2019XZ | GRMZM2G045435 (beta-1,4-N-acetylglucosaminyltransferase family protein) |
| FarmCPU | 3_122929643 | 3 | 1.93 × 10−5 | 4.01 | 2019XZ | - |
| FarmCPU/Blink | 3_125129946 | 3 | 1.88 × 10−5 | 8.12 | 2019XZ,2019XZ | - |
| FarmCPU | 3_229327953 | 3 | 2.22 × 10−6 | 0.01 | 2021YC | GRMZM2G093175 (Asparagine synthetase); GRMZM2G354558 (Omega-3 fatty acid desaturase) |
| FarmCPU | 3_36009926 | 3 | 1.74 × 10−5 | 2.22 | 2021YC | |
| FarmCPU/Blink | 4_238711294 | 4 | 8.27 × 10−6/1.47 × 10−8 | 7.44 | BLUP/2019YC | GRMZM2G060669 (Histone deacetylase complex subunit SAP18, GRMZM2G090156 (Methyltransferases) |
| FarmCPU/Blink | 5_142616635 | 5 | 7.45 × 10−10/1.27 × 10−6 | 9.05 | 2021XZ/2021YC | GRMZM2G162748 (Cytochrome b6-f complex iron-sulfur subunit) |
| FarmCPU | 5_18123084 | 5 | 1.63 × 10−6 | 1.32 | 2019XZ | GRMZM2G061292 (Dof transcription factor gene); GRMZM2G320754 (B3 domain transcription factors) |
| FarmCPU | 5_201092307 | 5 | 5.99 × 10−7 | 0.15 | 2020YC | GRMZM2G101287 (Adenine nucleotide alpha hydrolases-like superfamily protein); GRMZM2G347767 (Isoflavone reductase) |
| Blink | 1_50864066 | 1 | 5.66 × 10−8 | 7.17 | 2019XZ | - |
| Blink | 4_1490461 | 4 | 4.10 × 10−6 | 4.46 | 2019XZ | GRMZM2G015232 (Putative cytochrome P450 superfamily protein); GRMZM2G349791(O-methyltransferase ZRP4) |
| Blink | 5_210354193 | 5 | 9.97 × 10−6 | 1.06 | 2019XZ | GRMZM2G113418 (Glutaredoxin 2); GRMZM2G154332 (SAUR12-auxin-responsive SAUR family member) |
| Blink | 2_186957636 | 2 | 3.55 × 10−7 | 10.77 | 2019YC | GRMZM2G002075 (BZIP transcription factor;); GRMZM2G138454 (bHLH transcription factor) |
| Blink | 1_298487278 | 1 | 3.54 × 10−7 | 7.28 | 2021XZ | GRMZM2G159353 (putative cytochrome P450 superfamily protein) |
| Blink | 2_65602039 | 2 | 1.19 × 10−7 | 6.05 | 2021XZ | GRMZM2G015024 (50S ribosomal protein L22,ribosomal protein L22) |
| Blink | 2_79335071 | 2 | 2.076 × 10−6 | 6.55 | 2021XZ | - |
| Blink | 3_177543426 | 3 | 4.31 × 10−7 | 6.17 | 2021XZ | - |
| Blink | 4_188053528 | 4 | 1.16 × 10−6 | 1.34 | 2021YC | - |
| Blink | 3_178779239 | 3 | 1.35 × 10−8 | 7.31 | BLUP | - |
| Blink | 4_228498570 | 4 | 5.34 × 10−14 | 6.75 | BLUP | - |
| Blink | 5_68455590 | 5 | 1.12 × 10−9 | 4.95 | BLUP | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Cheng, D.; Ren, H.; Li, H.; Wang, R.; Dong, C.; Chang, J. Genome-Wide Association Analysis Identifies Loci and Candidate Genes for 100-Kernel Weight in Maize. Agronomy 2024, 14, 2954. https://doi.org/10.3390/agronomy14122954
Wang M, Cheng D, Ren H, Li H, Wang R, Dong C, Chang J. Genome-Wide Association Analysis Identifies Loci and Candidate Genes for 100-Kernel Weight in Maize. Agronomy. 2024; 14(12):2954. https://doi.org/10.3390/agronomy14122954
Chicago/Turabian StyleWang, Meixia, Danyang Cheng, Haojie Ren, Haoyang Li, Ruiyu Wang, Chunlin Dong, and Jianzhong Chang. 2024. "Genome-Wide Association Analysis Identifies Loci and Candidate Genes for 100-Kernel Weight in Maize" Agronomy 14, no. 12: 2954. https://doi.org/10.3390/agronomy14122954
APA StyleWang, M., Cheng, D., Ren, H., Li, H., Wang, R., Dong, C., & Chang, J. (2024). Genome-Wide Association Analysis Identifies Loci and Candidate Genes for 100-Kernel Weight in Maize. Agronomy, 14(12), 2954. https://doi.org/10.3390/agronomy14122954