
Integrated Bioinformatics and Multi-Omics Analyses Reveal Possible Molecular Mechanisms for Seed Starch Content Differences between Glycine max and Cicer arietinum



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Sources
2.1.1. Genomic and Proteomic Data
2.1.2. Transcriptomic Data
2.1.3. TFs and miRNAs in Soybean and Chickpea
2.1.4. Metabolomics Data of Soybean
2.2. Identification of Sucrose and Starch Metabolism-Related Genes in Arabidopsis and Their Homologous Genes in Soybean and Chickpea
2.3. Temporal Expression Analysis of Genes Involved in Sucrose and Starch Metabolism in Soybean and Chickpea
2.4. Differential Expression Analysis of Sucrose and Starch Metabolism-Related Genes in Soybean and Chickpea
2.5. Analysis of Sucrose and Starch Metabolism-Related Protein Interactions in Soybean and Chickpea
2.6. Co-Expression Network Analysis of Sucrose and Starch Metabolism-Related Genes in Soybean and Chickpea
2.7. Identification of Sucrose and Starch Metabolism-Related TFs and miRNAs in Soybean and Chickpea and Prediction of Their Target Genes
2.8. KEGG Enrichment and GO Annotation Analysis
2.9. Statistical Analysis
3. Results
3.1. Identification of Sucrose and Starch Metabolism-Related Genes in Soybean and Chickpea
3.2. Expression Trend Analysis of Sucrose and Starch Metabolism-Related Genes in Soybean and Chickpea
3.3. Differential Expression Analysis of Sucrose and Starch Metabolism-Related Genes in Soybean and Chickpea
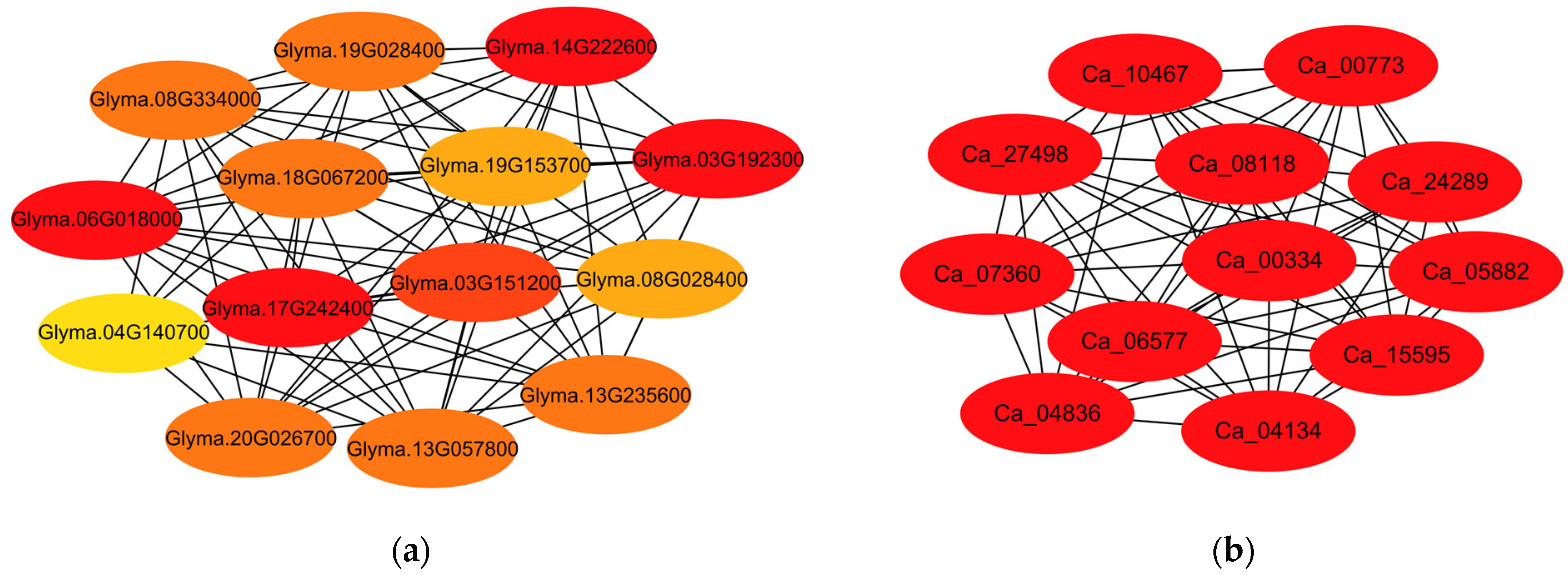
3.4. Interaction Analysis of Sucrose and Starch Metabolism-Related Proteins in Soybean and Chickpea
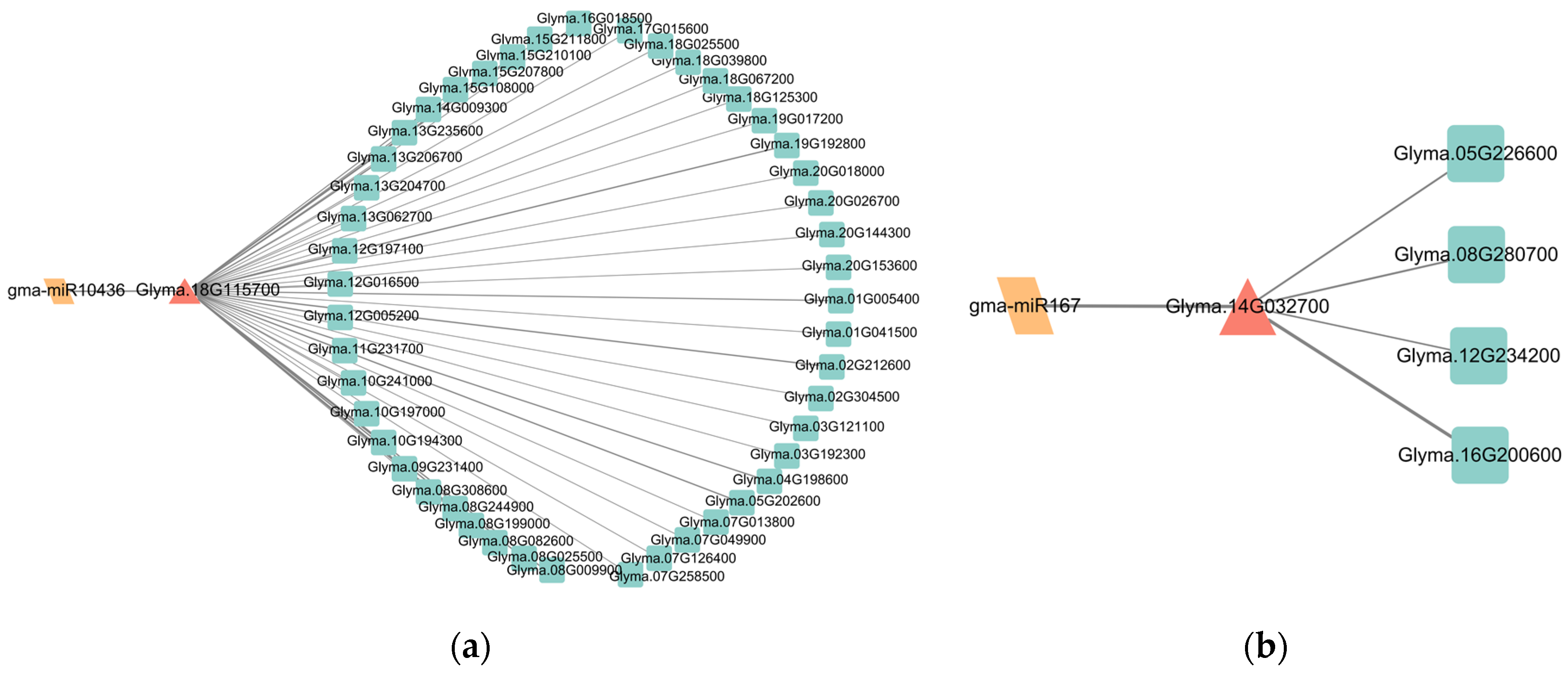
3.5. Identification of Sucrose and Starch Metabolism-Related miRNAs and Their Target Genes in Soybean and Chickpea
3.6. Co-Expression Analysis of Sucrose and Starch Metabolism-Related Genes and TFs in Soybean and Chickpea
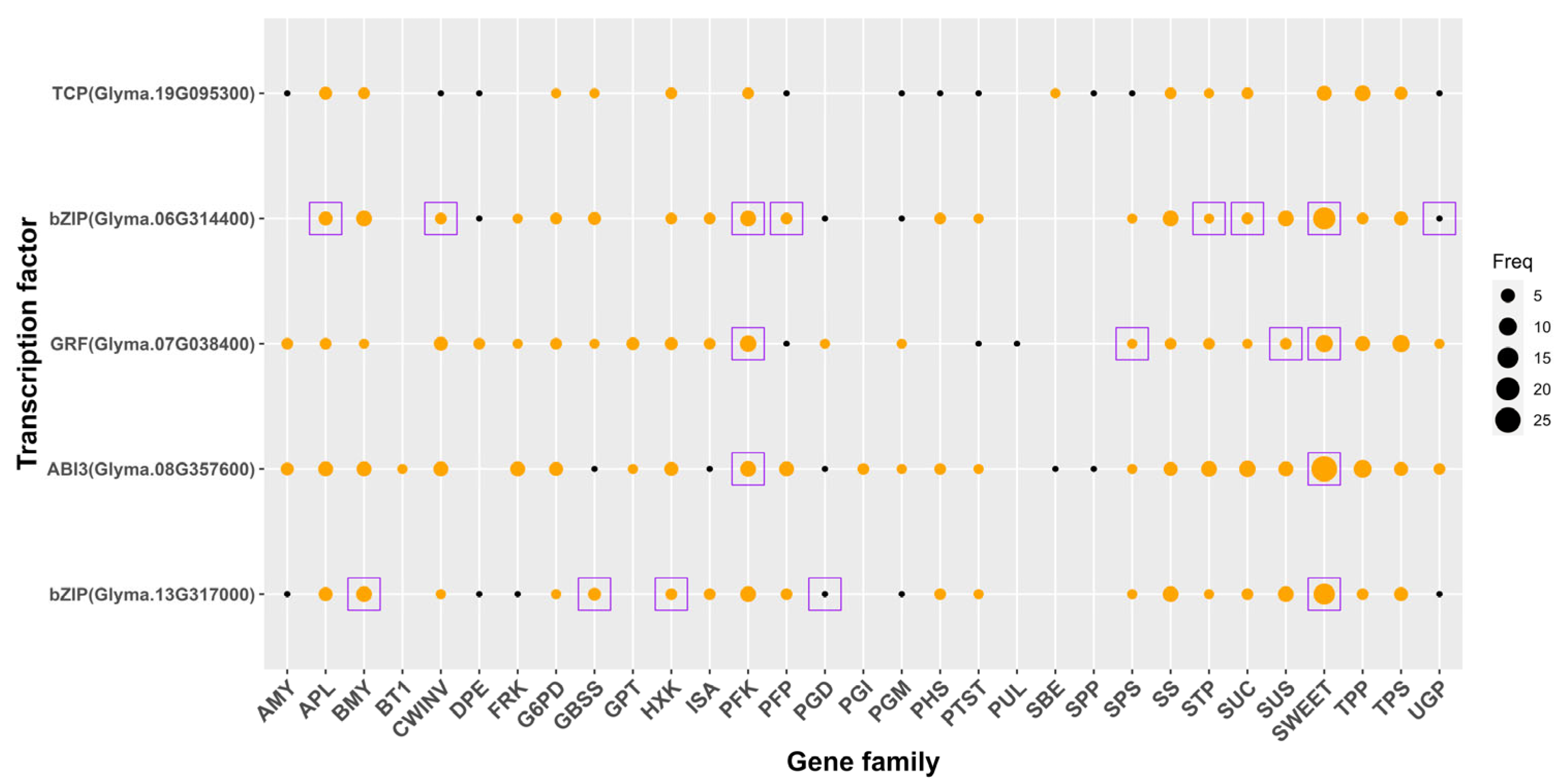
3.7. Identification of Sucrose and Starch Metabolism-Related TFs and Their Target Genes in Soybean and Chickpea
3.8. The Regulatory Network of Sucrose and Starch Metabolism-Related Genes in Soybean
4. Discussion
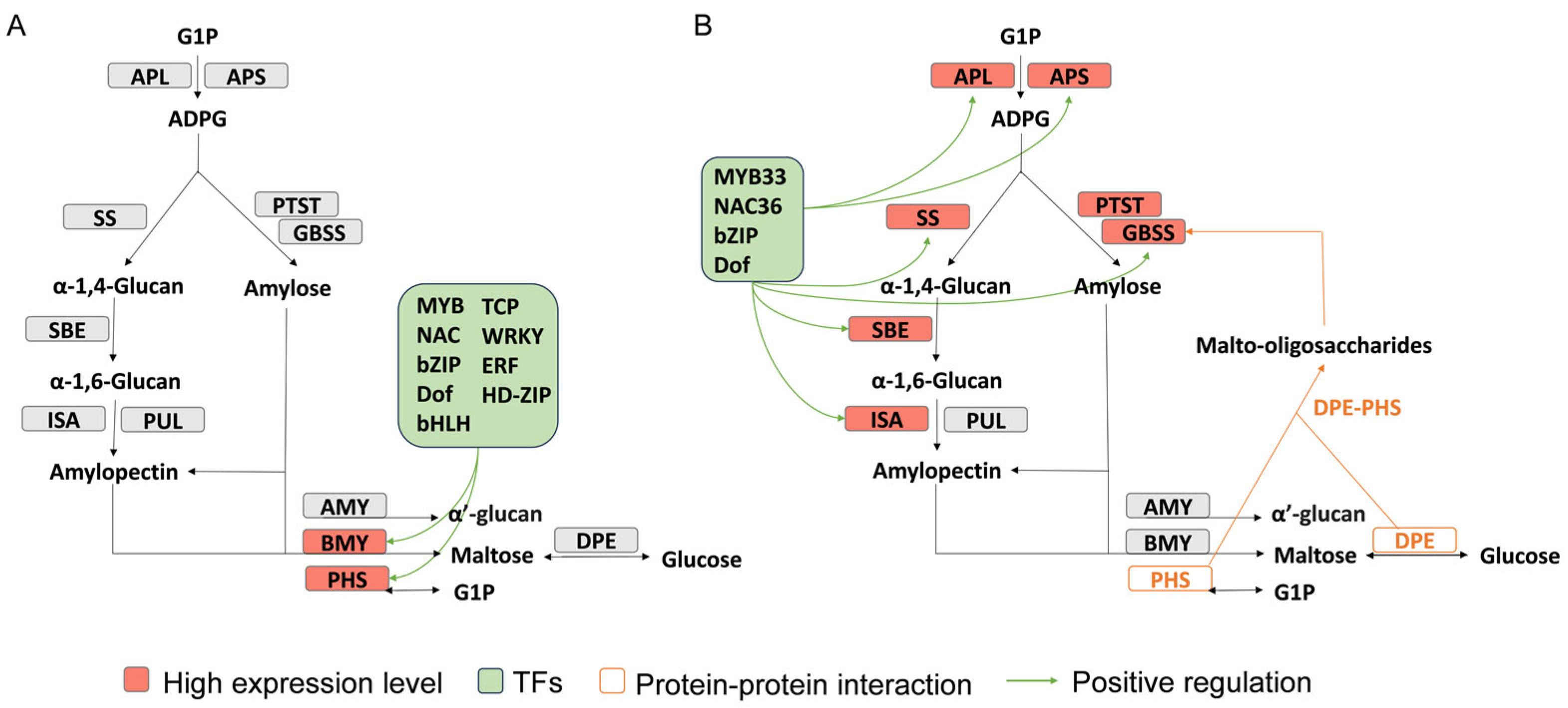
4.1. Different RELs of Starch Synthesis and Degradation-Related Genes in Soybean and Chickpea at the Nutrient Accumulation Stage May Lead to the Difference in Seed Starch Content between the Two Species
4.2. DPE and PHS Interaction in Chickpea May Increase Higher Seed Starch Content
4.3. miR167–ARF–Sucrose Metabolism Gene Pathway and TFs Positively Regulate Starch Degradation Genes BMY and PHS, Leading to Lower Seed Starch Content in Soybean
4.4. Improvement Strategies to Enhance Starch Content in Chickpea Seeds
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rachwa-Rosiak, D.; Nebesny, E.; Budryn, G. Chickpeas-composition, nutritional value, health benefits, application to bread and snacks: A review. Crit. Rev. Food Sci. Nutr. 2015, 55, 1137–1145. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, S.; Wang, Z.; Yuan, Y.; Zhang, Z.; Liang, Q.; Yang, X.; Duan, Z.; Liu, Y.; Kong, F.; et al. Progress in soybean functional genomics over the past decade. Plant Biotechnol. J. 2022, 20, 256–282. [Google Scholar] [CrossRef]
- Zeeman, S.C.; Kossmann, J.; Smith, A.M. Starch: Its metabolism, evolution, and biotechnological modification in plants. Annu. Rev. Plant Biol. 2010, 61, 209–234. [Google Scholar] [CrossRef]
- Huang, L.; Tan, H.; Zhang, C.; Li, Q.; Liu, Q. Starch biosynthesis in cereal endosperms: An updated review over the last decade. Plant Commun. 2021, 2, 100237. [Google Scholar] [CrossRef]
- Ruan, Y.L. Sucrose metabolism: Gateway to diverse carbon use and sugar signaling. Annu. Rev. Plant Biol. 2014, 65, 33–67. [Google Scholar] [CrossRef] [PubMed]
- Wingler, A. Transitioning to the next phase: The role of sugar signaling throughout the plant life cycle. Plant Physiol. 2018, 176, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Ma, S.; Wu, L.M.; Zhou, S.L.; Ruan, Y.L. Winners take all: Competition for carbon resource determines grain fate. Trends Plant Sci. 2023, 28, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Wang, J.; Zhu, X.D.; Hao, W.; Wang, L.Y.; Li, Q.; Zhang, L.X.; He, W.; Lu, B.R.; Lin, H.X.; et al. Control of rice grain-filling and yield by a gene with a potential signature of domestication. Nat. Genet. 2008, 40, 1370–1374. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Liu, H.; Zhang, Y.; Kang, T.; Zhang, L.; Tong, J.H.; Xiao, L.T.; Zhang, H.X. Constitutive expression of cell wall invertase genes increases grain yield and starch content in maize. Plant Biotechnol. J. 2013, 11, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Q.; Lin, I.W.N.; Qu, X.Q.; Sosso, D.; McFarlane, H.E.; Londoño, A.; Samuels, A.L.; Frommer, W.B. A cascade of sequentially expressed sucrose transporters in the seed coat and endosperm provides nutrition for the Arabidopsis embryo. Plant Cell 2015, 27, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, L.; Liu, H.; Yuan, M. Impaired SWEET-mediated sugar transportation impacts starch metabolism in developing rice seeds. Crop J. 2022, 10, 98–108. [Google Scholar] [CrossRef]
- Kotting, O.; Kossmann, J.; Zeeman, S.C.; Lloyd, J.R. Regulation of starch metabolism: The age of enlightenment? Curr. Opin. Plant Biol. 2010, 13, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.H.; Yu, W.W.; Zhang, C.Q.; Zhu, Y.J.; Xu, J.L.; Li, E.P.; Gilbert, R.G.; Liu, Q.Q. New insights into amylose and amylopectin biosynthesis in rice endosperm. Carbohydr. Polym. 2020, 230, 115656. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.K.; Nishi, A.; Satoh, H.; Okita, T.W. Rice endosperm-specific plastidial alpha-glucan phosphorylase is important for synthesis of short-chain malto-oligosaccharides. Arch. Biochem. Biophys. 2010, 495, 82–92. [Google Scholar] [CrossRef] [PubMed]
- van der Maarel, M.J.E.C.; Leemhuis, H. Starch modification with microbial alpha-glucanotransferase enzymes. Carbohydr. Polym. 2013, 93, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Damaris, R.N.; Lin, Z.Y.; Yang, P.F.; He, D.L. The rice alpha-amylase, conserved regulator of seed maturation and germination. Int. J. Mol. Sci. 2019, 20, 450. [Google Scholar] [CrossRef] [PubMed]
- Zhan, S.H.; Lukens, L. Identification of novel miRNAs and miRNA dependent developmental shifts of gene expression in Arabidoopsis thaliana. PLoS ONE 2010, 5, 1015710. [Google Scholar] [CrossRef]
- Peng, T.; Lv, Q.; Zhang, J.; Li, J.Z.; Du, Y.X.; Zhao, Q.Z. Differential expression of the microRNAs in superior and inferior spikelets in rice (Oryza sativa). J. Exp. Bot. 2011, 62, 4943–4954. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, Y.F.; Sunkar, R.; Zhang, W.X. SeqTar: An effective method for identifying microRNA guided cleavage sites from degradome of polyadenylated transcripts in plants. Nucleic Acids Res. 2012, 40, 2810. [Google Scholar] [CrossRef]
- Wu, Y.F.; Lee, S.K.; Yoo, Y.; Wei, J.; Kwon, S.Y.; Lee, S.W.; Jeon, J.S.; An, G. Rice transcription factor OsDOF11 modulates sugar transport by promoting expression of Sucrose Transporter and SWEET genes. Mol. Plant 2018, 11, 833–845. [Google Scholar] [CrossRef]
- Sun, W.J.; Gao, Z.Y.; Wang, J.; Huang, Y.Q.; Chen, Y.; Li, J.F.; Lv, M.L.; Wang, J.; Luo, M.; Zuo, K.J. Cotton fiber elongation requires the transcription factor GhMYB212 to regulate sucrose transportation into expanding fibers. New Phytol. 2019, 222, 864–881. [Google Scholar] [CrossRef]
- Mathan, J.; Singh, A.; Ranjan, A. Sucrose transport in response to drought and salt stress involves ABA-mediated induction of OsSWEET13 and OsSWEET15 in rice. Physiol. Plant. 2021, 171, 620–637. [Google Scholar] [CrossRef]
- Li, J.; Foster, R.; Ma, S.; Liao, S.J.; Bliss, S.; Kartika, D.; Wang, L.; Wu, L.; Eamens, A.L.; Ruan, Y.L. Identification of transcription factors controlling cell wall invertase gene expression for reproductive development via bioinformatic and transgenic analyses. Plant J. 2021, 106, 1058–1074. [Google Scholar] [CrossRef]
- Hu, Y.F.; Li, Y.P.; Zhang, J.; Liu, H.; Tian, M.; Huang, Y. Binding of ABI4 to a CACCG motif mediates the ABA-induced expression of the ZmSSI gene in maize (Zea mays L.) endosperm. J. Exp. Bot. 2012, 63, 5979–5989. [Google Scholar] [CrossRef]
- Wang, J.C.; Xu, H.; Zhu, Y.; Liu, Q.Q.; Cai, X.L. OsbZIP58, a basic leucine zipper transcription factor, regulates starch biosynthesis in rice endosperm. J. Exp. Bot. 2013, 64, 3453–3466. [Google Scholar] [CrossRef]
- Gao, Y.J.; An, K.X.; Guo, W.W.; Chen, Y.M.; Zhang, R.J.; Zhang, X.; Chang, S.Y.; Rossi, V.; Jin, F.M.; Cao, X.Y.; et al. The endosperm-specific transcription factor TaNAC019 regulates glutenin and starch accumulation and its elite allele improves wheat grain quality. Plant Cell 2021, 33, 603–622. [Google Scholar] [CrossRef]
- Hu, Y.F.; Li, Y.P.; Weng, J.F.; Liu, H.M.; Yu, G.W.; Liu, Y.H.; Xiao, Q.L.; Huang, H.N.; Wang, Y.B.; Wei, B.; et al. Coordinated regulation of starch synthesis in maize endosperm by microRNAs and DNA methylation. Plant J. 2021, 105, 108–123. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Pan, Y.F.; Liu, L.M.; Zhang, H.Q.; Zhang, Y.M. Integrated transcriptomic and bioinformatics analyses reveal the molecular mechanisms for the differences in seed oil and starch content between Glycine and Cicer arietinum. Front. Plant Sci. 2021, 12, 743680. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.Y.; Krishnakumar, V.; Chan, A.P.; Thibaud-Nissen, F.; Schobel, S.; Town, C.D. Araport11: A complete reannotation of the Arabidoopsis thaliana reference genome. Plant J. 2017, 89, 789–804. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.X.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.J.; Thelen, J.J.; Cheng, J.L.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 465, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Song, C.; Saxena, R.K.; Azam, S.; Yu, S.; Sharpe, A.G.; Cannon, S.; Baek, J.; Rosen, B.D.; Tar’an, B.; et al. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 2013, 31, 240–246. [Google Scholar] [CrossRef]
- Ouyang, S.; Zhu, W.; Hamilton, J.; Lin, H.; Campbell, M.; Childs, K.; Thibaud-Nissen, F.; Malek, R.L.; Lee, Y.; Zheng, L.; et al. The TIGR rice genome annotation resource: Improvements and new features. Nucleic Acids Res. 2007, 35, D883–D887. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.T.; Zhou, Z.K.; Wang, Z.; Li, W.Y.; Fang, C.; Wu, M.; Ma, Y.M.; Liu, T.F.; Kong, L.A.; Peng, D.L.; et al. Global dissection of alternative splicing in paleopolyploid soybean. Plant Cell 2014, 26, 996–1008. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Singh, V.K.; Rajkumar, M.S.; Kumar, V.; Jain, M. Global transcriptome and coexpression network analyses reveal cultivar-specific molecular signatures associated with seed development and seed size/weight determination in chickpea. Plant J. 2017, 91, 1088–1107. [Google Scholar] [CrossRef]
- Steven, W.R.; John, J.H.; Harvey, T. How a Soybean Plant Develops; Iowa State University, Cooperative Extension Service: Ames, IA, USA, 1967; pp. 1–20. [Google Scholar]
- Jain, M.; Chevala, V.V.; Garg, R. Genome-wide discovery and differential regulation of conserved and novel microRNAs in chickpea via deep sequencing. J. Exp. Bot. 2014, 65, 5945–5958. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhang, Y.W.; Liu, J.Y.; Zuo, J.F.; Zhang, Z.C.; Guo, L.; Zhang, Y.M. 4D genetic networks reveal the genetic basis of metabolites and seed oil-related traits in 398 soybean RILs. Biotechnol. Biofuels Bioprod. 2022, 15, 92. [Google Scholar] [CrossRef]
- Zuo, J.F.; Niu, Y.; Cheng, P.; Feng, J.Y.; Han, S.F.; Zhang, Y.H.; Shu, G.; Wang, Y.; Zhang, Y.M. Effect of marker segregation distortion on high density linkage map construction and QTL mapping in Soybean (Glycine max L.). Heredity 2019, 123, 579–592. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Zhang, Z.B.; Dunwell, J.M.; Zhang, Y.M. An integrated omics analysis reveals molecular mechanisms that are associated with differences in seed oil content between Glycine max and Brassica. BMC Plant Biol. 2018, 18, 328. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Yang, D.C.; Meng, Y.Q.; Jin, J.P.; Gao, G. PlantRegMap: Charting functional regulatory maps in plants. Nucleic Acids Res. 2020, 48, D1104–D1113. [Google Scholar] [CrossRef]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef]
- Dai, X.B.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, W155–W159. [Google Scholar] [CrossRef]
- Bu, D.C.; Luo, H.T.; Huo, P.P.; Wang, Z.H.; Zhang, S.; He, Z.H.; Wu, Y.; Zhao, L.H.; Liu, J.J.; Guo, J.C.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, F.M.; Wang, N.; Magee, G.B.; Nanduri, B.; Lawrence, M.L.; Camon, E.B.; Barrell, D.G.; Hill, D.P.; Dolan, M.E.; Williams, W.P.; et al. AgBase: A functional genomics resource for agriculture. BMC Genom. 2006, 7, 229. [Google Scholar] [CrossRef]
- Gámez-Arjona, F.M.; Raynaud, S.; Ragel, P.; Mérida, A. Starch synthase 4 is located in the thylakoid membrane and interacts with plastoglobule-associated proteins in Arabidopsis. Plant J. 2014, 80, 305–316. [Google Scholar] [CrossRef]
- McWhite, C.D.; Papoulas, O.; Drew, K.; Cox, R.M.; June, V.; Dong, O.X.; Kwon, T.; Wan, C.H.; Salmi, M.L.; Roux, S.J.; et al. A pan-plant protein complex map reveals deep conservation and novel assemblies. Cell 2020, 181, 460–474. [Google Scholar] [CrossRef]
- Meyer, T.; Hölscher, C.; Schwöppe, C.; von Schaewen, A. Alternative targeting of Arabidopsis plastidic glucose-6-phosphate dehydrogenase G6PD1 involves cysteine-dependent interaction with G6PD4 in the cytosol. Plant J. 2011, 66, 745–758. [Google Scholar] [CrossRef]
- Cho, Y.H.; Yoo, S.D.; Sheen, J. Regulatory functions of nuclear hexokinase1 complex in glucose signaling. Cell 2006, 127, 579–589. [Google Scholar] [CrossRef]
- Karve, A.; Xia, X.X.; Moore, B.D. Arabidopsis hexokinase-like1 and hexokinase1 form a critical node in mediating plant glucose and ethylene responses. Plant Physiol. 2012, 158, 1965–1975. [Google Scholar] [CrossRef]
- Seung, D.; Soyk, S.; Coiro, M.; Maier, B.A.; Eicke, S.; Zeeman, S.C. PROTEIN TARGETING TO STARCH is required for localising GRANULE-BOUND STARCH SYNTHASE to starch granules and for normal amylose synthesis in Arabidopsis. PLoS Biol. 2015, 13, 100208010. [Google Scholar] [CrossRef]
- Zhang, L.; Li, N.; Zhang, J.; Zhao, L.L.; Qiu, J.J.; Wei, C.X. The CBM48 domain-containing protein FLO6 regulates starch synthesis by interacting with SSIVb and GBSS in rice. Plant Mol. Biol. 2022, 108, 343–361. [Google Scholar] [CrossRef]
- Hwang, S.K.; Koper, K.; Satoh, H.; Okita, T.W. Rice Endosperm Starch Phosphorylase (Pho1) assembles with disproportionating enzyme (Dpe1) to form a protein complex that enhances synthesis of malto-oligosaccharides. J. Biol. Chem. 2016, 291, 19994–20007. [Google Scholar] [CrossRef]
- Nakamura, Y.; Ono, M.; Sawada, T.; Crofts, N.; Fujita, N.; Steup, M. Characterization of the functional interactions of plastidial starch phosphorylase and starch branching enzymes from rice endosperm during reserve starch biosynthesis. Plant Sci. 2017, 264, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Viola, I.L.; Uberti Manassero, N.G.; Ripoll, R.; Gonzalez, D.H. The Arabidopsis class I TCP transcription factor AtTCP11 is a developmental regulator with distinct DNA-binding properties due to the presence of a threonine residue at position 15 of the TCP domain. Biochem. J. 2011, 435, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Jo, L.; Pelletier, J.M.; Hsu, S.W.; Baden, R.; Goldberg, R.B.; Harada, J.J. Combinatorial interactions of the LEC1 transcription factor specify diverse developmental programs during soybean seed development. Proc. Natl. Acad. Sci. USA 2020, 117, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Omidbakhshfard, M.A.; Fujikura, U.; Olas, J.J.; Xue, G.P.; Balazadeh, S.; Mueller-Roeber, B. GROWTH-REGULATING FACTOR 9 negatively regulates Arabidopsis leaf growth by controlling ORG3 and restricting cell proliferation in leaf primordia. PLoS Genet. 2018, 14, 1007484. [Google Scholar] [CrossRef] [PubMed]
- Sasnauskas, G.; Kauneckaite, K.; Siksnys, V. Structural basis of DNA target recognition by the B3 domain of Arabidopsis epigenome reader VAL1. Nucleic Acids Res. 2018, 46, 4316–4324. [Google Scholar] [CrossRef]
- Liao, Y.; Zou, H.F.; Wei, W.; Hao, Y.J.; Tian, A.G.; Huang, J.; Liu, Y.F.; Zhang, J.S.; Chen, S.Y. Soybean GmbZIP44, GmbZIP62 and GmbZIP78 genes function as negative regulator of ABA signaling and confer salt and freezing tolerance in transgenic Arabidopsis. Planta 2008, 228, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Ohdan, T.; Francisco, P.B.; Sawada, T.; Hirose, T.; Terao, T.; Satoh, H.; Nakamura, Y. Expression profiling of genes involved in starch synthesis in sink and source organs of rice. J. Exp. Bot. 2005, 56, 3229–3244. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.Z.; Xu, S.T.; Zhang, Z.Q.; Chen, G.Z.; Zhong, Y.Y.; Liu, L.S.; Zhang, R.H.; Xue, J.Q.; Guo, D.W. Evolutionary, structural and expression analysis of core genes involved in starch synthesis. Sci. Rep. 2018, 8, 12736. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Tian, Z.X.; Chen, C.H.; Luo, L.H.; Zhao, B.; Wang, Z.; Yu, L.L.; Li, Y.S.; Sun, Y.D.; Li, W.Y.; et al. Genome sequencing of adzuki bean (Vignaangularis) provides insight into high starch and low fat accumulation and domestication. Proc. Natl. Acad. Sci. USA 2015, 112, 13213–13218. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhang, G.W.; Xu, S.C.; Mao, W.H.; Hu, Q.Z.; Gong, Y.M. Comparative transcriptomic analyses of vegetable and grain pea (Pisum sativum L.) seed development. Front. Plant Sci. 2015, 6, 1039. [Google Scholar] [CrossRef] [PubMed]
- Scheidig, A.; Fröhlich, A.; Schulze, S.; Lloyd, J.R.; Kossmann, J. Downregulation of a chloroplast-targeted beta-amylase leads to a starch-excess phenotype in leaves. Plant J. 2002, 30, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Zeeman, S.C.; Delatte, T.; Messerli, G.; Umhang, M.; Stettler, M.; Mettler, T.; Streb, S.; Reinhold, H.; Kötting, O. Starch breakdown: Recent discoveries suggest distinct pathways and novel mechanisms. Funct. Plant Biol. 2007, 34, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Andriotis, V.M.E.; Pike, M.J.; Kular, B.; Rawsthorne, S.; Smith, A.M. Starch turnover in developing oilseed embryos. New Phytol. 2010, 187, 791–804. [Google Scholar] [CrossRef]
- Dong, X.B.; Zhang, D.; Liu, J.; Liu, Q.Q.; Liu, H.L.; Tian, L.H.; Jiang, L.; Qu, L.Q. Plastidial disproportionating enzyme participates in starch synthesis in rice endosperm by transferring maltooligosyl groups from amylose and amylopectin to amylopectin. Plant Physiol. 2015, 169, 2496–2512. [Google Scholar] [CrossRef]
- Liu, H.; Jia, S.; Shen, D.; Liu, J.; Li, J.; Zhao, H.; Han, S.; Wang, Y. Four AUXIN RESPONSE FACTOR genes downregulated by microRNA167 are associated with growth and development in Oryza sativa. Funct. Plant Biol. 2012, 39, 736–744. [Google Scholar] [CrossRef]
- Ruuska, S.A.; Girke, T.; Benning, C.; Ohlrogge, J.B. Contrapuntal networks of gene expression during Arabidopsis seed filling. Plant Cell 2002, 14, 1191–1206. [Google Scholar] [CrossRef] [PubMed]
- Radchuk, V.V.; Borisjuk, L.; Sreenivasulu, N.; Merx, K.; Mock, H.P.; Rolletschek, H.; Wobus, U.; Weschke, W. Spatiotemporal profiling of starch biosynthesis and degradation in the developing barley grain. Plant Physiol. 2009, 150, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, K.; Küster, H.; Rutten, T.; Fait, A.; Fernie, A.R.; Miersch, O.; Wasternack, C.; Emery, R.J.N.; Desel, C.; Hosein, F.; et al. ADP-Glucose pyrophosphorylase-deficient pea embryos reveal specific transcriptional and metabolic changes of carbon-nitrogen metabolism and stress responses. Plant Physiol. 2009, 149, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, M.; Rai, S.; Venkadesan, S.; Sinha, S.K.; Mohan, S.; Mandal, P.K. Transcriptome analysis reveals important candidate genes related to nutrient reservoir, carbohydrate metabolism, and defence proteins during grain development of hexaploid bread wheat and its diploid progenitors. Genes 2020, 11, 509. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.D.; Wang, W.Q.; Tong, Y.; Li, M.J.; Grierson, D.; Ferguson, I.; Chen, K.S.; Yin, X.R. Transcriptome analysis identifies a zinc finger protein regulating starch degradation in kiwifruit. Plant Physiol. 2018, 178, 850–863. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, M.; Wang, J.; Zhang, J.; Miao, H.; Wang, Z.; Jia, C.; Zhang, J.; Xu, B.; Jin, Z. Transcription factor MaMADS36 plays a central role in regulating banana fruit ripening. J. Exp. Bot. 2021, 72, 7078–7091. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.Y. The Function and Mechanism of Transcription Factor MaAP2a-1 and MabHLH6 in Regulation of Postharvest Banana Starch Degradation. Ph.D. Dissertation, South China Agricultral University, Guangzhou, China, 2017. (In Chinese). [Google Scholar]
- Liu, L.; Wang, K.; Han, Y.L.; Chen, W.; Cao, S.F.; Shi, L.Y. Functional identification of AcbHLH137 and its transcriptional activation of starch degradation gene AcBAM3 in kiwifruit. Henong Xuebao 2022, 36, 544–553. (In Chinese) [Google Scholar] [CrossRef]
- Shoaib, N.; Liu, L.; Ali, A.; Mughal, N.; Yu, G.; Huang, Y. Molecular functions and pathways of plastidial starch phosphorylase (PHO1) in starch metabolism: Current and future perspectives. Int. J. Mol. Sci. 2021, 22, 10450. [Google Scholar] [CrossRef]
- Wang, J.Y.; Li, Y.H.; Li, A.; Liu, R.H.; Gao, X.; Li, D.; Kou, X.H.; Xue, Z.H. Nutritional constituent and health benefits of chickpea (Cicer arietinum L.): A review. Food Res. Int. 2021, 150 Pt A, 110790. [Google Scholar] [CrossRef]
- Chen, C.; He, B.S.; Liu, X.X.; Ma, X.D.; Liu, Y.J.; Yao, H.Y.; Zhang, P.; Yin, J.L.; Wei, X.; Koh, H.J.; et al. Pyrophosphate-fructose 6-phosphate 1-phosphotransferase (PFP1) regulates starch biosynthesis and seed development via heterotetramer formation in rice (Oryza sativa L.). Plant Biotechnol. J. 2020, 18, 83–95. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, Y.; Zheng, A.; Li, G.; Zhang, Y. Integrated Bioinformatics and Multi-Omics Analyses Reveal Possible Molecular Mechanisms for Seed Starch Content Differences between Glycine max and Cicer arietinum. Agronomy 2024, 14, 328. https://doi.org/10.3390/agronomy14020328
Pan Y, Zheng A, Li G, Zhang Y. Integrated Bioinformatics and Multi-Omics Analyses Reveal Possible Molecular Mechanisms for Seed Starch Content Differences between Glycine max and Cicer arietinum. Agronomy. 2024; 14(2):328. https://doi.org/10.3390/agronomy14020328
Chicago/Turabian StylePan, Yifan, Ao Zheng, Guiqi Li, and Yuanming Zhang. 2024. "Integrated Bioinformatics and Multi-Omics Analyses Reveal Possible Molecular Mechanisms for Seed Starch Content Differences between Glycine max and Cicer arietinum" Agronomy 14, no. 2: 328. https://doi.org/10.3390/agronomy14020328
APA StylePan, Y., Zheng, A., Li, G., & Zhang, Y. (2024). Integrated Bioinformatics and Multi-Omics Analyses Reveal Possible Molecular Mechanisms for Seed Starch Content Differences between Glycine max and Cicer arietinum. Agronomy, 14(2), 328. https://doi.org/10.3390/agronomy14020328