
Exploring the Roles of TALE Gene Family in Maize Drought Stress Responses

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Stress Treatment Procedures
2.2. TALE Family Member Identification in Maize
2.3. Phylogenetic Analysis and Categorization of ZmTALE Family Members
2.4. Structural and Motif Analysis of ZmTALE Family Genes
2.5. Chromosomal Localisation of the Maize ZmTALE Gene
2.6. ZmTALE Gene Collinearity Analysis within the Maize Genome
2.7. Expression Analysis of ZmTALE Family Genes
3. Results
3.1. Identification of the ZmTALE Family in Maize
3.2. Analysis of Physicochemical Properties of Proteins Encoded by the ZmTALE Gene Family
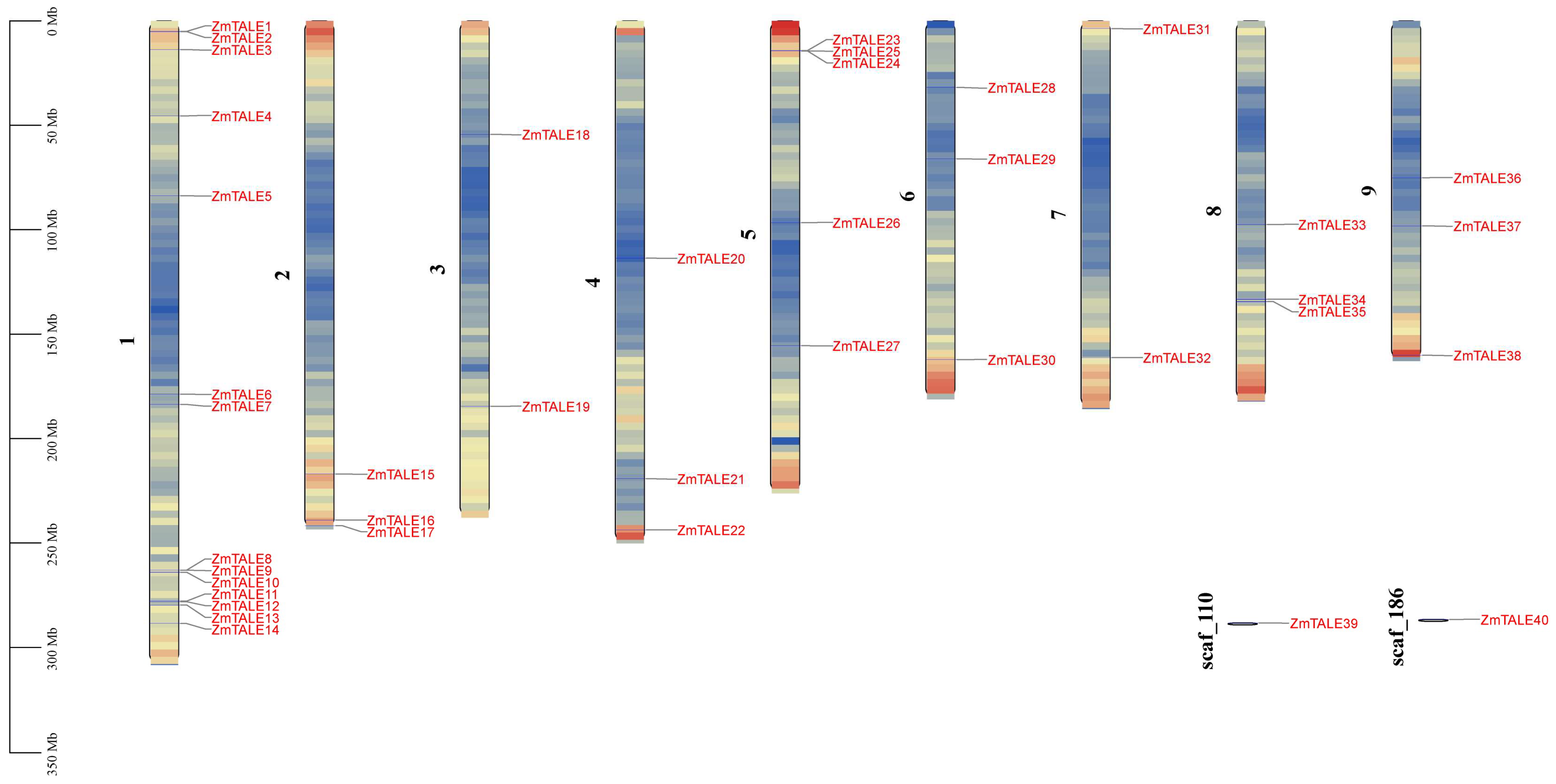
3.3. Distribution of ZmTALE Genes across Chromosomes
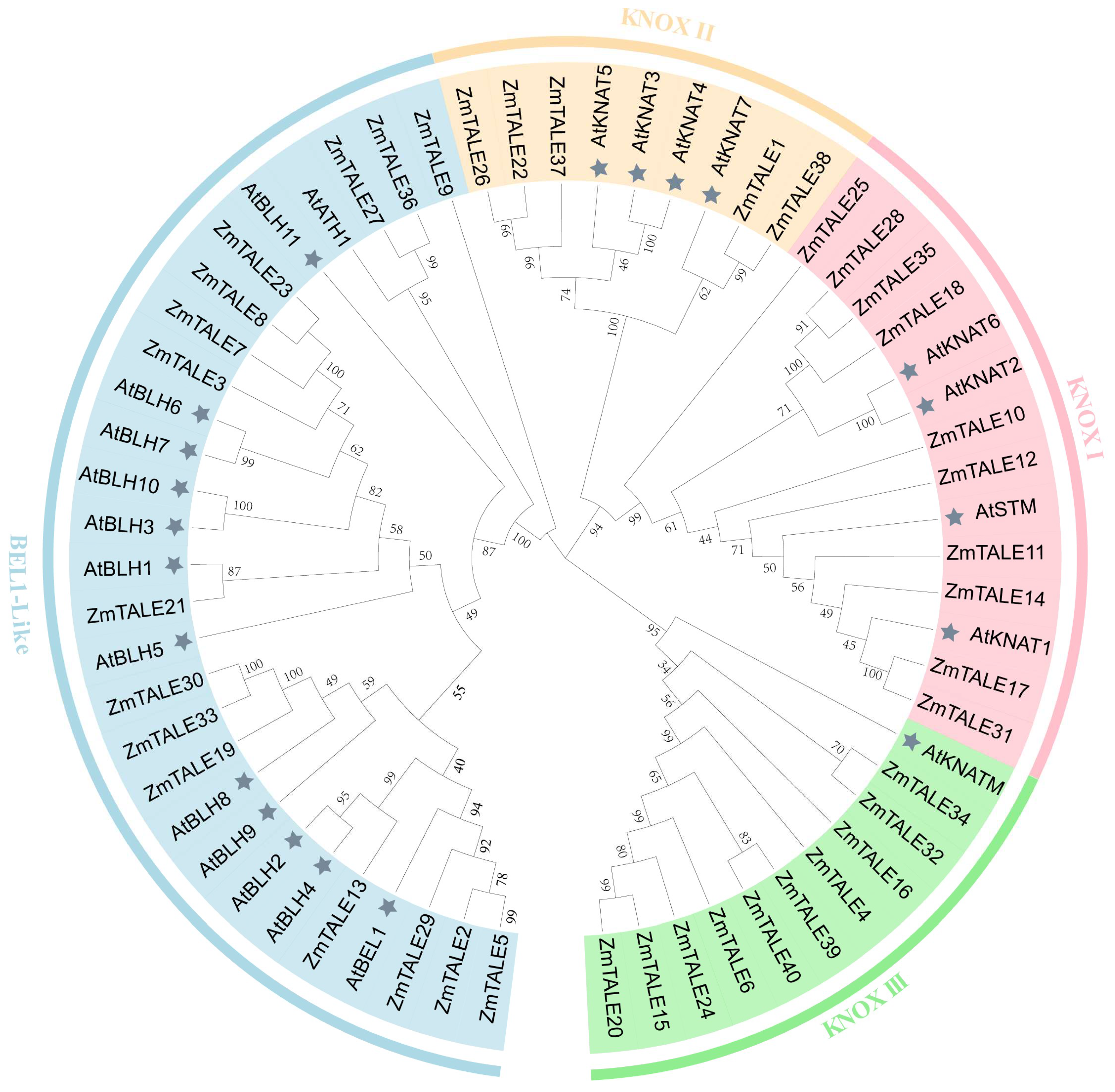
3.4. Phylogenetic Division and Classification of ZmTALE Family Members
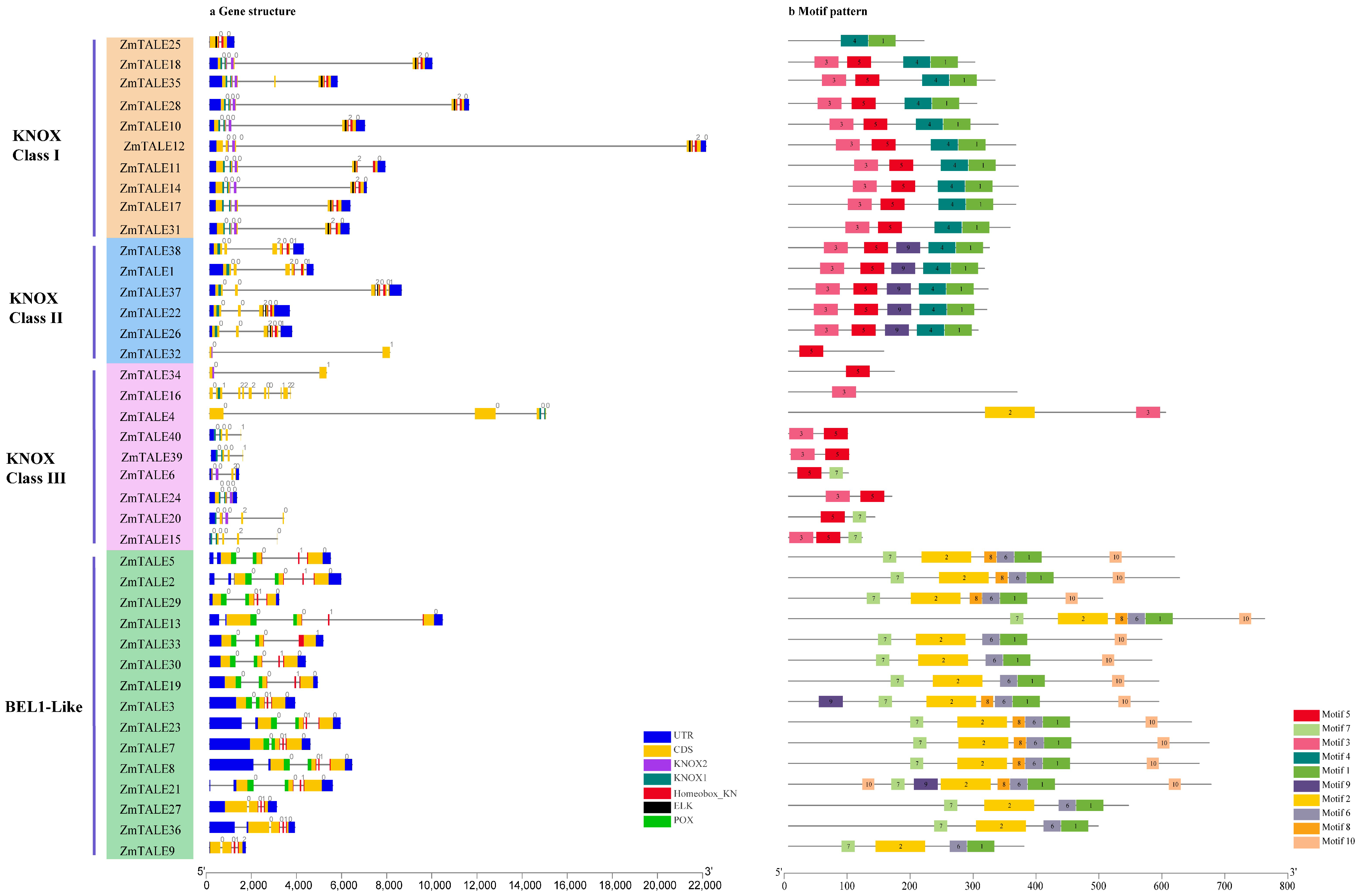
3.5. Gene Structure Analysis of ZmTALE Members
3.6. Collinearity Analysis of ZmTALE Gene Family
3.7. Analysis of ZmTALE Gene Expression
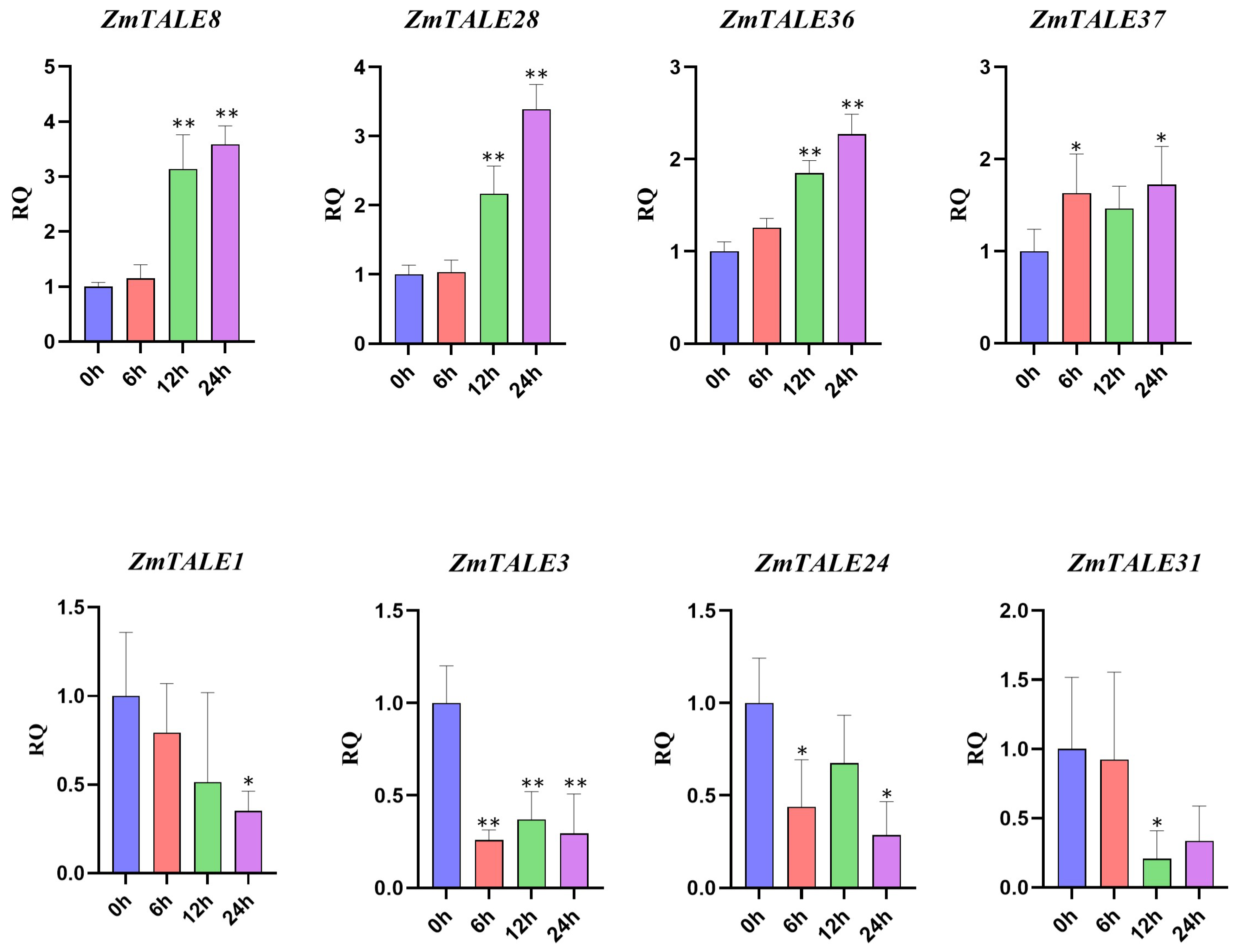
3.8. Analysis of ZmTALE Gene Expression under Drought Stress and qPCR Validation
4. Discussion
4.1. The Functional Domains and Evolutionary Dynamics of the ZmTALE Family
4.2. The TALE Gene Family as Transcription Factors in Plant Development
4.3. The Role of TALE Gene Family in Response to Abiotic Stress
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bürglin, T.R. Analysis of TALE superclass homeobox genes (MEIS, PBC, KNOX, Iroquois, TGIF) reveals a novel domain conserved between plants and animals. Nucleic Acids Res. 1997, 25, 4173–4180. [Google Scholar] [CrossRef]
- Hamant, O.; Pautot, V. Plant development: A TALE story. C. R. Biol. 2010, 333, 371–381. [Google Scholar] [CrossRef]
- Hake, S.; Smith, H.M.; Holtan, H.; Magnani, E.; Mele, G.; Ramirez, J. The role of knox genes in plant development. Annu. Rev. Cell Dev. Biol. 2004, 20, 125–151. [Google Scholar] [CrossRef]
- Gao, J.; Yang, X.; Zhao, W.; Lang, T.; Samuelsson, T. Evolution, diversification, and expression of KNOX proteins in plants. Front. Plant Sci. 2015, 6, 162656. [Google Scholar] [CrossRef] [PubMed]
- Bürglin, T.R. The PBC domain contains a MEINOX domain: Coevolution of Hox and TALE homeobox genes? Dev. Genes Evol. 1998, 208, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Truernit, E.; Siemering, K.R.; Hodge, S.; Grbic, V.; Haseloff, J. A map of KNAT gene expression in the Arabidopsis root. Plant Mol. Biol. 2006, 60, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Micol, J.L.; Hake, S. The development of plant leaves. Plant Physiol. 2003, 131, 389–394. [Google Scholar] [CrossRef]
- Peng, W.; Yang, Y.; Xu, J.; Peng, E.; Dai, S.; Dai, L.; Wang, Y.; Yi, T.; Wang, B.; Li, D.; et al. TALE Transcription Factors in Sweet Orange (Citrus sinensis): Genome-Wide Identification, Characterization, and Expression in Response to Biotic and Abiotic Stresses. Front. Plant Sci. 2022, 12, 814252. [Google Scholar] [CrossRef]
- Hung, F.-Y.; Feng, Y.-R.; Hsin, K.-T.; Shih, Y.-H.; Chang, C.-H.; Zhong, W.; Lai, Y.-C.; Xu, Y.; Yang, S.; Sugimoto, K. Arabidopsis histone H3 lysine 9 methyltransferases KYP/SUVH5/6 are involved in leaf development by interacting with AS1-AS2 to repress KNAT1 and KNAT2. Commun. Biol. 2023, 6, 219. [Google Scholar] [CrossRef]
- Gastaldi, V.; Alem, A.L.; Mansilla, N.; Ariel, F.D.; Viola, I.L.; Lucero, L.E.; Gonzalez, D.H. BREVIPEDICELLUS/KNAT1 targets TCP15 to modulate filament elongation during Arabidopsis late stamen development. Plant Physiol. 2023, 191, 29–34. [Google Scholar] [CrossRef]
- Wang, L.; Yang, X.; Gao, Y.; Yang, S. Genome-wide identification and characterization of TALE superfamily genes in soybean (Glycine max L.). Int. J. Mol. Sci. 2021, 22, 4117. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-J.; Wang, W.; Qin, W.-Q.; Men, S.-Z.; Li, H.-L.; Mitsuda, N.; Ohme-Takagi, M.; Wu, A.-M. Transcription factors KNAT3 and KNAT4 are essential for integument and ovule formation in Arabidopsis. Plant Physiol. 2023, 191, 463–478. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Tabb, P.; Hepworth, S.R. BLADE-ON-PETIOLE1 and 2 regulate Arabidopsis inflorescence architecture in conjunction with homeobox genes KNAT6 and ATH1. Plant Signal. Behav. 2012, 7, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Magnani, E.; Hake, S. KNOX lost the OX: The Arabidopsis KNATM gene defines a novel class of KNOX transcriptional regulators missing the homeodomain. Plant Cell 2008, 20, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Tanaka, W.; Toriba, T.; Suzuki, C.; Maeno, A.; Tsuda, K.; Shiroishi, T.; Kurata, T.; Sakamoto, T.; Murai, M. BELL 1-like homeobox genes regulate inflorescence architecture and meristem maintenance in rice. Plant J. 2019, 98, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, Y.; Yan, M.; Liu, C.; Yuan, Z. BELL1 interacts with CRABS CLAW and INNER NO OUTER to regulate ovule and seed development in pomegranate. Plant Physiol. 2023, 191, 1066–1083. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Yuan, C.; Cong, T.; Wang, J.; Zhang, Q. Genome-wide identification of three-amino-acid-loop-extension gene family and their expression profile under hormone and abiotic stress treatments during stem development of Prunus mume. Front. Plant Sci. 2022, 13, 1006360. [Google Scholar] [CrossRef]
- He, Y.; Yang, T.; Yan, S.; Niu, S.; Zhang, Y. Identification and characterization of the BEL1-like genes reveal their potential roles in plant growth and abiotic stress response in tomato. Int. J. Biol. Macromol. 2022, 200, 193–205. [Google Scholar] [CrossRef]
- Wang, G.; Yuan, Z.; Zhang, P.; Liu, Z.; Wang, T.; Wei, L. Genome-wide analysis of NAC transcription factor family in maize under drought stress and rewatering. Physiol. Mol. Biol. Plants 2020, 26, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; An, L.; Li, F.; Ahmad, W.; Aslam, M.; Ul Haq, M.Z.; Yan, Y.; Ahmad, R.M. Wide-range portrayal of AP2/ERF transcription factor family in maize (Zea mays L.) development and stress responses. Genes 2023, 14, 194. [Google Scholar] [CrossRef]
- Wang, Y.; Li, W.; Qu, J.; Li, F.; Du, W.; Weng, J. Genome-Wide Characterization of the Maize (Zea mays L.) WRKY Transcription Factor Family and Their Responses to Ustilago maydis. Int. J. Mol. Sci. 2023, 24, 14916. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Sun, X.; Chen, Z.; Wang, Q.; Zhang, C.; Liu, X.; Wu, W.; Yin, Y. Exploring the roles of ZmARM gene family in maize development and abiotic stress response. PeerJ 2023, 11, e16254. [Google Scholar] [CrossRef] [PubMed]
- Stelpflug, S.C.; Sekhon, R.S.; Vaillancourt, B.; Hirsch, C.N.; Buell, C.R.; de Leon, N.; Kaeppler, S.M. An expanded maize gene expression atlas based on RNA sequencing and its use to explore root development. Plant Genome 2015, 4, 0025. [Google Scholar] [CrossRef] [PubMed]
- Opitz, N.; Paschold, A.; Marcon, C.; Malik, W.A.; Lanz, C.; Piepho, H.P.; Hochholdinger, F. Transcriptomic complexity in young maize primary roots in response to low water potentials. BMC Genom. 2014, 15, 741. [Google Scholar] [CrossRef] [PubMed]
- Kerstetter, R.; Vollbrecht, E.; Lowe, B.; Veit, B.; Yamaguchi, J.; Hake, S. Sequence analysis and expression patterns divide the maize knotted1-like homeobox genes into two classes. Plant Cell 1994, 6, 1877–1887. [Google Scholar]
- Scofield, S.; Murray, J.A. KNOX gene function in plant stem cell niches. Plant Mol. Biol. 2006, 60, 929–946. [Google Scholar] [CrossRef] [PubMed]
- Nekrutenko, A.; Makova, K.D.; Li, W.H. The K(A)/K(S) ratio test for assessing the protein-coding potential of genomic regions: An empirical and simulation study. Genome Res. 2002, 12, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Hay, A.; Tsiantis, M. A KNOX family tale. Curr. Opin. Plant Biol. 2009, 12, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, N.; Yilmaz, A.; Mejia-Guerra, M.K.; Morohashi, K.; O’Connor, D.; Grotewold, E.; Hake, S. Unraveling the KNOTTED1 regulatory network in maize meristems. Gene Dev. 2012, 26, 1685–1690. [Google Scholar] [CrossRef]
- Vollbrecht, E.; Veit, B.; Sinha, N.; Hake, S. The developmental gene Knotted-1 is a member of a maize homeobox gene family. Nature 1991, 350, 241–243. [Google Scholar] [CrossRef]
- Zhou, R.; Fan, M.; Zhao, M.; Jiang, X.; Liu, Q. Overexpression of LtKNOX1 from Lilium tsingtauense in Nicotiana benthamiana affects the development of leaf morphology. Plant Signal. Behav. 2022, 17, 2031783. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Zhou, P.; Ni, Z.; Iqbal, S.; Ouma, K.O.; Huang, X.; Gao, F.; Ma, C.; Shi, T.; Gao, Z. AGAMOUS-LIKE24 controls pistil number in Japanese apricot by targeting the KNOTTED1-LIKE gene KNAT2/6-a. Plant Physiol. 2024, 195, 566–579. [Google Scholar] [CrossRef] [PubMed]
- Gubert, C.M.; Christy, M.E.; Ward, D.L.; Groner, W.D.; Liljegren, S.J. ASYMMETRIC LEAVES1 regulates abscission zone placement in Arabidopsis flowers. BMC Plant Biol. 2014, 14, 195. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zhao, Y.; Wang, J.; Lu, M.-Z. The transcription factor KNAT2/6b mediates changes in plant architecture in response to drought via down-regulating GA20ox1 in Populus alba× P. glandulosa. J. Exp. Bot. 2021, 72, 5625–5637. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Zheng, S.; Zhang, C.; Huang, R.; Yuan, L.; Tong, H. Identification and expression analysis of the KNOX genes during organogenesis and stress responseness in Camellia sinensis (L.) O. Kuntze. Mol. Genet. Genom. 2023, 298, 1559–1578. [Google Scholar] [CrossRef]
- Li, G.; Manzoor, M.A.; Wang, G.; Chen, C.; Song, C. Comparative analysis of KNOX genes and their expression patterns under various treatments in Dendrobium huoshanense. Front. Plant Sci. 2023, 14, 1258533. [Google Scholar] [CrossRef] [PubMed]
- Razzaq, A.; Ashraf, J.; Malik, W.; Shaban, M.; Zhang, R.; Liang, C.-Z.; Hanif, M.; Abid, M.A.; Qayyum, A. In silico analyses of TALE transcription factors revealed its potential role for organ development and abiotic stress tolerance in cotton. Int. J. Agric. Biol. 2020, 23, 1083–1093. [Google Scholar]
- Qin, W.; Yin, Q.; Chen, J.; Zhao, X.; Yue, F.; He, J.; Yang, L.; Liu, L.; Zeng, Q.; Lu, F. The class II KNOX transcription factors KNAT3 and KNAT7 syner-gistically regulate monolignol biosynthesis in Arabidopsis. J. Exp. Bot. 2020, 71, 5469–5483. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Aznar, A.; Chalvin, C.; Birdseye, D.S.; Baidoo, E.E.; Eudes, A.; Shih, P.M.; Loqué, D.; Zhang, A.; Scheller, H.V. Increased drought tolerance in plants engineered for low lignin and low xylan content. Biotechnol. Biofuels 2018, 11, 195. [Google Scholar] [CrossRef]
- Zhang, Q.; Ahmad, N.; Li, Z.; He, J.; Wang, N.; Naeem, M.; Jin, L.; Yao, N.; Liu, X. CtCYP71A1 promotes drought stress tolerance and lignin accumulation in safflower and Arabidopsis. Environ. Exp. Bot. 2023, 213, 105430. [Google Scholar] [CrossRef]
- Felipo-Benavent, A.; Úrbez, C.; Blanco-Touriñán, N.; Serrano-Mislata, A.; Baumberger, N.; Achard, P.; Agustí, J.; Blázquez, M.A.; Alabadí, D. Regulation of xylem fiber differentiation by gibberellins through DELLA-KNAT1 interaction. Development 2018, 145, dev164962. [Google Scholar] [CrossRef] [PubMed]
- Jang, G.; Choi, Y.D. Drought stress promotes xylem differentiation by modulating the interaction between cytokinin and jasmonic acid. Plant Signal. Behav. 2018, 13, e1451707. [Google Scholar] [CrossRef] [PubMed]
- Lens, F.; Gleason, S.M.; Bortolami, G.; Brodersen, C.; Delzon, S.; Jansen, S. Functional xylem characteristics associated with drought-induced embolism in angiosperms. New Phytol. 2022, 236, 2019–2036. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Sequence ID | Length (aa) | WM (Da) | pI | GRAVY | Aliphatic Index | Predicted Location (s) |
|---|---|---|---|---|---|---|---|
| ZmTALE1 | Zm00001eb001720_T001 | 310 | 34,336.78 | 5.73 | −0.555 | 75.61 | nucleus |
| ZmTALE2 | Zm00001eb001770_T001 | 620 | 66,061.11 | 6.16 | −0.486 | 68.18 | nucleus |
| ZmTALE3 | Zm00001eb005020_T005 | 587 | 62,181.77 | 5.82 | −0.439 | 67.31 | nucleus |
| ZmTALE4 | Zm00001eb013560_T001 | 598 | 64,715.13 | 6.69 | −0.21 | 88.63 | nucleus |
| ZmTALE5 | Zm00001eb021970_T001 | 612 | 64,715.25 | 6.34 | −0.338 | 69.69 | cytosol |
| ZmTALE6 | Zm00001eb031950_T001 | 95 | 10,998.39 | 4.88 | −0.489 | 78.11 | cytosol |
| ZmTALE7 | Zm00001eb032780_T001 | 668 | 72,679.92 | 5.76 | −0.627 | 64.64 | cytosol |
| ZmTALE8 | Zm00001eb051900_T002 | 651 | 71,815.74 | 5.68 | −0.686 | 63.16 | cytosol |
| ZmTALE9 | Zm00001eb051910_T002 | 374 | 40,996.23 | 9.07 | −0.523 | 84.47 | cytosol |
| ZmTALE10 | Zm00001eb052120_T001 | 332 | 36,589.86 | 5.96 | −0.726 | 65.66 | nucleus |
| ZmTALE11 | Zm00001eb055920_T001 | 359 | 39,826.68 | 6.41 | −0.635 | 73.43 | nucleus |
| ZmTALE12 | Zm00001eb055940_T001 | 360 | 39,643.75 | 6.03 | −0.52 | 69 | nucleus |
| ZmTALE13 | Zm00001eb056260_T001 | 755 | 79,167.79 | 6.49 | −0.495 | 62.07 | nucleus |
| ZmTALE14 | Zm00001eb058930_T002 | 364 | 40,276.27 | 5.91 | −0.642 | 62.88 | nucleus |
| ZmTALE15 | Zm00001eb108700_T001 | 116 | 13,112.02 | 4.97 | −0.231 | 91.72 | nucleus |
| ZmTALE16 | Zm00001eb116630_T001 | 362 | 40,246.68 | 8.32 | −0.55 | 79.5 | chloroplast |
| ZmTALE17 | Zm00001eb117820_T001 | 360 | 39,215.02 | 6.23 | −0.626 | 64.72 | nucleus |
| ZmTALE18 | Zm00001eb130180_T001 | 295 | 32,493.47 | 5.51 | −0.554 | 68.51 | nucleus |
| ZmTALE19 | Zm00001eb147970_T001 | 587 | 63,158.83 | 7.29 | −0.441 | 68.07 | nucleus |
| ZmTALE20 | Zm00001eb181490_T001 | 136 | 15,591.79 | 4.91 | −0.207 | 96.76 | nucleus |
| ZmTALE21 | Zm00001eb202140_T001 | 671 | 69,914.65 | 6.06 | −0.413 | 62.43 | nucleus |
| ZmTALE22 | Zm00001eb206760_T001 | 314 | 34,111.33 | 6.02 | −0.514 | 71.34 | nucleus |
| ZmTALE23 | Zm00001eb217350_T002 | 639 | 70,848.35 | 5.46 | −0.71 | 63.87 | nucleus |
| ZmTALE24 | Zm00001eb217470_T001 | 163 | 18,005.31 | 4.59 | −0.325 | 77.98 | mitochondrion |
| ZmTALE25 | Zm00001eb217480_T001 | 214 | 24,066.87 | 7.77 | −0.762 | 67.57 | nucleus |
| ZmTALE26 | Zm00001eb234270_T001 | 300 | 32,669.76 | 5.89 | −0.544 | 70.73 | nucleus |
| ZmTALE27 | Zm00001eb239680_T002 | 539 | 56,452.11 | 6.73 | −0.251 | 68.78 | nucleus |
| ZmTALE28 | Zm00001eb264910_T001 | 298 | 32,662.69 | 5.32 | −0.579 | 65.91 | nucleus |
| ZmTALE29 | Zm00001eb268690_T001 | 498 | 54,229.07 | 6.68 | −0.381 | 73.78 | nucleus |
| ZmTALE30 | Zm00001eb289480_T001 | 576 | 61,910.33 | 8.29 | −0.45 | 68.87 | nucleus |
| ZmTALE31 | Zm00001eb299420_T001 | 351 | 38,800.64 | 6.4 | −0.677 | 64.7 | nucleus |
| ZmTALE32 | Zm00001eb322410_T001 | 150 | 16,469 | 6.29 | −0.038 | 104.07 | nucleus |
| ZmTALE33 | Zm00001eb347710_T001 | 592 | 64,138.12 | 9.24 | −0.449 | 70.12 | nucleus |
| ZmTALE34 | Zm00001eb354670_T001 | 167 | 18,305.62 | 5.48 | −0.423 | 81.8 | nucleus |
| ZmTALE35 | Zm00001eb354880_T002 | 327 | 35,593.02 | 5.27 | −0.512 | 67.25 | nucleus |
| ZmTALE36 | Zm00001eb384240_T002 | 491 | 52,469.65 | 7.19 | −0.317 | 66.86 | nucleus |
| ZmTALE37 | Zm00001eb386830_T001 | 316 | 34,522.64 | 5.69 | −0.563 | 69.05 | nucleus |
| ZmTALE38 | Zm00001eb403720_T001 | 318 | 34,768.18 | 5.64 | −0.458 | 80.13 | nucleus |
| ZmTALE39 | Zm00001eb437470_T001 | 93 | 10,577.17 | 9.23 | −0.605 | 82.9 | nucleus |
| ZmTALE40 | Zm00001eb438600_T001 | 93 | 10,547.08 | 9.23 | −0.633 | 82.9 | cytosol |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qian, B.; Wang, Q.; Zhang, C.; Guo, J.; Yu, Z.; Han, J.; Xia, H.; Zhao, R.; Yin, Y. Exploring the Roles of TALE Gene Family in Maize Drought Stress Responses. Agronomy 2024, 14, 1267. https://doi.org/10.3390/agronomy14061267
Qian B, Wang Q, Zhang C, Guo J, Yu Z, Han J, Xia H, Zhao R, Yin Y. Exploring the Roles of TALE Gene Family in Maize Drought Stress Responses. Agronomy. 2024; 14(6):1267. https://doi.org/10.3390/agronomy14061267
Chicago/Turabian StyleQian, Buxuan, Qi Wang, Chuang Zhang, Jia Guo, Zhijia Yu, Jiarui Han, Hanchao Xia, Rengui Zhao, and Yuejia Yin. 2024. "Exploring the Roles of TALE Gene Family in Maize Drought Stress Responses" Agronomy 14, no. 6: 1267. https://doi.org/10.3390/agronomy14061267
APA StyleQian, B., Wang, Q., Zhang, C., Guo, J., Yu, Z., Han, J., Xia, H., Zhao, R., & Yin, Y. (2024). Exploring the Roles of TALE Gene Family in Maize Drought Stress Responses. Agronomy, 14(6), 1267. https://doi.org/10.3390/agronomy14061267