
Strigolactone and Karrikin Signaling Influence the Recruitment of Wild Tobacco’s Root Microbiome in the Desert

 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
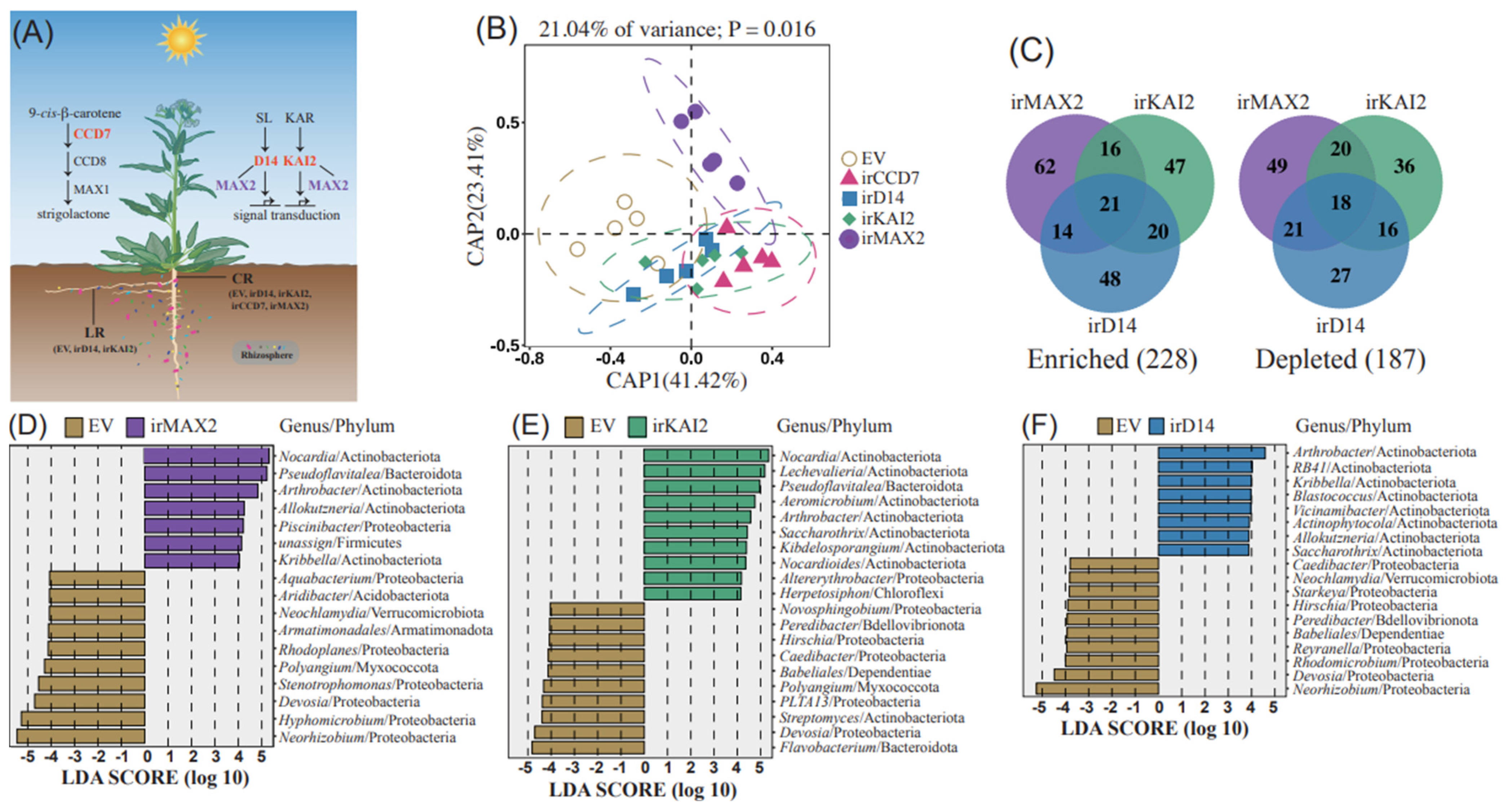
2.1. Distinct Roles of SL and KAR Signaling in Main Root Microbiome Recruitment
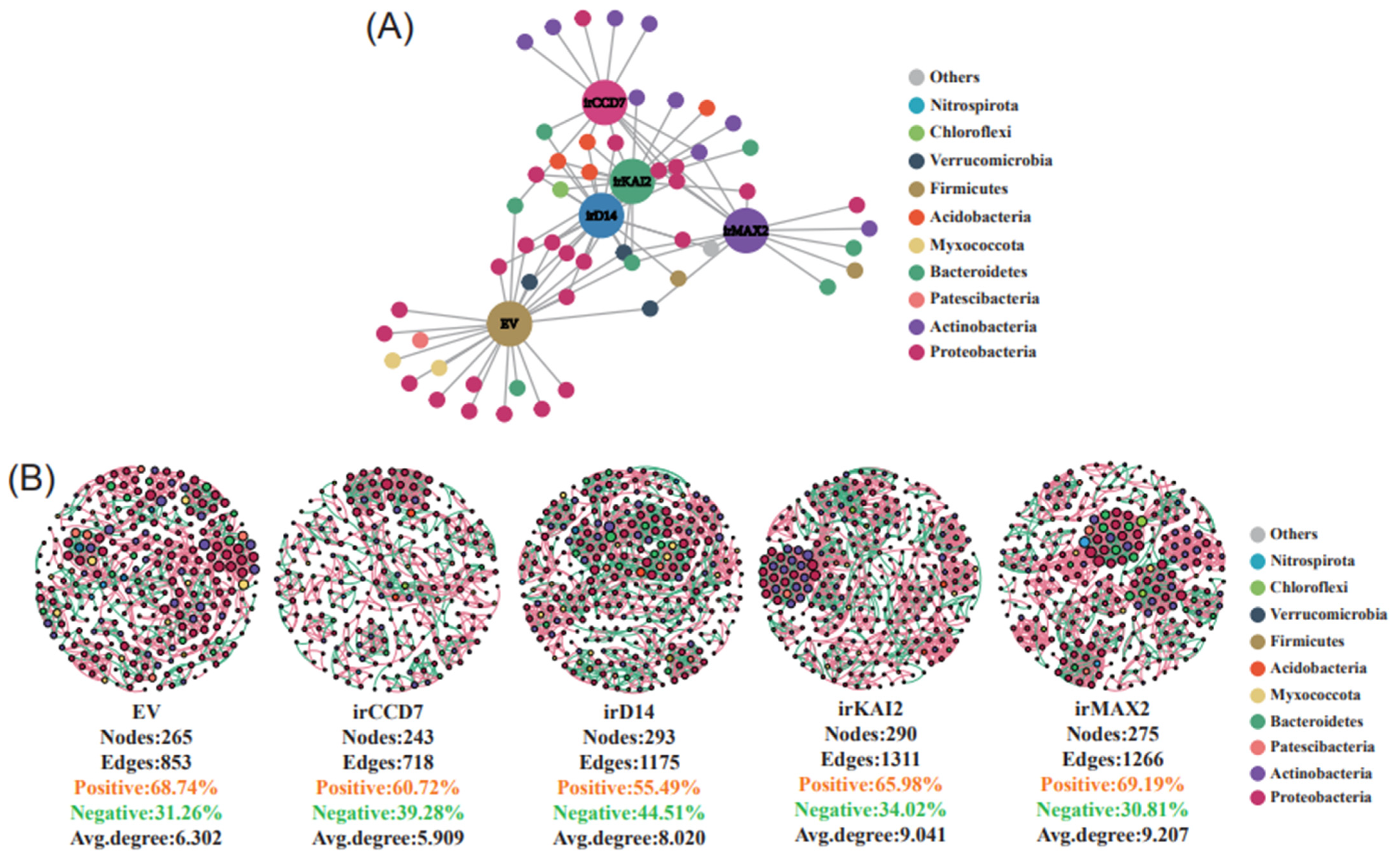
2.2. Distinct Roles of SL and KAR Signaling on Microbiome Network Connections
2.3. SL and KAR Signaling Play Critical Roles in LR Microbiome Recruitment
3. Discussion
4. Methods
4.1. Plant Growth in Desert
4.2. Sample Collection
4.3. DNA Extraction, Library Preparation, and Sequencing
4.4. Sequence Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alsharif, W.; Saad, M.M.; Hirt, H. Desert microbes for boosting sustainable agriculture in extreme environments. Front. Microbiol. 2020, 11, 1666. [Google Scholar] [CrossRef]
- De Zelicourt, A.; Al-Yousif, M.; Hirt, H. Rhizosphere microbes as essential partners for plant stress tolerance. Mol. Plant. 2013, 6, 242–245. [Google Scholar] [CrossRef]
- Na, X.; Xu, T.; Li, M.; Zhou, Z.; Ma, S.; Wang, J.; He, J.; Jiao, B.; Ma, F. Variations of bacterial community diversity within the rhizosphere of three phylogenetically related perennial shrub plant species across environmental gradients. Front. Microbiol. 2018, 9, 709. [Google Scholar] [CrossRef]
- Booker, J.; Auldridge, M.; Wills, S.; McCarty, D.; Klee, H.; Leyser, O. MAX3/CCD7 is a carotenoid cleavage dioxygenase required for the synthesis of a novel plant signaling molecule. Curr. Biol. 2004, 14, 1232–1238. [Google Scholar] [CrossRef]
- Waters, M.T.; Nelson, D.C.; Scaffidi, A.; Flematti, G.R.; Sun, Y.K.; Dixon, K.W.; Smith, S.M. Specialisation within the DWARF14 protein family confers distinct responses to karrikins and strigolactones in Arabidopsis. Development 2012, 139, 1285–1295. [Google Scholar] [CrossRef]
- Yao, R.; Ming, Z.; Yan, L.; Li, S.; Wang, F.; Ma, S.; Yu, C.; Yang, M.; Chen, L.; Chen, L.; et al. DWARF14 is a non-canonical hormone receptor for strigolactone. Nature 2016, 536, 469–473. [Google Scholar] [CrossRef]
- Nelson, D.C.; Scaffidi, A.; Dun, E.A.; Waters, M.T.; Flematti, G.R.; Dixon, K.W.; Beveridge, C.A.; Ghisalberti, E.L.; Smith, S.M. F-box protein MAX2 has dual roles in karrikin and strigolactone signaling in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2011, 108, 8897–8902. [Google Scholar] [CrossRef]
- Gomez-Roldan, V.; Fermas, S.; Brewer, P.B.; Puech-Pagès, V.; Dun, E.A.; Pillot, J.-P.; Letisse, F.; Matusova, R.; Danoun, S.; Portais, J.-C.; et al. Strigolactone inhibition of shoot branching. Nature 2008, 455, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.-C.; Li, C.; Flint-Garcia, S.; Suzuki, M.; Wu, S.; Saunders, J.W.; Dong, L.; Bouwmeester, H.J.; McCarty, D.R.; Koch, K.E. Maize domestication phenotypes reveal strigolactone networks coordinating grain size evolution with kernel-bearing cupule architecture. Plant Cell 2022, 35, 1013–1037. [Google Scholar] [CrossRef]
- Li, S.; Joo, Y.; Cao, D.; Li, R.; Lee, G.; Halitschke, R.; Baldwin, G.; Baldwin, I.T.; Wang, M. Strigolactone signaling regulates specialized metabolism in tobacco stems and interactions with stem-feeding herbivores. PLoS Biol. 2020, 18, e3000830. [Google Scholar] [CrossRef]
- Akiyama, K.; Matsuzaki, K.-I.; Hayashi, H. Plant sesquiterpenes induce hyphal branching in arbuscular mycorrhizal fungi. Nature 2005, 435, 824–827. [Google Scholar] [CrossRef] [PubMed]
- Cook, C.W.; Whichard, L.P.; Turner, B.; Wall, M.E.; Egley, G.H. Germination of witchweed (Striga lutea Lour.): Isolation and properties of a potent stimulant. Science 1966, 154, 1189–1190. [Google Scholar] [CrossRef]
- Krügel, T.; Lim, M.; Gase, K.; Halitschke, R.; Baldwin, I.T. Agrobacterium-mediated transformation of Nicotiana attenuata, a model ecological expression system. Chemoecology 2002, 12, 177–183. [Google Scholar] [CrossRef]
- Li, S.; Baldwin, G.; Yang, C.; Lu, R.; Meng, S.; Huang, J.; Wang, M.; Baldwin, I.T. Field-work reveals a novel function for MAX2 in a native tobacco’s high-light adaptions. Plant Cell Environ. 2024, 47, 230–245. [Google Scholar] [CrossRef]
- Mitra, D.; Mondal, R.; Khoshru, B.; Senapati, A.; Radha, T.K.; Mahakur, B.; Uniyal, N.; Myo, E.M.; Boutaj, H.; Sierra, B.E.G.; et al. Actinobacteria-enhanced plant growth, nutrient acquisition, and crop protection: Advances in soil, plant, and microbial multifactorial interactions. Pedosphere 2022, 32, 149–170. [Google Scholar] [CrossRef]
- Glick, B.R. Plant growth-promoting bacteria: Mechanisms and applications. Scientifica 2012, 2012, 79–86. [Google Scholar] [CrossRef]
- Simkin, A.J. Carotenoids and apocarotenoids in planta: Their role in plant development, contribution to the flavour and aroma of fruits and flowers, and their nutraceutical benefits. Plants 2021, 10, 2321. [Google Scholar] [CrossRef]
- Yalamanchili, K.; Vermeer, J.E.M.; Scheres, B.; Willemsen, V. Shaping root architecture: Towards understanding the mechanisms involved in lateral root development. Biol. Direct 2024, 19, 87. [Google Scholar] [CrossRef]
- Gao, Y.-Q.; Su, Y.; Chao, D.-Y. Exploring the function of plant root diffusion barriers in sealing and shielding for environmental adaptation. Nat. Plants 2024, 10, 1865–1874. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Liu, Y.; Geng, J.; Lü, H.; Zhao, H.-M.; Xiang, L.; Li, H.; Mo, C.-H.; Li, Y.-W.; Cai, Q.-Y. Maize root-associated niches determine the response variation in bacterial community assembly and function to phthalate pollution. J. Hazard. Mater. 2022, 429, 128280. [Google Scholar] [CrossRef]
- Waidmann, S.; Sarkel, E.; Kleine-Vehn, J. Same same, but different: Growth responses of primary and lateral roots. J. Exp. Bot. 2020, 71, 2397–2411. [Google Scholar] [CrossRef] [PubMed]
- Thorup-Kristensen, K.; Halberg, N.; Nicolaisen, M.; Olesen, J.E.; Crews, T.E.; Hinsinger, P.; Kirkegaard, J.; Pierret, A.; Dresbøll, D.B. Digging deeper for agricultural resources, the value of deep rooting. Trends Plant Sci. 2020, 25, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Gutjahr, C.; Gobbato, E.; Choi, J.; Riemann, M.; Johnston, M.G.; Summers, W.; Carbonnel, S.; Mansfield, C.; Yang, S.Y.; Nadal, M.; et al. Rice perception of symbiotic arbuscular mycorrhizal fungi requires the karrikin receptor complex. Science 2015, 350, 1521–1524. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.Z.; Rehman, N.U.; Yu, S.; Zhou, Y.; Haq, B.U.; Wang, J.; Li, P.; Zeng, Z.; Zhao, J. GmMAX2-D14 and -KAI interaction-mediated SL and KAR signaling play essential roles in soybean root nodulation. Plant J. 2020, 101, 334–351. [Google Scholar] [CrossRef]
- Kim, B.; Westerhuis, J.A.; Smilde, A.K.; Floková, K.; Suleiman, A.K.A.; Kuramae, E.E.; Bouwmeester, H.J.; Zancarini, A. Effect of strigolactones on recruitment of the rice root-associated microbiome. FEMS Microbiol. Ecol. 2022, 98, fiac010. [Google Scholar] [CrossRef] [PubMed]
- Nasir, F.; Shi, S.; Tian, L.; Chang, C.; Ma, L.; Li, X.; Gao, Y.; Tian, C. Strigolactones shape the rhizomicrobiome in rice (Oryza sativa). Plant Sci. 2019, 286, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Rice, J.H.; Lopes, V.; Grewal, P.; Lebeis, S.L.; Hewezi, T.; Staton, M.E. Overexpression of strigolactone-associated genes exerts fine-tuning selection on soybean rhizosphere bacterial and fungal microbiome. Phytobiomes J. 2020, 4, 239–251. [Google Scholar] [CrossRef]
- Arite, T.; Iwata, H.; Ohshima, K.; Maekawa, M.; Nakajima, M.; Kojima, M.; Sakakibara, H.; Kyozuka, J. DWARF10, an RMS1/MAX4/DAD1 ortholog, controls lateral bud outgrowth in rice. Plant J. 2007, 51, 1019–1029. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Wagner, H.H. Vegan Community Ecology Package Version 2.5–7 November 2020. Available online: https://github.com/vegandevs/vegan (accessed on 9 September 2024).
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Nearing, J.T.; Douglas, G.M.; Hayes, M.G.; MacDonald, J.; Desai, D.K.; Allward, N.; Jones, C.M.A.; Wright, R.J.; Dhanani, A.S.; Comeau, A.M.; et al. Microbiome differential abundance methods produce different results across 38 datasets. Nat. Commun. 2022, 13, 342–357. [Google Scholar] [CrossRef]
- De Cáceres, M.; Legendre, P.; Moretti, M. Improving indicator species analysis by combining groups of sites. Oikos 2010, 119, 1674–1684. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Peng, C.; Chen, Q.; Tan, S.; Shen, X.; Jiang, C. Generalized reporter score-based enrichment analysis for omics data. Brief. Bioinform. 2024, 25, bbae116. [Google Scholar] [CrossRef]
- Wen, T.; Xie, P.; Yang, S.; Niu, G.; Liu, X.; Ding, Z.; Xue, C.; Liu, Y.-X.; Shen, Q.; Yuan, J. ggclusternet: An R package for microbiome network analysis and modularity-based multiple network layouts. iMeta 2022, 1, e32. [Google Scholar] [CrossRef] [PubMed]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks. Proc. Int. AAAI Conf. Web Soc. Media 2009, 3, 361–362. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, J.; Luo, S.; Baldwin, G.; Cheng, X.; Baldwin, I.T.; Li, S. Strigolactone and Karrikin Signaling Influence the Recruitment of Wild Tobacco’s Root Microbiome in the Desert. Agronomy 2025, 15, 44. https://doi.org/10.3390/agronomy15010044
Cheng J, Luo S, Baldwin G, Cheng X, Baldwin IT, Li S. Strigolactone and Karrikin Signaling Influence the Recruitment of Wild Tobacco’s Root Microbiome in the Desert. Agronomy. 2025; 15(1):44. https://doi.org/10.3390/agronomy15010044
Chicago/Turabian StyleCheng, Jie, Shuai Luo, Gundega Baldwin, Xu Cheng, Ian T. Baldwin, and Suhua Li. 2025. "Strigolactone and Karrikin Signaling Influence the Recruitment of Wild Tobacco’s Root Microbiome in the Desert" Agronomy 15, no. 1: 44. https://doi.org/10.3390/agronomy15010044
APA StyleCheng, J., Luo, S., Baldwin, G., Cheng, X., Baldwin, I. T., & Li, S. (2025). Strigolactone and Karrikin Signaling Influence the Recruitment of Wild Tobacco’s Root Microbiome in the Desert. Agronomy, 15(1), 44. https://doi.org/10.3390/agronomy15010044