
Integrative Transcriptomic and Metabolomic Analysis Reveals Regulatory Networks and Metabolite Dynamics in Gastrodia elata Flower Development


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Extraction and Illumina Sequencing
2.3. Quantitative Real-Time PCR Analysis
2.4. Widely Targeted Metabolic Profiling
3. Results
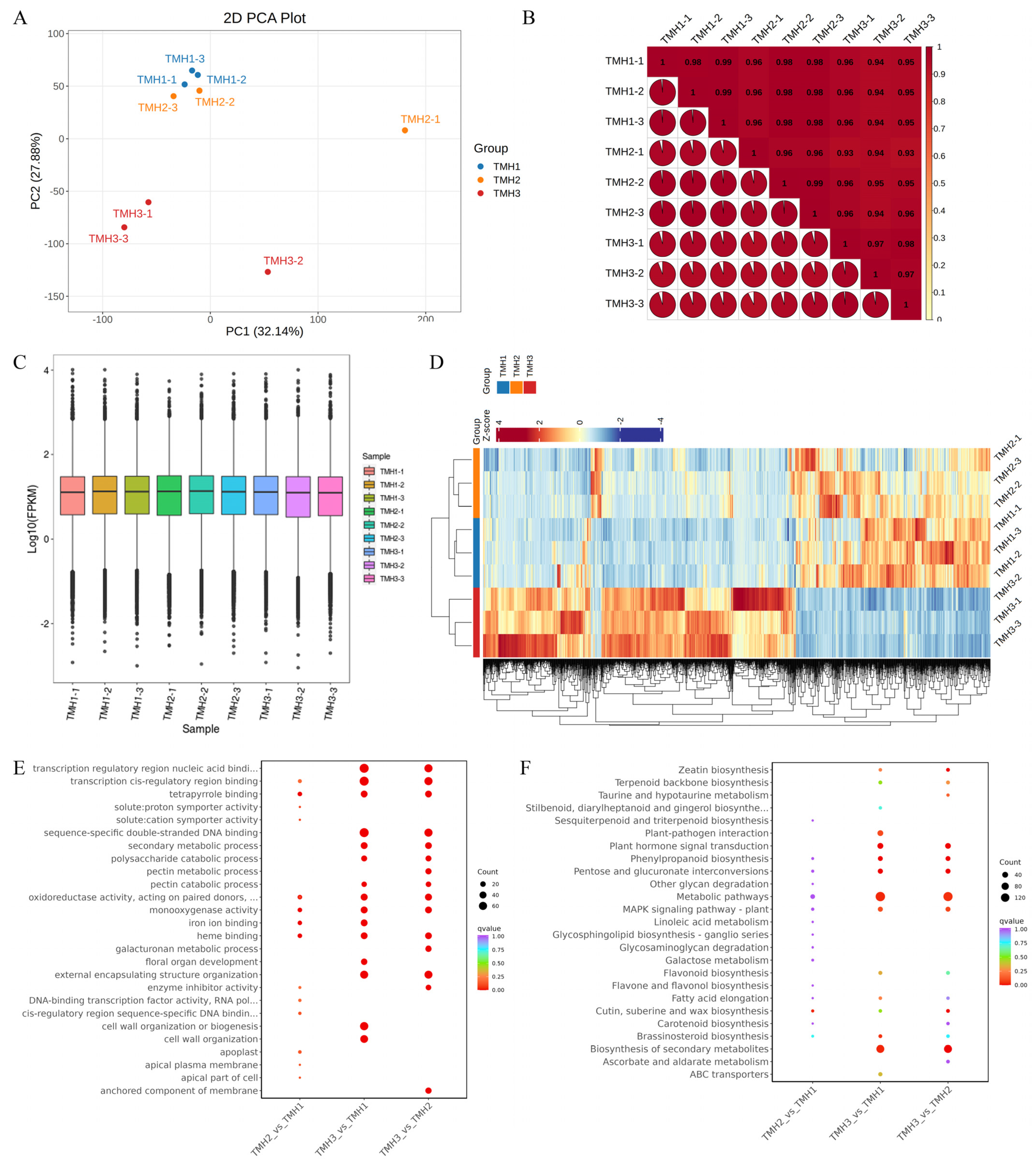
3.1. DEG Analysis in G. elata Flowers During Three Developmental Stages
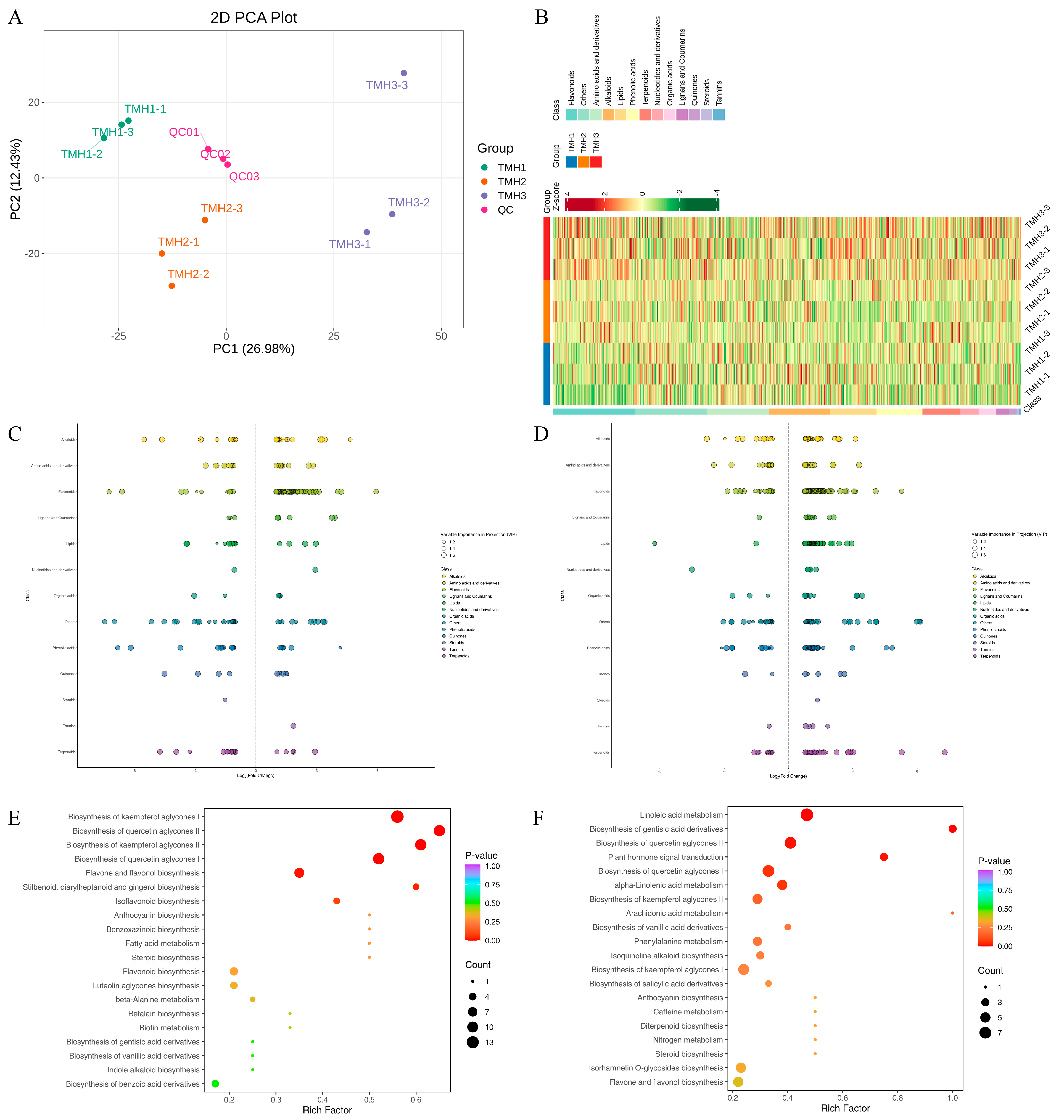
3.2. Metabolic Differences Observed Between Three G. elata Flower Developmental Stages
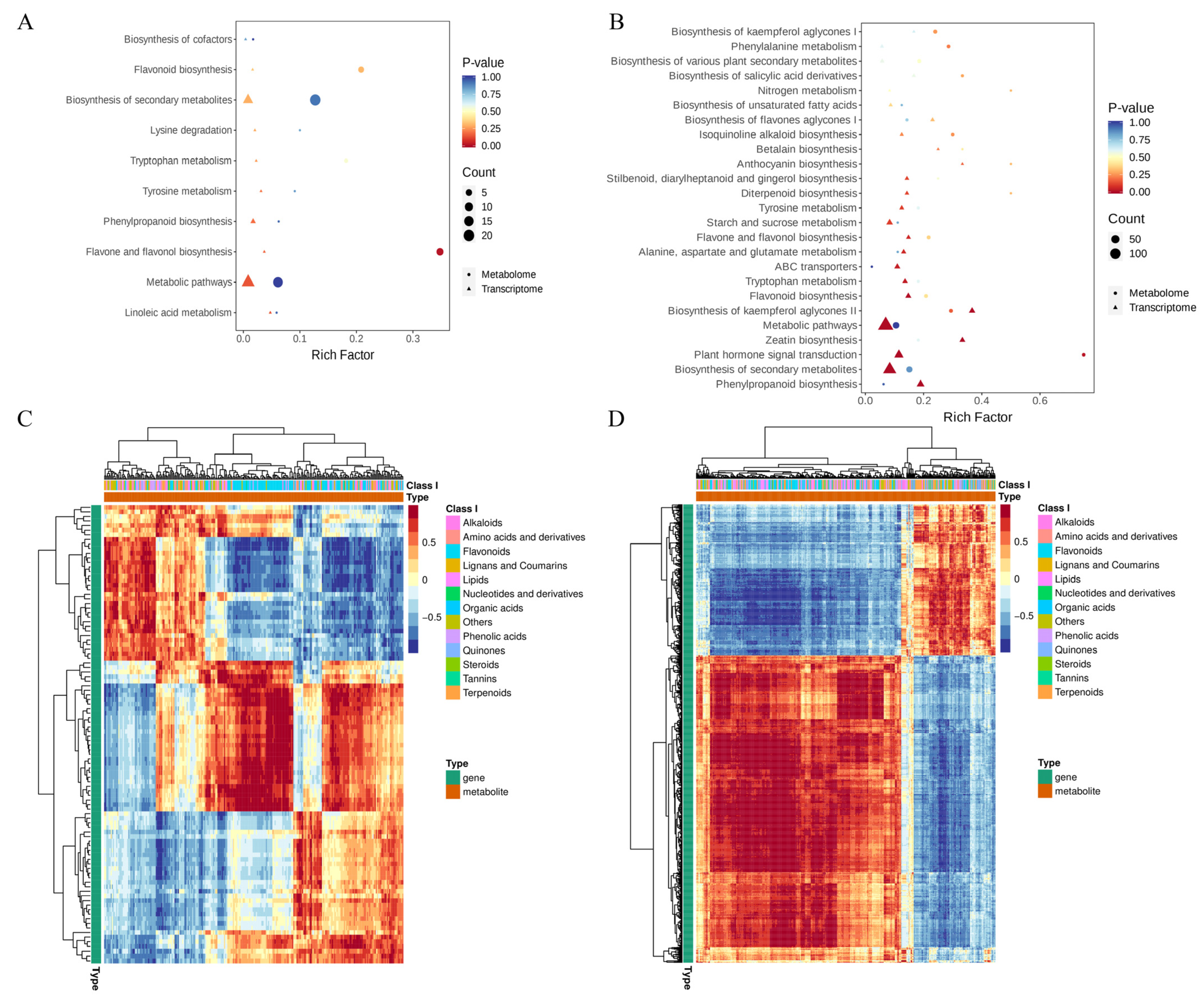
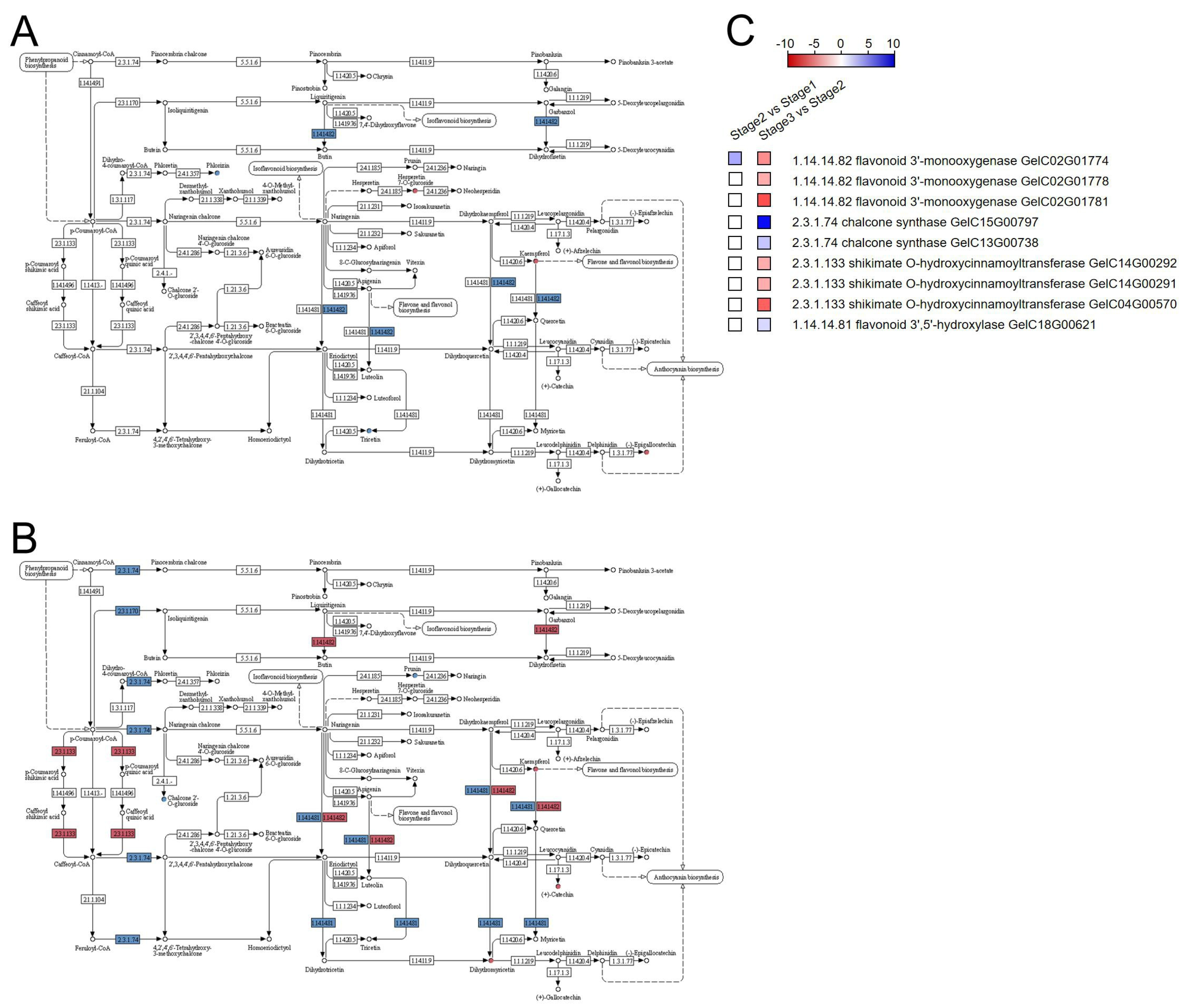
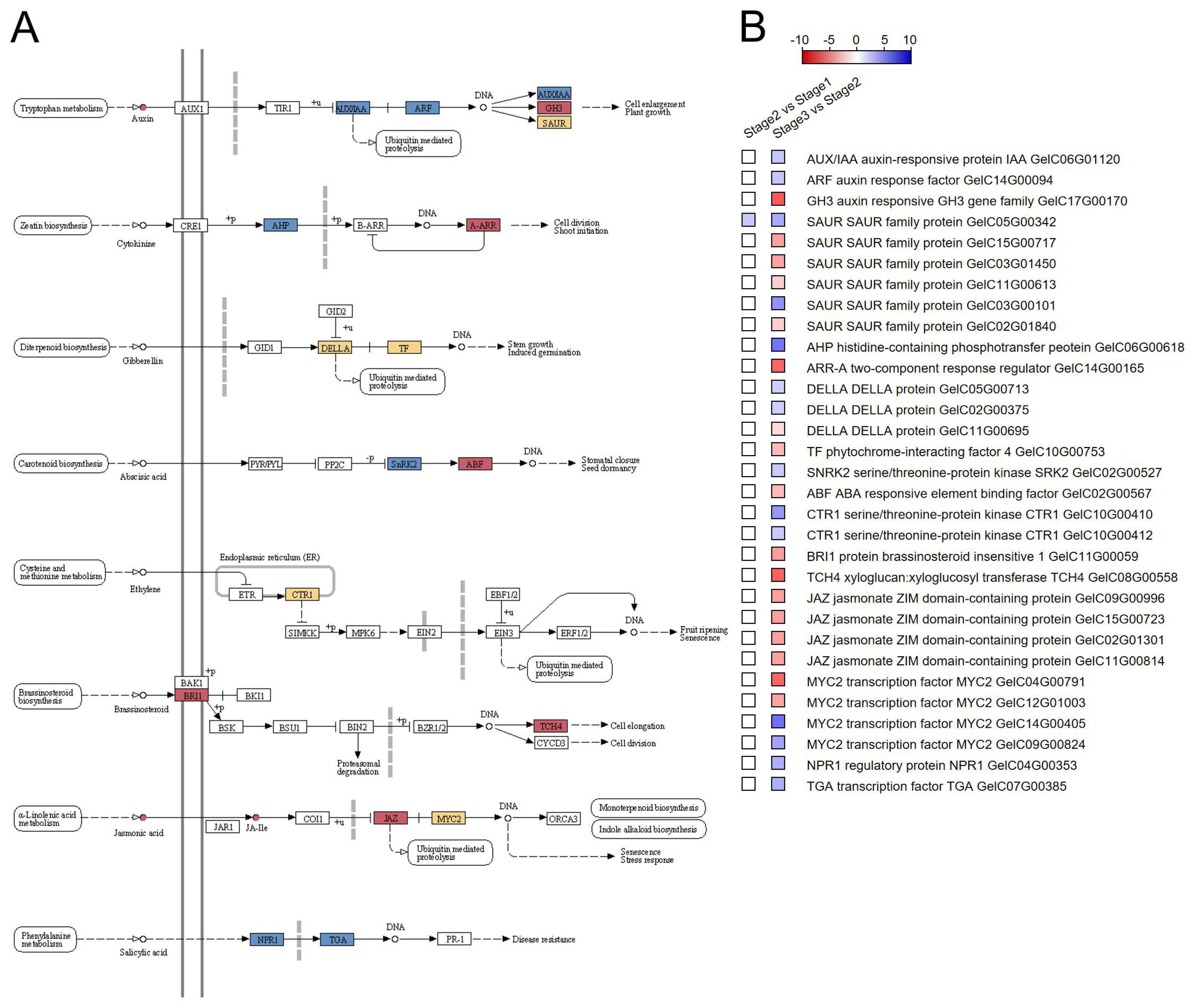
3.3. Integrative Transcriptome and Metabolome Analysis Related to “Flavonoid Biosynthesis” and “Plant Hormone Signal Transduction” Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| PSM | plant secondary metabolite |
| NGS | next-generation sequencing |
| CTAB | cetyltrimethylammonium bromide |
| FPKM | fragments per kilobase per million mapped reads |
| DEGs | differentially expressed genes |
| KEGG | genes and genomes |
| GO | gene ontology |
| CT | cycle threshold |
| PCA | principal component analysis |
| PCCs | Pearson correlation coefficients |
| HCA | hierarchical cluster analysis |
| SCMs | significant changes in metabolites |
References
- Zhang, M.J.; Xu, H.D.; An, X.G. Research on ultrasonic wave extraction of Gastrodia elata Bl. polysaccharides. J. Nat. Sci. Ed. 2007, 35, 91–95. [Google Scholar]
- Wu, Y.N.; Wen, S.H.; Zhang, W. Gastrodia elata BI.:A comprehensive review of its traditional use, botany, phytochemistry, pharmacology, and pharmacokinetics. Evid. Based Complement. Alternat. Med. 2023, 18, 5606021. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, G.L. The chemical composition, pharmacological effects, clinical applications and market analysis of Gastrodia elata. Pharm. Chem. J. 2017, 51, 211–215. [Google Scholar] [CrossRef]
- Ho, L.H.; Lee, Y.I.; Hsieh, S.Y.; Lin, I.S.; Wu, Y.C.; Ko, H.Y.; Klemens, P.A.; Neuhaus, H.E.; Chen, Y.M.; Huang, T.P.; et al. GeSUT4 mediates sucrose import at the symbiotic interface for carbon allocation of heterotrophic Gastrodia elata (Orchidaceae). Plant Cell Environ. 2021, 44, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Shan, T.Y.; Yin, M.Z.; Wu, J.X.; Yu, H.W.; Liu, M.L.; Xu, R.; Wang, J.T.; Peng, H.S.; Zha, L.P.; Gui, S.Y. Comparative transcriptome analysis of tubers, stems, and flowers of Gastrodia elata Blume reveals potential genes involved in the biosynthesis of phenolics. Fitoterapia 2021, 153, 104988. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Yang, X.Q.; Li, Z.Y.; Miao, J.Y.; Li, S.B.; Zhang, W.P.; Lin, Y.C.; Lin, L.B. The role of symbiotic fungi in the life cycle of Gastrodia elata Blume (Orchidaceae): A comprehensive review. Front. Plant Sci. 2024, 14, 1309038. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, J.Z. Flowering and flower development in plants. Agronomy 2024, 15, 256. [Google Scholar] [CrossRef]
- Irish, V. The ABC model of floral development. Curr. Biol. 2017, 27, 887–890. [Google Scholar] [CrossRef]
- Li, X.M.; Fan, T.; Zou, P.; Zhang, W.H.; Wu, X.J.; Zhang, Y.X.; Liao, J.P. Can the anatomy of abnormal flowers elucidate relationships of the androecial members in the ginger (Zingiberaceae)? Evodevo 2020, 11, 12. [Google Scholar] [CrossRef]
- Luo, H.H.; Wang, Q.; Zhang, J.K.; Wang, L.S.; Li, Y.B.; Yang, G.Z. Minimum fertilization at the appearance of the first flower benefits cotton nutrient utilization of nitrogen, phosphorus and potassium. Sci. Rep. 2020, 10, 6815. [Google Scholar] [CrossRef]
- Gase, K.B.I. Transformational tools for next-generation plant ecology: Manipulation of gene expression for the functional analysis of genes. Plant Ecol. Divers. 2012, 5, 485–490. [Google Scholar] [CrossRef]
- Morozova, O.; Marra, M.A. Applications of next-generation sequencing technologies in functional genomics. Genomics 2008, 92, 255–264. [Google Scholar] [CrossRef]
- Bae, E.K.; An, C.; Kang, M.J.; Lee, S.A.; Lee, S.J.; Kim, K.T.; Park, E.J. Chromosome-level genome assembly of the fully mycoheterotrophic orchid Gastrodia elata. G3 2022, 12, jkab433. [Google Scholar] [CrossRef]
- Tsai, C.C.; Wu, K.M.; Chiang, T.Y.; Huang, C.Y.; Chou, C.H.; Li, S.J.; Chiang, Y.C. Comparative transcriptome analysis of Gastrodia elata (Orchidaceae) in response to fungus symbiosis to identify gastrodin biosynthesis-related genes. BMC Genom. 2016, 17, 212. [Google Scholar] [CrossRef]
- Zeng, X.; Li, Y.Y.; Ling, H.; Liu, S.S.; Liu, M.M.; Chen, J.; Guo, S.X. Transcriptomic analyses reveal clathrin-mediated endocytosis involved in symbiotic seed germination of Gastrodia elata. Bot. Stud. 2017, 58, 31. [Google Scholar] [CrossRef]
- Wang, Y.S.; Qasim Shahid, M.; Ghouri, F.; Ercişli, S.; Baloch, F.S. Development of EST-based SSR and SNP markers in Gastrodia elata (herbal medicine) by sequencing, de novo assembly and annotation of the transcriptome. 3 Biotech 2019, 9, 292. [Google Scholar] [CrossRef]
- Wang, Y.H.; Gao, Y.G.; Zang, P.; Xu, Y. Transcriptome analysis reveals underlying immune response mechanism of fungal (Penicillium oxalicum) disease in Gastrodia elata Bl. f. glauca S. chow (Orchidaceae). BMC Plant Biol. 2020, 20, 445. [Google Scholar] [CrossRef]
- Ren, L.Y.; Zhao, H.; Liu, X.L.; Zong, T.K.; Qiao, M.; Liu, S.Y.; Liu, X.Y. Transcriptome reveals roles of lignin-modifying enzymes and abscisic acid in the symbiosis of mycena and Gastrodia elata. Int. J. Mol. Sci. 2021, 22, 6557. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.F.; Sui, S.Z.; Ma, J.; Li, Z.N.; Guo, Y.L.; Luo, D.P.; Yang, J.F.; Li, M.Y. Transcriptomic analysis of flower development in wintersweet (Chimonanthus praecox). PLoS ONE 2014, 9, e86976. [Google Scholar] [CrossRef]
- Nakamura, Y.; Teo, N.Z.W.; Shui, G.H.; Chua, C.H.L.; Cheong, W.F.; Parameswaran, S.; Koizumi, R.; Ohta, H.; Wenk, M.R.; Ito, T. Transcriptomic and lipidomic profiles of glycerolipids during Arabidopsis flower development. New Phytol. 2014, 203, 310–322. [Google Scholar] [CrossRef]
- Liu, F.; Wang, Y.; Ding, Z.T.; Zhao, L.; Xiao, J.; Wang, L.J.; Ding, S.B. Transcriptomic analysis of flower development in tea (Camellia sinensis (L.)). Gene 2017, 631, 39–51. [Google Scholar] [CrossRef]
- Jiao, Y.L.; Hu, Q.T.; Zhu, Y.; Zhu, L.F.; Ma, T.F.; Zeng, H.Y.; Zang, Q.L.; Li, X.; Lin, X.C. Comparative transcriptomic analysis of the flower induction and development of the Lei bamboo (Phyllostachys violascens). BMC Bioinform. 2019, 20, 687. [Google Scholar] [CrossRef]
- Yang, L.; Liu, L.M.; Wang, Z.Y.; Zong, Y.; Yu, L.; Li, Y.Q.; Liao, F.L.; Chen, M.M.; Cai, K.L.; Guo, W.D. Comparative anatomical and transcriptomic insights into Vaccinium corymbosum flower bud and fruit throughout development. BMC Plant Biol. 2021, 21, 289. [Google Scholar] [CrossRef]
- Wang, N.; Shu, X.C.; Zhang, F.J.; Zhuang, W.B.; Wang, T.; Wang, Z. Comparative transcriptome analysis identifies key regulatory genes involved in anthocyanin metabolism during flower development in Lycoris radiata. Front. Plant Sci. 2021, 12, 761862. [Google Scholar] [CrossRef]
- Suetsugu, K.; Fukushima, K.; Makino, T.; Ikematsu, S.; Sakamoto, T.; Kimura, S. Transcriptomic heterochrony and completely cleistogamous flower development in the mycoheterotrophic orchid Gastrodia. New Phytol. 2023, 237, 323–338. [Google Scholar] [CrossRef]
- Choi, D.Y.; Kim, Y. Transcriptome analysis of female western flower thrips, Frankliniella occidentalis, exhibiting neo-panoistic ovarian development. PLoS ONE 2022, 17, e0272399. [Google Scholar] [CrossRef]
- Shi, Z.Y.; Zhao, W.Q.; Li, Z.A.; Kang, D.R.; Ai, P.H.; Ding, H.X.; Wang, Z.C. Development and validation of SSR markers related to fower color based on full-length transcriptome sequencing in Chrysanthemum. Sci. Rep. 2022, 12, 22310. [Google Scholar] [CrossRef]
- Liu, L.M.; Zheng, Y.Q.; Feng, S.J.; Yu, L.; Li, Y.Q.; Zong, Y.; Chen, W.R.; Liao, F.L.; Yang, L.; Guo, W.D. Transcriptomic and physiological analysis reveals the responses to auxin and abscisic acid accumulation during Vaccinium corymbosum flower bud and fruit development. Front. Plant Sci. 2022, 13, 818233. [Google Scholar] [CrossRef]
- Wei, Y.; Li, A.H.; Zhao, Y.R.; Li, W.Q.; Dong, Z.Y.; Zhang, L.; Zhu, Y.T.; Zhang, H.; Gao, Y.K.; Zhang, Q.X. Time-course transcriptomic analysis reveals molecular insights into the inflorescence and flower development of Cardiocrinum giganteum. Plants 2024, 13, 649. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.F.; Liu, Y.; Yu, J.; Yao, G.L.; Yang, H.G.; Yang, D.M.; Wu, Y.G. An integrated transcriptome and metabolome analysis reveals the gene network regulating flower development in Pogostemon cablin. Front. Plant Sci. 2023, 14, 1201486. [Google Scholar] [CrossRef]
- Zhu, L.; Liao, J.H.; Liu, Y.F.; Zhou, C.M.; Wang, X.; Hu, Z.G.; Huang, B.S.; Zhang, J.J. Integrative metabolome and transcriptome analyses reveal the molecular mechanism underlying variation in floral scent during flower development of Chrysanthemum indicum var. aromaticum. Front. Plant Sci. 2022, 13, 919151. [Google Scholar] [CrossRef]
- Sun, Y.; Hu, P.L.; Jiang, Y.N.; Li, J.; Chang, J.X.; Zhang, H.H.; Shao, H.J.; Zhou, Y.W. Integrated metabolome and transcriptome analysis of petal anthocyanin accumulation mechanism in Gloriosa superba ‘rothschildiana’ during different flower development stages. Int. J. Mol. Sci. 2023, 24, 15034. [Google Scholar] [CrossRef]
- Long, Y.Q.; Zeng, J.; Liu, X.R.; Wang, Z.H.; Tong, Q.Z.; Zhou, R.B.; Liu, X.D. Transcriptomic and metabolomic profling reveals molecular regulatory network involved in fower development and phenotypic changes in two Lonicera macranthoides varieties. 3 Biotech 2024, 14, 174. [Google Scholar] [CrossRef]
- Ahmad, S.; Lu, C.Q.; Gao, J.; Wei, Y.L.; Xie, Q.; Jin, J.P.; Zhu, G.F.; Yang, F.X. Integrated proteomic, transcriptomic, and metabolomic profiling reveals that the gibberellin–abscisic acid hub runs flower development in the Chinese orchid Cymbidium sinense. Hortic. Res. 2024, 11, uhae073. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nature methods. 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Varet, H.; Brillet-Guéguen, L.; Coppée, J.Y.; Dillies, M.A. SARTools: A DESeq2- and EdgeR-based rpipeline for comprehensive differential analysis of RNA-seq data. PLoS ONE 2016, 11, e0157022. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Wu, Z.; Fang, X.; Zhu, D.; Dean, C. Autonomous pathway: FLOWERING LOCUS C repression through an antisense-mediated chromatin-silencing mechanism. Plant Physiol. 2020, 182, 27–37. [Google Scholar] [CrossRef]
- Wickland, D.P.; Hanzawa, Y. The FLOWERING LOCUS T/TERMINAL FLOWER 1 gene family: Functional evolution and molecular mechanisms. Mol. Plant 2015, 8, 983–997. [Google Scholar] [CrossRef]
- Coelho, C.P.; Minow, M.A.A.; Chalfun-Júnior, A.; Colasanti, J. Putative sugarcane FT/TFL1 genes delay flowering time and alter reproductive architecture in Arabidopsis. Front. Plant Sci. 2014, 5, 221. [Google Scholar] [CrossRef]
- Schilling, S.; Kennedy, A.; Pan, S.; Jermiin, L.S.; Melzer, R. Genome-wide analysis of MIKC-type MADS-box genes in wheat: Pervasive duplications, functional conservation and putative neofunctionalization. New Phytol. 2020, 225, 511–529. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Raw Reads | Clean Reads | Clean Bases (Gb) | Clean Reads Q20 (%) | Clean Reads Q30 (%) |
|---|---|---|---|---|---|
| Stage1-1 | 48,258,880 | 47,134,884 | 7.07 | 97.56 | 93.18 |
| Stage1-2 | 51,444,202 | 50,169,530 | 7.53 | 97.59 | 93.22 |
| Stage1-3 | 54,631,210 | 53,461,502 | 8.02 | 97.63 | 93.38 |
| Stage2-1 | 61,991,110 | 55,062,098 | 8.26 | 98.19 | 94.88 |
| Stage2-2 | 49,126,842 | 48,052,418 | 7.21 | 97.60 | 93.30 |
| Stage2-3 | 54,897,790 | 53,795,776 | 8.07 | 97.59 | 93.17 |
| Stage3-1 | 45,013,372 | 44,198,806 | 6.63 | 97.64 | 93.26 |
| Stage3-2 | 60,224,282 | 59,050,464 | 8.86 | 97.80 | 93.83 |
| Stage3-3 | 54,954,658 | 53,808,556 | 8.07 | 97.57 | 93.21 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Yu, Y.; Zhao, J.; Zhang, J. Integrative Transcriptomic and Metabolomic Analysis Reveals Regulatory Networks and Metabolite Dynamics in Gastrodia elata Flower Development. Agronomy 2025, 15, 441. https://doi.org/10.3390/agronomy15020441
Chen H, Yu Y, Zhao J, Zhang J. Integrative Transcriptomic and Metabolomic Analysis Reveals Regulatory Networks and Metabolite Dynamics in Gastrodia elata Flower Development. Agronomy. 2025; 15(2):441. https://doi.org/10.3390/agronomy15020441
Chicago/Turabian StyleChen, Hongyu, Ying Yu, Jiehong Zhao, and Jian Zhang. 2025. "Integrative Transcriptomic and Metabolomic Analysis Reveals Regulatory Networks and Metabolite Dynamics in Gastrodia elata Flower Development" Agronomy 15, no. 2: 441. https://doi.org/10.3390/agronomy15020441
APA StyleChen, H., Yu, Y., Zhao, J., & Zhang, J. (2025). Integrative Transcriptomic and Metabolomic Analysis Reveals Regulatory Networks and Metabolite Dynamics in Gastrodia elata Flower Development. Agronomy, 15(2), 441. https://doi.org/10.3390/agronomy15020441