
Transcriptome and Proteome Reveal Heat Shock Promotes Haploid Induction Rate via Activating ABA Signal Transduction in Watermelon
Abstract
1. Introduction
2. Materials and Methods
2.1. Unfertilized Ovary Collection and Ovule Culture
2.2. Library Construction, Transcriptome Sequencing and Expression Analysis
2.3. Peptide Preparation
2.4. Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) Analysis and DIA Mode
2.5. The Identification and Quantitation of Protein
2.6. Functional Analysis of Differentially Expressed Proteins and Genes
2.7. Differentially Expressed Genes Analysis by Quantitative Real-Time PCR (qRT-PCR)
2.8. Abscisic Acid Content Detection
3. Results
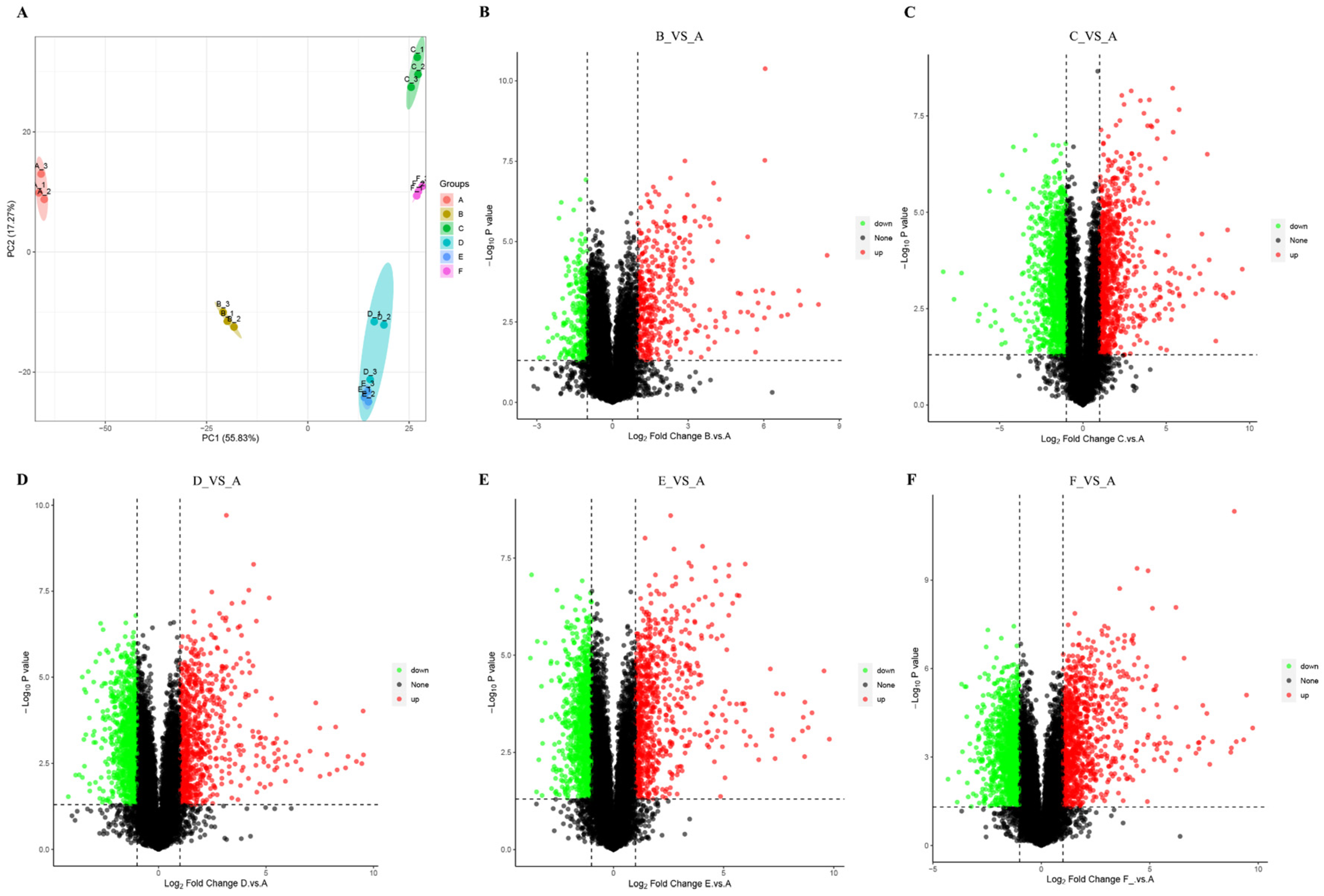
3.1. Phenotype Differences, Transcriptome and Proteome Profiles of Unfertilized Ovules
3.2. Transcriptome and Proteome Profiles of Unfertilized Ovules
3.3. Differentially Expressed Genes from Unfertilized Ovary After Heat Shock
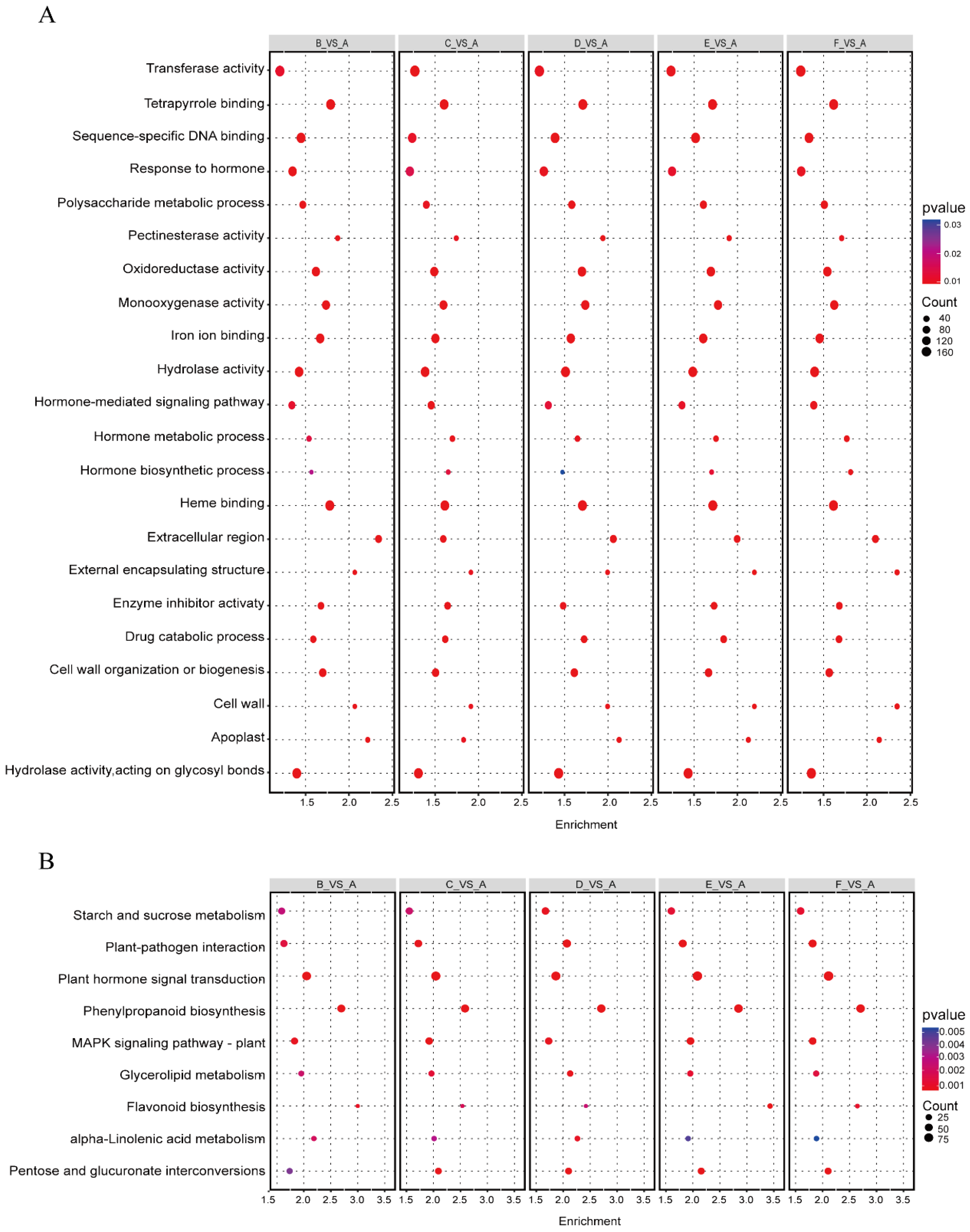
3.4. Differentially Expressed Genes Enrichment in Unfertilized Ovules After Heat Shock
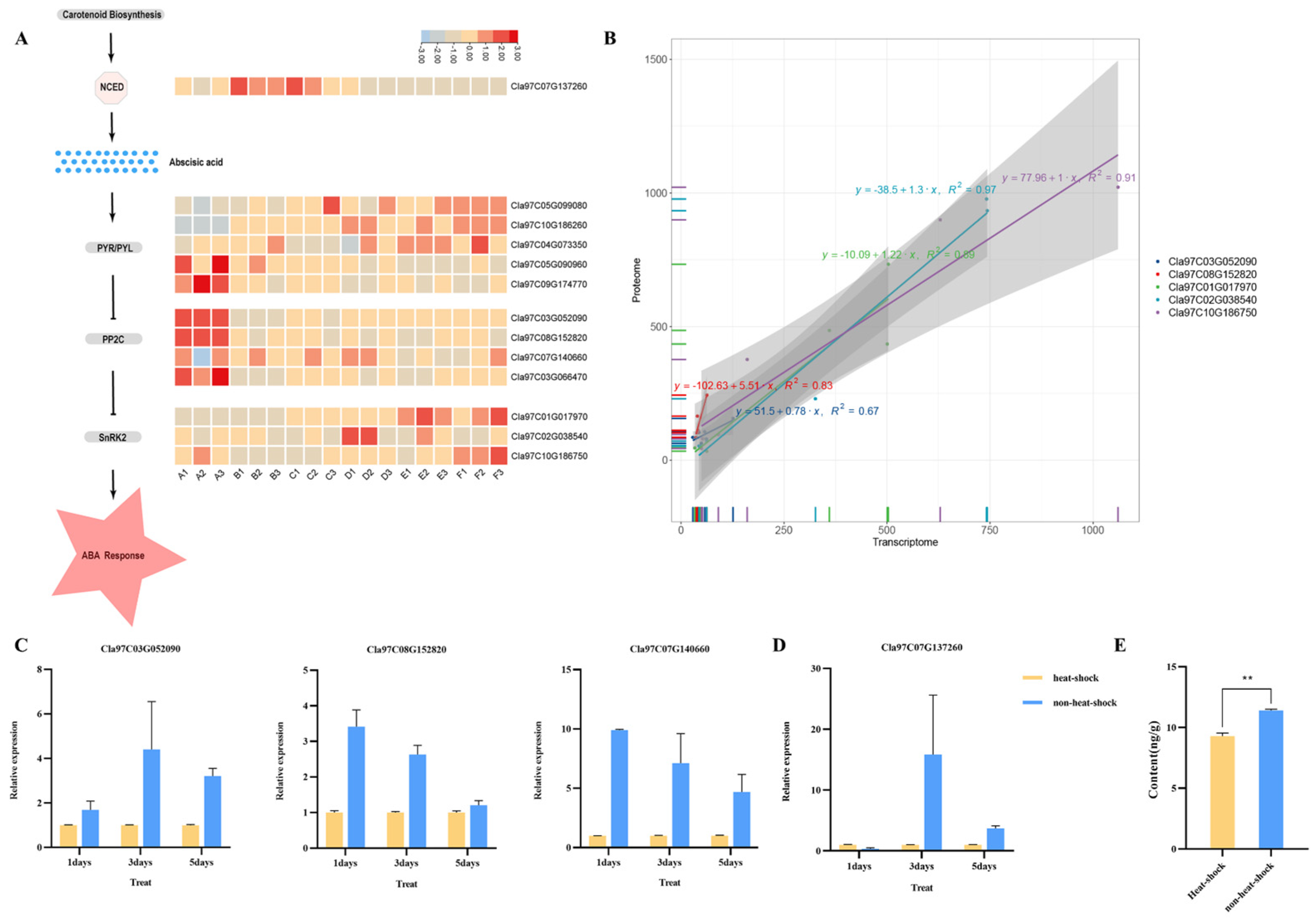
3.5. Expression of Genes Involved in Biosynthesis and Signal Transduction of ABA
3.6. Expression of Genes Involved in Biosynthesis of Amino Acids and Unsaturated Fatty Acids
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, S.; Wang, X.; Reddy, U.; Sun, H.; Bao, K.; Gao, L.; Mao, L.; Patel, T.; Ortiz, C.; Abburi, V.L.; et al. Genome of ‘Charleston Gray’, the principal American watermelon cultivar, and genetic characterization of 1365 accessions in the U.S. National Plant Germplasm System watermelon collection. Plant Biotechnol. J. 2019, 17, 2246–2258. [Google Scholar] [CrossRef]
- Guo, S.; Zhao, S.; Sun, H.; Wang, X.; Wu, S.; Lin, T.; Ren, Y.; Gao, L.; Deng, Y.; Zhang, J.; et al. Resequencing of 414 cultivated and wild watermelon accessions identifies selection for fruit quality traits. Nat. Genet. 2019, 51, 1616–1623. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Liu, S.; Zhang, Y.; Tan, J.; Li, X.; Chu, X.; Xu, B.; Tian, Y.; Sun, Y.; Li, B.; et al. A telomere-to-telomere gap-free reference genome of watermelon and its mutation library provide important resources for gene discovery and breeding. Mol. Plant 2022, 15, 1268–1284. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Xiao, L.; He, Y.; Liu, M.; Wang, J.; Tian, S.; Zhang, X.; Yuan, L. Highly efficient, genotype-independent transformation and gene editing in watermelon (Citrullus lanatus) using a chimeric ClGRF4-GIF1 gene. J. Integr. Plant Biol. 2021, 63, 2038–2042. [Google Scholar] [CrossRef]
- Tian, J.; Wang, C.; Chen, F.; Qin, W.; Yang, H.; Zhao, S.; Xia, J.; Du, X.; Zhu, Y.; Wu, L.; et al. Maize smart-canopy architecture enhances yield at high densities. Nature 2024, 632, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Liu, C.; Chen, S.; Jin, W. Haploid induction and its application in maize breeding. Mol. Breed. 2021, 41, 20. [Google Scholar] [CrossRef]
- Wang, L.; Huang, Y.; Liu, Z.; He, J.; Jiang, X.; He, F.; Lu, Z.; Yang, S.; Chen, P.; Yu, H.; et al. Somatic variations led to the selection of acidic and acidless orange cultivars. Nat. Plants 2021, 7, 954–965. [Google Scholar] [CrossRef]
- Liu, C.; Li, X.; Meng, D.; Zhong, Y.; Chen, C.; Dong, X.; Xu, X.; Chen, B.; Li, W.; Li, L.; et al. A 4-bp Insertion at ZmPLA1 Encoding a Putative Phospholipase a Generates Haploid Induction in Maize. Mol. Plant 2017, 10, 520–522. [Google Scholar] [CrossRef]
- Yao, L.; Zhang, Y.; Liu, C.; Liu, Y.; Wang, Y.; Liang, D.; Liu, J.; Sahoo, G.; Kelliher, T. OsMATL mutation induces haploid seed formation in indica rice. Nat. Plants 2018, 4, 530–533. [Google Scholar] [CrossRef]
- Zhong, Y.; Liu, C.; Qi, X.; Jiao, Y.; Wang, D.; Wang, Y.; Liu, Z.; Chen, C.; Chen, B.; Tian, X.; et al. Mutation of ZmDMP enhances haploid induction in maize. Nat. Plants 2019, 5, 575–580. [Google Scholar] [CrossRef]
- Malik, A.A.; Cui, L.; Zhang, S.; Chen, J.-F. Efficiency of SSR markers for determining the origin of melon plantlets derived through unfertilized ovary culture. Hortic. Sci. 2011, 38, 27–34. [Google Scholar] [CrossRef]
- Rakha, M.T.; Metwally, E.I.; Moustafa, S.A.; Etman, A.A.; Dewir, Y.H. Evaluation of regenerated strains from six Cucurbita interspecific hybrids obtained through anther and ovule in vitro cultures. Aust. J. Crop Sci. 2012, 6, 23–30. [Google Scholar]
- Zou, T.; Su, H.-N.; Wu, Q.; Sun, X.-W. Haploid induction via unfertilized ovary culture in watermelon. Plant Cell Tissue Organ Cult. (PCTOC) 2018, 135, 179–187. [Google Scholar] [CrossRef]
- Shalaby, T.A. Factors affecting haploid induction through in vitro gynogenesis in summer squash (Cucurbita pepo L.). Sci. Hortic. 2007, 115, 1–6. [Google Scholar] [CrossRef]
- Zou, T.; Min, Z.; Song, H.; Gong, H.; Tong, L.; Sun, L.; Yang, H.; Zhang, J.; Sun, X. Whole-transcriptome sequence analysis of pumpkin ovules in response to heat shock during unfertilized ovary culture. Sci. Hortic. 2024, 329, 113007. [Google Scholar] [CrossRef]
- Zhu, Y.-c.; Sun, D.-x.; Deng, Y.; An, G.-l.; Li, W.-h.; Si, W.-j.; Liu, J.-p.; Sun, X.-w. Comparative transcriptome analysis of the effect of different heat shock periods on the unfertilized ovule in watermelon (Citrullus lanatus). J. Integr. Agric. 2020, 19, 528–540. [Google Scholar] [CrossRef]
- Lou, R.; Shui, W. Acquisition and Analysis of DIA-Based Proteomic Data: A Comprehensive Survey in 2023. Mol. Cell. Proteom. 2024, 23, 100712. [Google Scholar] [CrossRef]
- Wang, Y.; Dai, M.; Cai, D.; Shi, Z. Proteome and transcriptome profile analysis reveals regulatory and stress-responsive networks in the russet fruit skin of sand pear. Hortic. Res. 2020, 7, 16. [Google Scholar] [CrossRef]
- Zhang, C.; Deng, Y.; Zhang, G.; Li, J.; Xiao, A.; Zhao, L.; Chen, A.; Tang, H.; Chang, L.; Pan, G.; et al. Comparative Transcriptome and Proteome Analysis Provides New Insights Into the Mechanism of Protein Synthesis in Kenaf (Hibiscus cannabinus L.) Leaves. Front. Plant Sci. 2022, 13, 879874. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Kumar, L.; Matthias, E.F. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Abdi, H.; Williams, L.J. Principal component analysis. Wiley Interdiscip. Rev. Comput. Stat. 2010, 2, 433–459. [Google Scholar] [CrossRef]
- Niu, L.; Zhang, H.; Wu, Z.; Wang, Y.; Liu, H.; Wu, X.; Wang, W. Modified TCA/acetone precipitation of plant proteins for proteomic analysis. PLoS ONE 2018, 13, e0202238. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- da Huang, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2 (-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- González-Curbelo, M.Á.; Socas-Rodríguez, B.; Herrera-Herrera, A.V.; González-Sálamo, J.; Hernández-Borges, J.; Rodríguez-Delgado, M.Á. Evolution and applications of the QuEChERS method. TrAC Trends Anal. Chem. 2015, 71, 169–185. [Google Scholar] [CrossRef]
- Song, H.; Zou, T.; Gong, S.; Zhang, C.; Sun, X.; Luo, Y.; Dai, S.; Sun, L.; Yang, H.; Sun, X. Research on the Creation and Application of a Doubled Haploid for Distant Hybrid Pumpkin. Hortic. Plant J. 2025, in press. [Google Scholar] [CrossRef]
- Gilles, L.M.; Martinant, J.-P.; Rogowsky, P.M.; Widiez, T. Haploid induction in plants. Curr. Biol. 2017, 27, R1095–R1097. [Google Scholar] [CrossRef] [PubMed]
- Prem, D.; Solís, M.-T.; Bárány, I.; Rodríguez-Sanz, H.; Risueño, M.C.; Testillano, P.S. A new microspore embryogenesis system under low temperature which mimics zygotic embryogenesis initials, expresses auxin and efficiently regenerates doubled-haploid plants in Brassica napus. BMC Plant Biol. 2012, 12, 127. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Li, G.J.; Bressan, R.A.; Song, C.P.; Zhu, J.K.; Zhao, Y. Abscisic acid dynamics, signaling, and functions in plants. J. Integr. Plant Biol. 2020, 62, 25–54. [Google Scholar] [CrossRef]
- Gosti, F.; Beaudoin, N.; Serizet, C.; Webb, A.A.; Vartanian, N.; Giraudat, J. ABI1 protein phosphatase 2C is a negative regulator of abscisic acid signaling. Plant Cell 1999, 11, 1897–1910. [Google Scholar] [CrossRef]
- Cai, Y. Lipid Synthesis and Beyond: SAD Fatty Acid Desaturases Contribute to Seed Development. Plant Cell 2020, 32, 3386–3387. [Google Scholar] [CrossRef]
- Amir, R.; Galili, G.; Cohen, H. The metabolic roles of free amino acids during seed development. Plant Sci. Int. J. Exp. Plant Biol. 2018, 275, 11–18. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Gene ID | Primer |
|---|---|---|
| Biosynthesis of ABA | Cla97C07G137260 | CTTACCGGTGATTGGGAAAG |
| CAGAGAATTCCACAGCGTGA | ||
| ABA signal transduction (PYL/PYR) | Cla97C05G099080 | CAGCCTTGCAGGGATTAGAA |
| CATCGATGCGTTGGGTAATA | ||
| Cla97C10G186260 | GGAGATGACGGAGCACCA | |
| GCTACCAAGTCCTTCAACTCG | ||
| Cla97C04G073350 | AATCCCCCTAAATCCTCTGC | |
| CAACAGCGTGGTTGTGGTAA | ||
| Cla97C05G090960 | AGCTCCGTCCTAATCAAGCA | |
| ATTCCCCTGCACTACACACC | ||
| ABA signal transduction (PP2C) | Cla97C03G052090 | ATGGCGGAGATTTGCTGTA |
| CTCCGGCGACAAATTTACAC | ||
| Cla97C08G152820 | GGTTGTGGTGCCATTTAGGT | |
| ACAGAATCAGATAACAAACCAGCA | ||
| Cla97C07G140660 | TCTCCTGCTGTTTCTTTGACC | |
| GGGGACGATGCAGAGAATAA | ||
| ABA signal transduction (SnRK2) | Cla97C01G017970 | TTGGTGTTGCCAAACTTGTG |
| TTGGGATGCTTCAAAGACCT | ||
| Cla97C10G186750 | AGGACCTTGGCTCTGGAAAT | |
| TCCCTCTGAACTTTCTCATCG | ||
| Biosynthesis of unsaturated acid | Cla97C04G071820 | GCCACGCTTCCGTTAGAC |
| TGGAAGCGTAGTTGATGACG | ||
| Cla97C08G149220 | TCGGGTCGATATGATGATGA | |
| ATATGAACGTGGCTCGCTCT | ||
| Cla97C05G081950 | CTTGGCCAATTGAGATACGG | |
| GTGCAAGGCTTGATCATCTG | ||
| Cla97C05G084200 | TCCAAAATGCTCAGAACTGC | |
| TGCAGCCTTCCTGTGAGAC | ||
| Cla97C07G136380 | TTAAGATGGGCGTCCAGTTC | |
| TCATAGCAAAACAGCCAGGA | ||
| Cla97C10G200350 | TGGAGGTCGAATGTCCTCTC | |
| GAACGTTGAAAGCAATGTGG | ||
| Biosynthesis of amino acid | Cla97C01G006270 | ACGTCTTTCTGTTCCGATCC |
| CAAGCCAATGGATGCTAACC | ||
| Cla97C09G174910 | TGTTCGGGGAAAAGTTGTCT | |
| CAATCTGTAGTCCCACGACCT | ||
| Cla97C04G076580 | ACCAAGATGACTGGGAGCAC | |
| CCTTGAGAAGAAGGGCATTG |
| Sample | Raw_Reads | Raw_Rases | Clean_Reads | Clean_Bases | Mapped Reads | Mapping Rate | Q20 | Q30 |
|---|---|---|---|---|---|---|---|---|
| A1 | 44,778,116 | 6.72 G | 43,909,136 | 6.59 G | 42,129,431 | 95.95% | 96.84 | 91.72 |
| A2 | 45,116,264 | 6.77 G | 44,273,292 | 6.64 G | 42,477,376 | 95.94% | 97.29 | 92.68 |
| A3 | 43,285,380 | 6.49 G | 42,572,246 | 6.39 G | 40,958,803 | 96.21% | 96.89 | 91.74 |
| B1 | 45,404,288 | 6.81 G | 44,454,896 | 6.67 G | 42,724,587 | 96.11% | 97.23 | 92.57 |
| B2 | 47,347,854 | 7.1 G | 46,330,350 | 6.95 G | 44,550,651 | 96.16% | 97.35 | 92.72 |
| B3 | 51,724,206 | 7.76 G | 50,458,778 | 7.57 G | 48,450,099 | 96.02% | 96.9 | 91.86 |
| C1 | 44,974,742 | 6.75 G | 43,807,364 | 6.57 G | 42,096,256 | 96.09% | 97.27 | 92.6 |
| C2 | 45,738,284 | 6.86 G | 44,775,652 | 6.72 G | 43,009,048 | 96.05% | 96.95 | 91.92 |
| C3 | 43,450,532 | 6.52 G | 42,449,150 | 6.37 G | 40,536,723 | 95.49% | 96.62 | 91.24 |
| D1 | 46,381,630 | 6.96 G | 45,147,020 | 6.77 G | 43,544,922 | 96.45% | 97.27 | 92.55 |
| D2 | 50,617,116 | 7.59 G | 49,620,388 | 7.44 G | 46,992,450 | 94.70% | 97.2 | 92.5 |
| D3 | 58,917,740 | 8.84 G | 57,662,538 | 8.65 G | 54,607,524 | 94.70% | 97.05 | 92.19 |
| E1 | 47,949,030 | 7.19 G | 47,160,738 | 7.07 G | 45,251,327 | 95.95% | 97.09 | 92.22 |
| E2 | 50,399,036 | 7.56 G | 48,975,548 | 7.35 G | 46,655,744 | 95.26% | 96.99 | 92.04 |
| E3 | 48,461,724 | 7.27 G | 47,519,596 | 7.13 G | 45,538,955 | 95.83% | 97.21 | 92.49 |
| F1 | 56,076,354 | 8.41 G | 54,869,120 | 8.23 G | 52,307,674 | 95.33% | 97.08 | 92.2 |
| F2 | 47,838,450 | 7.18 G | 46,586,662 | 6.99 G | 44,277,422 | 95.04% | 96.92 | 91.88 |
| F3 | 50,067,968 | 7.51 G | 48,707,704 | 7.31 G | 45,836,370 | 94.10% | 96.85 | 91.73 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, S.; Tang, B.; Dai, Y.; Sun, X.; Song, H.; Xiong, C.; Zou, T.; Sun, L.; Liu, G.; Yang, H.; et al. Transcriptome and Proteome Reveal Heat Shock Promotes Haploid Induction Rate via Activating ABA Signal Transduction in Watermelon. Agronomy 2025, 15, 1063. https://doi.org/10.3390/agronomy15051063
Gong S, Tang B, Dai Y, Sun X, Song H, Xiong C, Zou T, Sun L, Liu G, Yang H, et al. Transcriptome and Proteome Reveal Heat Shock Promotes Haploid Induction Rate via Activating ABA Signal Transduction in Watermelon. Agronomy. 2025; 15(5):1063. https://doi.org/10.3390/agronomy15051063
Chicago/Turabian StyleGong, Shiqi, Bingqian Tang, Yujuan Dai, Xiangyu Sun, Huijuan Song, Cheng Xiong, Tian Zou, Longjun Sun, Guang Liu, Hongbo Yang, and et al. 2025. "Transcriptome and Proteome Reveal Heat Shock Promotes Haploid Induction Rate via Activating ABA Signal Transduction in Watermelon" Agronomy 15, no. 5: 1063. https://doi.org/10.3390/agronomy15051063
APA StyleGong, S., Tang, B., Dai, Y., Sun, X., Song, H., Xiong, C., Zou, T., Sun, L., Liu, G., Yang, H., Zhu, S., Dai, S., & Sun, X. (2025). Transcriptome and Proteome Reveal Heat Shock Promotes Haploid Induction Rate via Activating ABA Signal Transduction in Watermelon. Agronomy, 15(5), 1063. https://doi.org/10.3390/agronomy15051063