1. Introduction
Sugarcane is a major crop cultivated in tropical and subtropical fields for sugar production. sugarcane mosaic virus (SCMV) is one of the main pathogens that substantially affect sugarcane productivity. Recently, it has been reported that this pathogen has spread in East Java, Indonesia [
1], with an incidence rate of 78% on the sugarcane-cultivated area, and reduced the sugar production by 20% [
2,
3]. The infected sugarcane plant shows symptoms such as a yellowish, chlorotic leaf area, which could be accompanied by varying degrees of necrosis. The chlorotic areas are mostly observed in young rapidly growing leaves and are particularly distinct in the basal portion of the leaves [
4]. However, this chlorotic symptom can be the result of a failure in chlorophyll development, due to a virus infection, or a lack of an essential mineral nutrient. Furthermore, the mosaic symptoms caused by sugarcane viruses—such as sugarcane streak mosaic virus (SCSMV), sugarcane mosaic virus (SCMV), and sorghum mosaic virus (SrMV)—are difficult to distinguish by morphological observation of the symptoms [
5]. The symptoms are also unstable, since symptoms are influenced by the time of infection, environment, and cultivar [
6]. Thus, the identification of the causal agent of mosaic diseases is an important step in determining the appropriate control management.
Sugarcane mosaic virus belongs to the
Potyviridae family, genus
Potyvirus, found worldwide in over 70 countries [
7], and infects some host plants—such as maize, sorghum, and several
Poaceae—and is transmitted by aphids [
8]. The virion is comprised of flexuous rods, about 750 nm in length, and has a genome of positive-sense RNA, approximately 10 kb long [
7]. The complete genome sequences of SCMV isolated from several geographic regions have been published in the NCBI GenBank database, such isolates from Argentina (JX237862.1); Australia (AJ278405); China (AF494510.1; JN021933.1); Iran (KT895081.1); and Mexico (GU474635.1). The SCMV genome contains a single long open reading frame (ORF), which encodes a polyprotein that is post-translationally processed into the individual gene products by viral proteases, and the coat protein is encoded at the 3′-end terminus [
9]. This coat protein has been studied intensively and highly conserved among SCMV isolates, thus facilitating detection of the virus by serological methods [
10].
Several immunodiagnostic methods, as well as molecular methods, have been widely used to detect plant viruses. Traditionally, a polyclonal antibody against a purified virion from infected plant material was developed for immunodiagnostics [
11]. However, the traditional preparation of a polyclonal antibody has its limitations, such as the purity of the virion and low viral titer in the infected plant tissue. This procedure is lengthy, requiring virus propagation in a suitable host, followed by virion purification [
12]. To overcome this problem, the serological method using a polyclonal antibody against a recombinant coat protein, was commonly used in immunodiagnostics [
11]. Several studies have reported on the production of polyclonal antibodies against recombinant proteins, such as the antibodies against coat protein of Pelargonium zonate spot virus [
13], cucumber mosaic virus [
14], or alfalfa mosaic virus [
15].
The recombinant viral proteins that are expressed in bacterial cells have great potential as a source of antigen to produce a specific antibody. The bacterial expression system is simple, fast, and inexpensive for producing a large amount of purified proteins as antigens to produce antibodies [
16]. In addition, a fusion tag added to the protein sequence and expressed in
Escherichia coli, improves protein expression, solubility, and stability, and facilitates a faster purification procedure [
17]. The combination of the bacterial expression system and the fusion tag protein purification has been routinely used in protein production and purification for the development of antibody [
18].
In this study, we have developed a polyclonal antibody against a recombinant coat protein of the SCMV and successfully tested its sensitivity for detection of SCMV infection in sugarcane by western blot and enzyme-linked immunosorbent assay (ELISA).
2. Materials and Methods
2.1. Cloning and Sequencing cDNA of SCMV Coat Protein
Field grown sugarcane cultivar PS-881 (4–5 months) with mosaic disease symptoms was used as the virus source. The yellowing and chlorosis symptom leaves were harvested and used for total RNA isolation using the guanidinium thiocyanate method, as described by Sambrook et al. [
19]. The published CP gene sequences were used for designing the primer F1; 5′-GTTTYCACCAAGCTGGAACAGTC-3′ and R1; 5′-AGCTGTGTGTCTCTCTGTATTCTCT-3′. First-strand cDNA was synthesized using a template from total RNA, with oligo dT primer and reverse transcriptase (Roche, Mannheim, Germany). Targeted coat protein cDNA was amplified using a pair of primers F1 and R1. The PCR reaction consisted of one cycle pre-denaturation at 94 °C for 5 min, 30 cycles at 94 °C denaturation for 30 s, 56 °C for 30 s, 68 °C extension for 60 s, and the final extension step at 68 °C for 5 min. The amplified cDNA was visualized using 1% agarose gel electrophoresis and purified using a gel extraction kit (Macherey-Nagel, Duren, Germany) according to the manufacturer’s instruction. The purified cDNA fragment was ligated into a pJET1.2 vector (ThermoFisher Scientific, Waltham, MA, USA) and transformed into the
E. coli strain XL10-Gold. The transformants were screened by colony PCR using F1, R1 set of primers.
The nucleotides sequence of the cDNA was determined bi-directionally, and the resulting sequence was analyzed using software Genetyx Ver.8 for Windows. The deduced amino acids sequences were compared with the GenBank NCBI database using BLASTp. The corresponding cDNA was named SCMVCp and used for the production of the recombinant protein.
2.2. Plasmid Construction for Production of the Recombinant Protein
To produce the recombinant protein, the
SCMVCp was amplified using PCR with specific primers F2: 5′-GCGGATCCGTCGATGCAGGTG-3′ and R2: 5′-GTGCTCGAGCAGAGAGAGTGCAT-3′ to produce a 924 bp DNA fragment, which encodes a recombinant protein (308 amino acids) starting with the DAG amino acids sequence. The DAG amino acids sequence was identified as the motif for SCMV transmission by aphid [
20]. The amplified cDNA was then digested with
BamHI and
XhoI restriction enzymes and ligated into a pET28a expression vector (Invitrogen, Carlsbad, CA, USA). The vector contains a fusion hexa-histidine (6-His) tag at the N terminal for purification by affinity chromatography. The pET28a construct was subsequently transformed into
E. coli XL10-Gold. The positive transformant colonies were selected by the PCR method using a set of primers for T7 promoter (5′-TAATACGACTCACTATAGGG-3′) and T7 terminator (5′-GCTAGTTATTGCTCAGCGGG-3′). Recombinant plasmids were isolated from the transformant using a Plasmid Miniprep Kit (Tiangen Co., Beijing, China) and the orientation of cDNA insertion was confirmed by restriction enzymes digestion and nucleotide sequence determination. The expression plasmid vector was transformed into
E. coli BL21, and the selected transformant was used for the production of the recombinant SCMVCp fusion protein.
2.3. Production and Purification of the Recombinant SCMVCp Fusion Protein
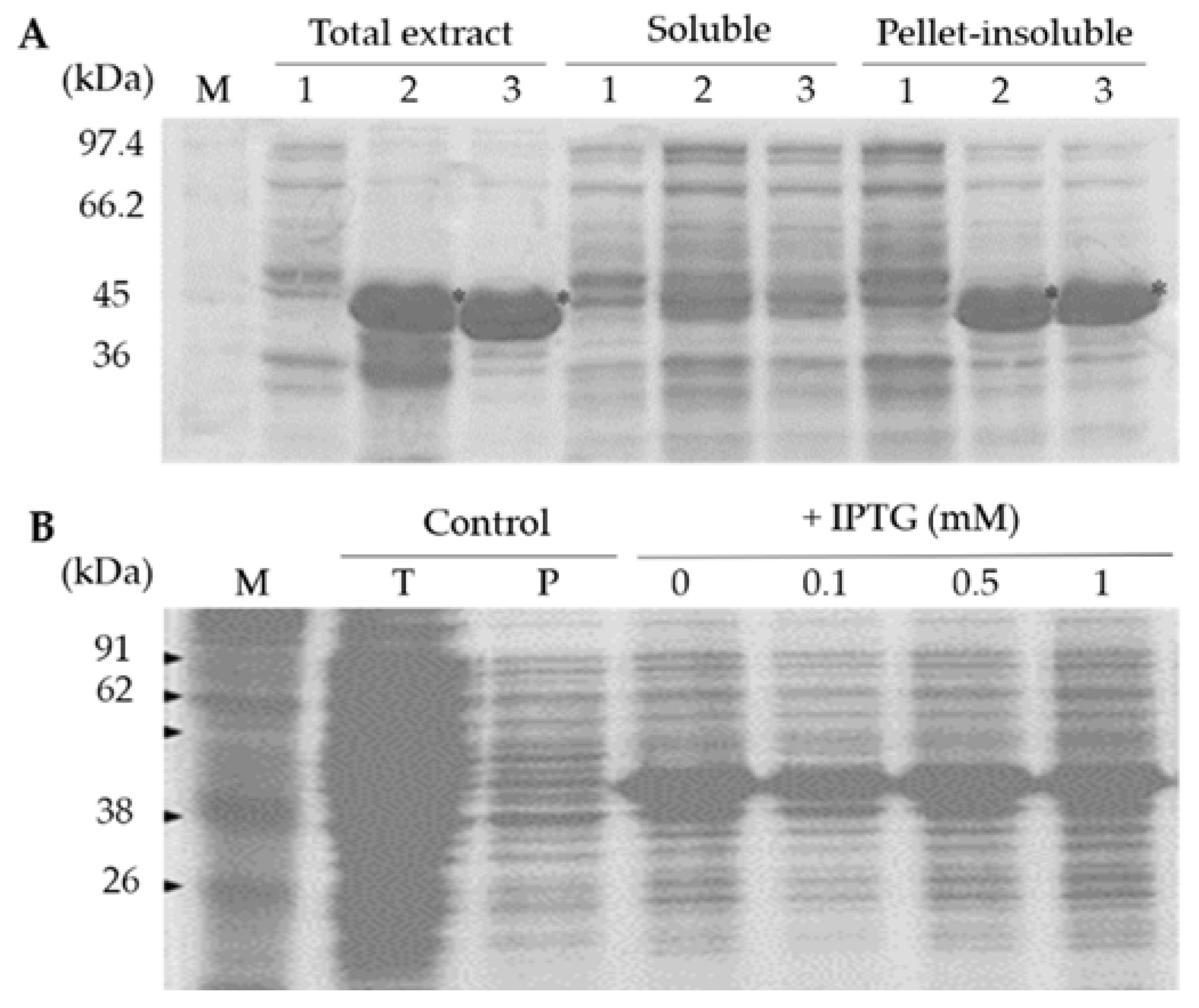
To analyze expression of the recombinant SCMVCp fusion protein, the E. coli BL21 transformant carrying pET28-SCMVCp construct was cultured in 0.1 L of LB (Luria-Bertani) medium containing 50 ppm kanamycin at 37 °C overnight on a shaking incubator (150 rpm). The expression of the recombinant SCMVCp fusion protein was induced by addition of IPTG at 0, 0.1, 0.3, 0.5, 1.0 mM. The bacterial cells were harvested by centrifugation at 6000 rpm 4 °C for 10 min, and the pellets were suspended in an extraction buffer containing 50 mM NaH2PO4 and 300 mM NaCl (pH 8). The recombinant protein was extracted by sonication and a soluble protein fraction was separated by centrifugation at 12,000 rpm 4 °C for 10 min. The proteins in total extract (before centrifugation), soluble fraction (supernatant), and insoluble fraction (pellet) were separated by SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) and then visualized by Coomassie Brilliant Blue (CBB) staining.
The production of the recombinant SCMVCp fusion protein was conducted by culturing the E. coli BL21 transformant in 5 L of LB medium containing 50 ppm kanamycin at 37 °C overnight. The bacterial cells were harvested by centrifugation and the pellet was used for protein extraction and purification. The purification of the recombinant fusion protein was conducted under denaturing condition, as the recombinant SCMVCp fusion protein was expressed in an insoluble fraction. The harvested bacterial cell was disrupted in the extraction buffer with sonication and the supernatant was separated by centrifugation at 12,000 rpm 4 °C for 10 min. The pellet cell debris was washed once with the extraction buffer, and the insoluble proteins fraction was resuspended in the solubilization buffer containing 50 mM NaH2PO4, 300 mM NaCl, 8 M Urea (pH 8). The recombinant protein was then solubilized by incubation at room temperature with gentle shaking and then separated by centrifugation at 12,000 rpm for 20 min. The recombinant His-tag fusion protein was purified by affinity column chromatography containing Ni-NTA resin equilibrated with the solubilized buffer according to the manufacturer’s instructions (Macherey-Nagel). Unbound proteins were washed with 50 mM NaH2PO4, 300 mM NaCl, 8 M Urea (pH 8) containing 20 mM imidazole, and the bounded-protein was eluted from the resin with 50 mM NaH2PO4, 300 mM NaCl, 8 M Urea (pH 8) containing 250 mM imidazole.
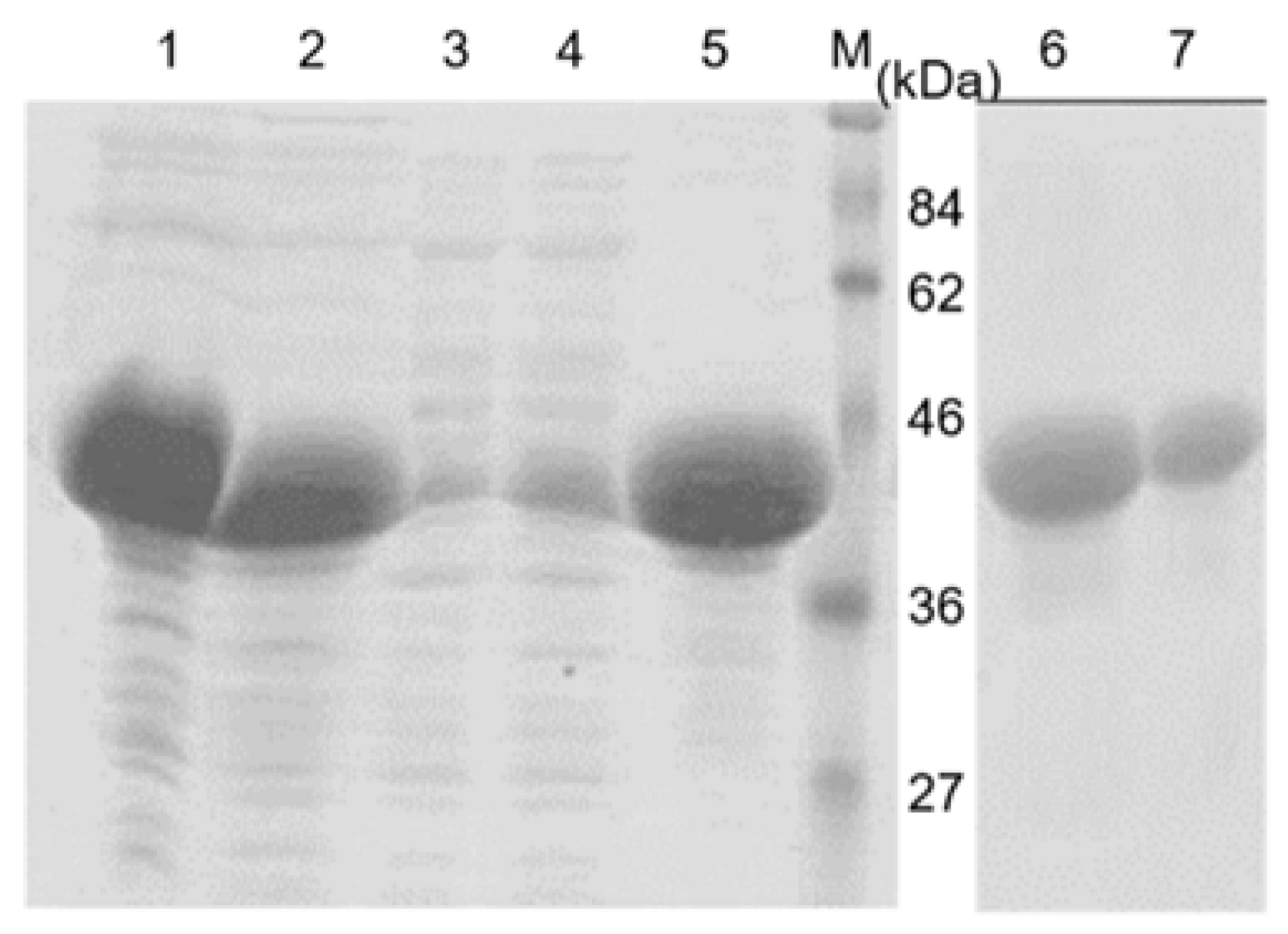
A further purification step was conducted by separation the eluted protein with SDS-PAGE and the targeted protein band was excised and recovered from the gels by electroelution (Bio-Rad, Hercules, CA, USA). The recovered protein was dialyzed against a phosphate buffer saline (PBS) overnight at 4 °C to remove SDS from the protein mixture. After evaluating the purity with SDS-PAGE and measuring the protein concentration with the Bradford reagent (Biorad), the recombinant protein was used as an antigen for the development of a polyclonal antibody.
2.4. Production of a Polyclonal Antibody and Estimation of the Antibody Development
Two female New Zealand White rabbits (two months’ age) were immunized subcutaneously with 500 µg of purified recombinant SCMVCp fusion protein emulsified with 500 µL of Freund’s complete adjuvant at a ratio of 1:1 (v/v). Two weeks after the first immunization, the rabbits were boosted by five additional subcutaneous injections with 100 µg of the purified protein mixed with 500 µL of Freund’s incomplete adjuvant per injection at a ratio of 1:1 every week. The rabbits were bled five times before and after injections to collect the antiserum, which were designated as pre-immune (before immunization) or S1 (after first boosted injection) to S5 (after fifth boosted injection) antiserum and then stored at −20 °C. The raised antiserum was monitored by Ouchterlony double diffusion analysis.
2.5. Western Blot Analysis
The polyclonal antiserums against the recombinant coat protein were tested by western blot and ELISA to evaluate their sensitivity and specificity. The purified SCMVCp protein, and either crude extracts of healthy or symptomatic sugarcane leaves, were used as test materials. To determine the sensitivity of the antiserum, a serial dilution of the purified SCMVCp protein was separated by SDS-PAGE. The separated proteins were electroblotted onto an Immobilon-P transfer membrane (Millipore) using semi-dry transblotter (Biorad) at a constant current of 250 mA for 60 min at room temperature. The membrane was removed from the transblotter and washed with TBS (Tris Buffer Saline) three times. After being blocked with a TBS solution containing 0.5% skim milk, the membrane was incubated with antiserum against the SCMVCp protein diluted in TBS containing 0.5% skim milk (1:3000) for overnight at room temperature with gentle agitation. The membrane was further washed three times in TBS, followed by incubation with the secondary antibody, goat anti-rabbit IgG alkaline phosphatase (AP)-conjugate (Biorad), at 1:3000 dilutions for 60 min at room temperature. The expected protein band was visualized by incubation in BCIP/NBT substrate (5-bromo-4-chloro 3-indolyl-phosphate/nitro blue tetrazolium) (Biorad).
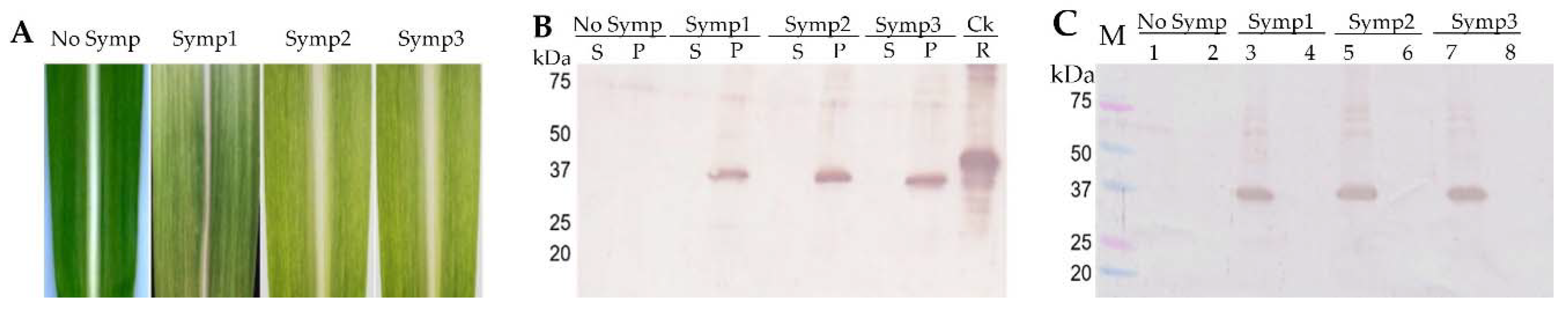
Crude leaf extracts were prepared by grinding 2 g of the healthy and symptomatic sugarcane leaf tissues in liquid nitrogen, and the frozen leaf powder was melted with a 6 mL extraction buffer containing 25 mM Tris-HCl (pH 7.5), 1 mM EDTA, 0.01% PMSF, 10% polyvinylpolypyrrolidone (PVP), and 10 mM 2-mercaptoethanol, and centrifuged at 12,000 rpm for 10 min at 4 °C. The supernatant containing soluble protein was stored at −80 °C, and the pellet debris containing insoluble protein was washed with the extraction buffer once. The insoluble proteins were solubilized with a buffer containing 50 mM Tris-HCl (pH 8.5), 1 mM EDTA, 2% SDS, and 30% sucrose. The soluble and insolubilize proteins were separated with SDS-PAGE and subjected to western blot analysis using the raised antiserum.
2.6. Indirect ELISA
Approximately one gram of the tissue of healthy and symptomatic sugarcane leaves was ground in liquid nitrogen, and the protein was extracted by continues grinding in three times extraction buffer containing 50 mM Mops-NaOH (pH 7.5), 10 mM MgCl2, 1 mM EDTA, and 2% PVP at 4 °C. The leaf extracts were centrifuged at 8000 rpm 4 °C for 10 min, and the supernatant was used for ELISA. The supernatant (100 µL) was loaded into immunoplate wells and coated by an additional 100 µL of coating buffer (15 mM Na2CO3, 35 NaHCO3, 2% PVP, pH 9.6), and then incubated at 4 °C overnight. The plate wells were washed tree times with TBS-T (tris-buffered saline plus 0.05% tween-20). The plate was blocked by addition 100 µL of the blocking solution TBS-T, containing 1% skim milk and 0.5% BSA, incubated at room temperature for an hour, and then washed with TBS-T once. Amount of 100 µL of diluted polyclonal antibody against recombinant coat protein with the blocking solution (1:1000) was added to the plate and incubated at room temperature for an hour. After washing the plate five times with TBS-T, 100 µL goat anti-rabbit IgG-AP conjugate (Sigma-Aldrich, St. Louis, MO, USA) at a dilution of 1:3000 in conjugate buffer was added and the plate was incubated at room temperature for an hour. The plate was further washed three times with TBS-T and then 100 µL p-nitrophenyl phosphate (PNP) substrate (0.6 mg/mL PNP 9.7% in diethanolamine buffer, pH 9.6) was added. After an hour of incubation at room temperature, the absorbance was measured at A405 nm. For analyzing the ELISA results, the S/H ratio (sample A405/TBS-T buffer A405) was calculated. A reaction was considered positive if the A405 of sample was 1.75 greater than the A405 of the TBS-T buffer.
The sensitivity of the polyclonal antiserum against the recombinant coat protein was compared with a DAS-ELISA test using reagent set for sugarcane mosaic virus (Agdia, Inc., Elkhart, IN, USA). To conduct an analysis using the ELISA reagent set, approximately one gram of healthy and symptomatic sugarcane leaves was ground in liquid nitrogen, and the pulverized leaves powder was homogenized with three times volume of the sample buffer 1 (SB1) provided by the ELISA reagent set. After centrifugation, the supernatant was used for DAS-ELISA analysis according to the manufacturer’s instructions (Agdia, Inc., Elkhart, IN, USA).
4. Discussion
The SCMV is one among many viruses that infect and cause yield loss of sugarcane cultivated in East Java Indonesia [
1]. Since the morphological symptom of SCMV is similar to symptoms caused by other diseases or the depletion of plant nutrition, the development of a serological method for the detection of SCMV infection is one of the most common methods to identify the disease. In addition, as a propagated vegetative plant, sugarcane is susceptible to contamination by virus through lateral buds during propagation. Therefore, it is valuable to establish a rapid, convenient, and reliable methods using the serological technique for viral detection. This method will facilitate the management for the prevention of the spread of the SCMV disease.
The alignment of nucleotides sequence showed that the
SCMVCp-cDNA isolate PS881-Jember has a high similarity to the isolate from Argentine (GenBank: JX237868.1), China (GenBank: AY590778.1), and Australia (GenBank: AJ278405.1). In this study, the
SCMVCp-cDNA was constructed in the plasmid with a His-tag fusion and expressed in
E. coli BL21. The expression of the recombinant SCMVCp fusion protein was detected as an insoluble protein in the transformed
E. coli BL21. Similarly, the insoluble expression of the recombinant coat protein has been reported for potato mop-top virus [
10] and Pelargonium zonate spot virus [
13]. Interestingly, the recombinant SCMVCp protein was strongly expressed and not induced by IPTG concentration (
Figure 2B). The presence of lactose as well as galactose in complex LB media, even in minute amounts, might act as an inducer of the recombinant protein expression [
21]. This leads to the fast and efficient production of the recombinant protein in the
E. coli cell, which is subsequently purified using the affinity Ni-NTA resin. Since the His-tag fused in the recombinant protein was expected not to have immunogenic activity, the polyclonal antibody against the recombinant CP was successfully developed.
The polyclonal antibody developed against the recombinant coat protein virus has been widely employed in virus detection using western blot or ELISA methods. Our results clearly demonstrated that the polyclonal antibody against the recombinant SCMVCp fusion protein shows a high sensitivity and be able to detect the antigen as low as 10 ng of the recombinant protein using western blot analysis (
Figure 4B). The antibody is highly specific as it detects a single band of the SCMV coat protein extracted from the infected sugarcane leaves, and no visible protein band observed from leaves with no symptom (healthy) was detected. These results imply that the antibody is suitable for the detection of the SCMV virion using the ELISA method, since the technique requires an antibody that only matches with a single targeted protein and does not cross-react with other proteins. A sensitivity comparison has shown that the antibody is more sensitive to SCMV detection than the DAS-ELISA reagent kit for sugarcane mosaic virus (Agdia, Inc., Elkhart, IN, USA) (
Table 2). Indirect ELISA analysis using the polyclonal antibody detected the SCMV in a 50 times diluted crude extract (
Table 1), but the DAS-ELISA detected only three times diluted crude extract of symptomatic sugarcane leaves (
Table 2). These results indicated differences in the specificity of the SCMV strain and the antibody sensitivity. The polyclonal antibody clearly detected the SCMV isolate PS881-Jember, but the ELISA reagent kit may work well with the SCMV strain from the USA and Australia. In addition, western blot analysis showed that the antibody detected a viral protein in a soluble fraction when the crude extract of infected leaves was centrifuged at below 10,000 rpm, but with ultra-centrifugation at 40,000 rpm, the protein was detected in an insoluble protein fraction. Thus, these results facilitate the easy detection of SCMV using ELISA method with low-speed centrifugation to maintain the virion in the soluble fraction.
The use of next-generation sequencing (NGS) for the identification and characterization of maize and sugarcane viruses has been reported [
25,
26]. It was claimed that the NGS method provides a powerful and rapid identifying potential diseases caused by virus, compared to the molecular and serological techniques. However, the NGS method needs sophisticated tools and bioinformatics expertise to process the NGS data [
26]. Since the antibody against the targeted virus has been developed, virus detection by serological methods, such as western blot and ELISA, are widely accepted. In this study, the antibody prepared against recombinant the SCMVCp protein was demonstrated to be efficient for the detection of SCMV using both western blot and ELISA methods. The availability of this antibody facilitates the screening of sugarcane in sanitation programs and the development of SCMV-resistant sugarcane.

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}