Seeding Propensity and Characteristics of Pathogenic αSyn Assemblies in Formalin-Fixed Human Tissue from the Enteric Nervous System, Olfactory Bulb, and Brainstem in Cases Staged for Parkinson’s Disease

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Autopsy Samples
2.2. Tissue Fixation, Embedding, and Sectioning
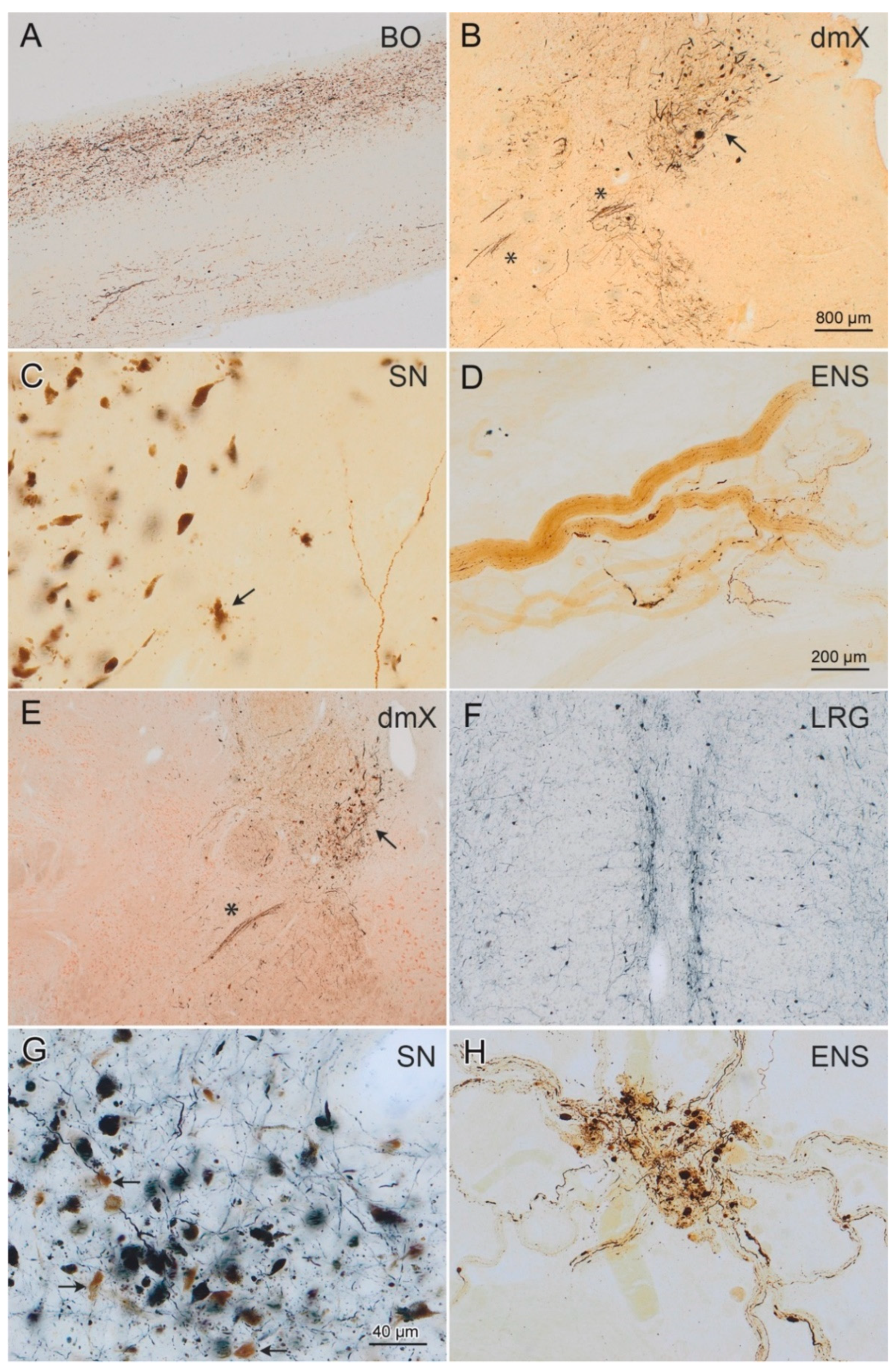
2.3. Immunohistochemistry and Neuropathological Staging
2.4. Punch Biopsies for Biochemical Analysis
2.5. Brain Tissue Homogenization
2.6. Protein Misfolding Cyclic Amplification (PMCA) Assay
2.7. Proteolytic Digestion
2.8. Transmission Electron Microscopy (TEM)
2.9. Aggregated αSyn Quantification in Patients’ Brain Homogenates
2.10. Frozen Brain Tissue Preparation
2.11. Preparation of Protein Assemblies
2.12. Statistical Analyses
3. Results
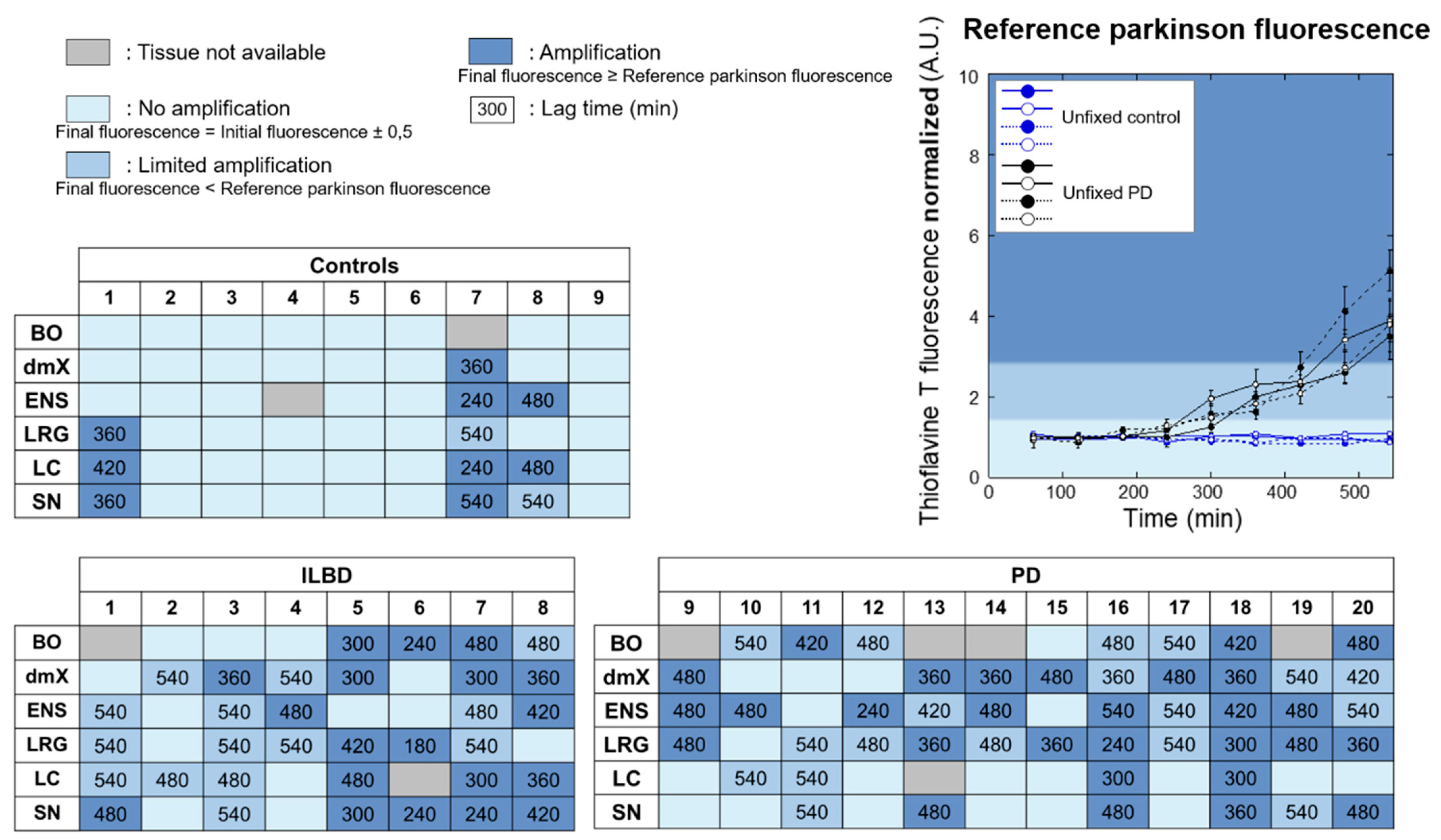
3.1. HTRF Cisbio Assay
3.2. PMCA Amplification
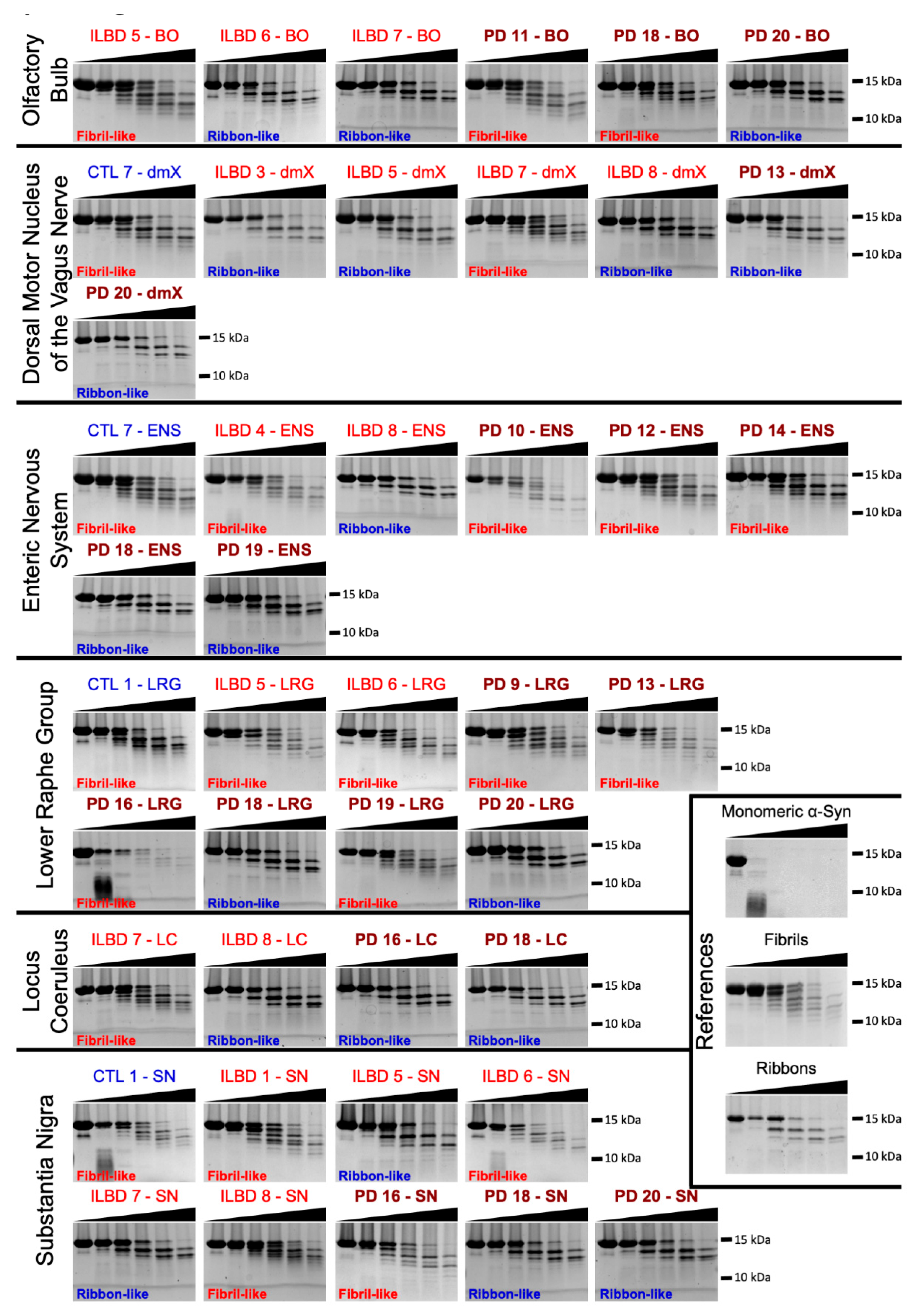
3.3. PMCA-Amplified Assemblies Characterization by TEM
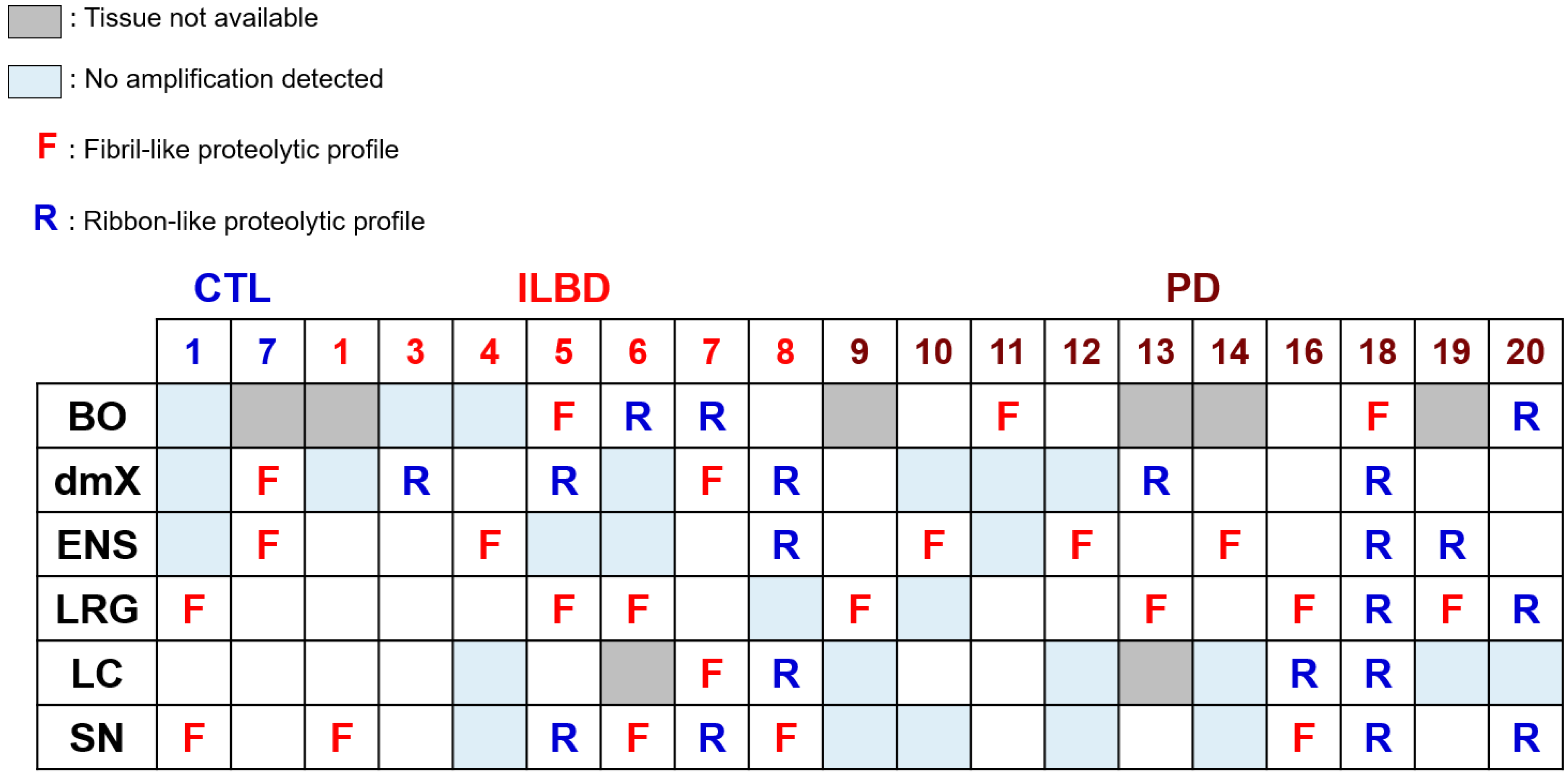
3.4. PMCA-Amplified Assemblies and Proteolytic Profiling
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braak, H.; Rüb, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Trans. 2003, 110, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Del Tredici, K.; Braak, H. Review: Sporadic Parkinson’s disease: Development and distribution of α-synuclein pathology. Neuropathol. Appl. Neurobiol. 2016, 42, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Björklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. NEURODEGENERATION. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 2015, 349, 1255555. [Google Scholar] [CrossRef]
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, J.H.; Paik, S.R.; Lee, S.J. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int. J. Biochem. Cell. Biol. 2008, 40, 1835–1849. [Google Scholar] [CrossRef]
- Danzer, K.M.; Krebs, S.K.; Wolff, M.; Birk, G.; Hengerer, B. Seeding induced by alpha-synuclein oligomers provides evidence for spreading of alpha synuclein pathology. J. Neurochem. 2009, 111, 192–203. [Google Scholar] [CrossRef]
- Desplats, P.; Lee, H.J.; Bae, E.J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef]
- Luk, K.C.; Song, C.; O’Brien, P.; Stieber, A.; Branch, J.R.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20051–20056. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.M.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.; Angot, E.; Bergström, A.L.; Steiner, J.A.; Pieri, L.; Paul, G.; Outeiro, T.F.; Melki, R.; Kallunki, P.; Fog, K.; et al. α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Investig. 2012, 121, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M. Exogenous α-synuclein fibrils induce Lewy Body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Dodiya, H.B.; Kordower, A.M.; Terpstra, B.; Paumier, K.; Madhavan, L.; Sortwell, C.; Steece-Collier, K.; Collier, T.J. Transfer of host derived alpha synuclein to grafted dopaminergic neurons in rat. Neurobiol. Dis. 2011, 43, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Angot, E.; Steiner, J.A.; Lema Tomé, C.M.; Ekström, P.; Mattsson, B.; Björklund, A.; Brundin, P. Alpha-synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons in vivo. PLoS ONE 2012, 7, e39465. [Google Scholar] [CrossRef]
- Mougenot, A.L.; Nicot, S.; Bencsik, A.; Morignat, E.; Verchère, J.; Lakhdar, L.; Legastelois, S.; Baron, T. Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol. Aging 2012, 33, 2225–2228. [Google Scholar] [CrossRef]
- Freundt, E.C.; Maynard, N.; Clancy, E.K.; Roy, S.; Bousset, L.; Sourigues, Y.; Covert, M.; Melki, R.; Kirkegaard, K.; Brahic, M. Neuron-to-neuron transmission of α-synuclein fibrils through axonal transport. Ann. Neurol. 2012, 72, 517–524. [Google Scholar] [CrossRef]
- Bousset, L.; Pieri, L.; Ruiz-Arlandis, G.; Gath, J.; Jensen, P.H.; Habenstein, B.; Madiona, K.; Olieric, V.; Böckmann, A.; Meier, B.H.; et al. Structural and functional characterization of two alpha-synuclein strains. Nat. Commun. 2013, 4, 2575. [Google Scholar] [CrossRef]
- Rey, N.L.; Petit, G.H.; Bousset, L.; Melki, R.; Brundin, P. Transfer of human α-synuclein from the olfactory bulb to interconnected brain regions in mice. Acta Neuropathol. 2013, 126, 555–573. [Google Scholar] [CrossRef]
- Rey, N.L.; Steiner, J.A.; Maroof, N.; Luk, K.C.; Madaj, Z.; Trojanowski, J.Q.; Lee, V.M.; Brundin, P. Widespread transneuronal propagation of α-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J. Exp. Med. 2016, 213, 1759–1778. [Google Scholar] [CrossRef] [PubMed]
- Sacino, A.N.; Brooks, M.; McGarvey, N.H.; McKinney, A.B.; Thomas, M.A.; Levites, Y.; Ran, Y.; Golde, T.E.; Giasson, B.I. Induction of CNS α-synuclein pathology by fibrillar and non-amyloidogenic recombinant α-synuclein. Acta Neuropathol. Commun. 2013, 1, 38. [Google Scholar] [CrossRef] [PubMed]
- Sacino, A.N.; Brooks, M.; Thomas, M.A.; McKinney, A.B.; Lee, S.; Regenhardt, R.W.; McGarvey, N.H.; Ayers, J.I.; Notterpek, L.; Borchelt, D.R.; et al. Intramuscular injection of α-synuclein induces CNS α-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc. Natl. Acad. Sci. USA 2014, 111, 10732–10737. [Google Scholar] [CrossRef] [PubMed]
- Aulić, S.; Le, T.T.; Moda, F.; Abounit, S.; Corvaglia, S.; Casalis, L.; Gustincich, S.; Zurzolo, C.; Tagliavini, F.; Legname, G. Defined α-synuclein prionlike molecular assemblies spreading in cell culture. BMC Neurosci. 2014, 15, 69. [Google Scholar] [CrossRef]
- Tran, H.T.; Chung, C.H.; Iba, M.; Zhang, B.; Trojanowski, J.Q.; Luk, K.C.; Lee, V.M. α-Synuclein immunotherapy blocks uptake and templated propagation of misfolded α-synuclein and neurodegeneration. Cell Rep. 2014, 7, 2054–2065. [Google Scholar] [CrossRef]
- Holmqvist, S.; Chutna, O.; Bousset, L.; Aldrin-Kirk, P.; Li, W.; Björklund, T.; Wang, Z.Y.; Roybon, L.; Melki, R.; Li, J.Y. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014, 128, 805–820. [Google Scholar] [CrossRef]
- Reyes, J.F.; Olsson, T.T.; Lamberts, J.T.; Devine, M.J.; Kunath, T.; Brundin, P. A cell culture model for monitoring α-synuclein cell-to-cell transfer. Neurobiol. Dis. 2015, 77, 266–275. [Google Scholar] [CrossRef]
- Peelaerts, W.; Bousset, L.; Van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van den Haute, C.; Melki, R.; Baekelandt, V. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef]
- Brahic, M.; Bousset, L.; Bieri, G.; Melki, R.; Gitler, A.D. Axonal transport and secretion of fibrillar forms of α-synuclein, Aβ42 peptide and HTTExon1. Acta Neuropathol. 2016, 131, 539–548. [Google Scholar] [CrossRef]
- Shimozawa, A.; Ono, M.; Takahara, D.; Tarutani, A.; Imura, S.; Masuda-Suzukake, M.; Higuchi, M.; Yanai, K.; Hisanaga, S.I.; Hasegawa, M. Propagation of pathological α-synuclein in marmoset brain. Acta Neuropathol. Commun. 2017, 5, 12. [Google Scholar] [CrossRef]
- Van der Perren, A.; Gelders, G.; Fenyi, A.; Bousset, L.; Brito, F.; Peelaerts, W.; Van den Haute, C.; Gentleman, S.; Melki, R.; Baekelandt, V. The structural differences between patient-derived α-synuclein strains dictate characteristics of Parkinson’s disease, multiple system atrophy and dementia with Lewy bodies. Acta Neuropathol. 2020, 139, 977–1000. [Google Scholar] [CrossRef] [PubMed]
- Fenyi, A.; Leclair-Visonneau, L.; Clairembault, T.; Coron, E.; Neunlist, M.; Melki, R.; Derkinderen, P.; Bousset, L. Detection of alpha-synuclein aggregates in gastrointestinal biopsies by protein misfolding cyclic amplification. Neurobiol. Dis. 2019, 129, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Candelise, N.; Schmitz, M.; Llorens, F.; Villar-Pique, A.; Cramm, M.; Thom, T.; da Silva Correia, S.M.; da Cunha, J.E.G.; Möbius, W.; Outeiro, T.F.; et al. Seeding variability of different alpha synuclein strains in synucleinopathies. Ann. Neurol. 2019, 85, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Strohäker, T.; Jung, B.C.; Liou, S.H.; Fernandez, C.O.; Riedel, D.; Becker, S.; Halliday, G.M.; Bennati, M.; Kim, W.S.; Lee, S.J.; et al. Structural heterogeneity of alpha-synuclein fibrils amplified from patient brain extracts. Nat. Commun. 2019, 10, 5535. [Google Scholar] [CrossRef] [PubMed]
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020, 578, 273–277. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1991, 1, 213–216. [Google Scholar] [CrossRef]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012, 8, 1–13. [Google Scholar] [CrossRef]
- Braak, H.; de Vos, R.A.I.; Bohl, J.; Del Tredici, K. Gastric α-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 2006, 396, 67–72. [Google Scholar] [CrossRef]
- Smithson, K.G.; MacVicar, B.A.; Hatton, G.I. Polyethylene glycol embedding: A technique compatible with immunocytochemistry, enzyme histochemistry, histofluorescence and intracellular staining. J. Neurosci. Methods 1983, 7, 27–41. [Google Scholar] [CrossRef]
- Mercken, M.; Vandermeeren, M.; Lübke, U.; Six, J.; Boons, J.; Van de Voorde, A.; Martin, J.J.; Gheuens, J. Monoclonal antibodies with selective specificity for Alzheimer tau are directed against phosphatase-sensitive epitopes. Acta Neuropathol. 1992, 84, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E.; Grundke-Iqbal, I.; Iqbal, K. Occurrence of neuropil threads in the senile human brain and in Alzheimer’s disease: A third location of paired helical filaments outside of neurofibrillary tangles and neuritic plaques. Neurosci. Lett. 1986, 65, 351–355. [Google Scholar] [CrossRef]
- Ashford, J.W.; Soultanian, N.S.; Zhang, S.X.; Geddes, J.W. Neuropil threads are collinear with MAP2 immunostaining in neuronal dendrites of Alzheimer brain. J. Neuropathol. Exp. Neurol. 1998, 57, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Del Tredici, K.; Braak, H. To stage, or not to stage. Curr. Opin. Neurobiol. 2020, 61, 10–22. [Google Scholar] [CrossRef]
- Fenyi, A.; Coens, A.; Bellande, T.; Melki, R.; Bousset, L. Assessment of the efficacy of different procedures that remove and disassemble alpha-synuclein, tau and A-beta fibrils from laboratory material and surfaces. Sci. Rep. 2018, 8, 10788. [Google Scholar] [CrossRef]
- Ghee, M.; Melki, R.; Michot, N.; Mallet, J. PA700, the regulatory complex of the 26S proteasome, interferes with alpha-synuclein assembly. FEBS J. 2005, 272, 4023–4033. [Google Scholar] [CrossRef]
- Degorce, F.; Card, A.; Soh, S.; Trinquet, E.; Knapik, G.P.; Xie, B. HTRF: A technology tailored for drug discovery—a review of theoretical aspects and recent applications. Curr. Chem. Genom. 2009, 3, 22–32. [Google Scholar] [CrossRef]
- Schweighauser, M.; Bacioglu, M.; Fritschi, S.K.; Shimshek, D.R.; Kahle, P.J.; Eisele, Y.S.; Jucker, M. Formaldehyde-fixed brain tissue from spontaneously ill α-synuclein transgenic mice induces fatal α-synucleinopathy in transgenic hosts. Acta Neuropathol. 2015, 129, 157–159. [Google Scholar] [CrossRef]
- Brown, P.; Rohwer, R.G.; Green, E.M.; Gajdusek, D.C. Effect of chemicals, heat, and histopathologic processing on high-infectivity hamster-adapted scrapie virus. J. Infect. Dis. 1982, 145, 683–687. [Google Scholar] [CrossRef]
- Brown, P.; Liberski, P.P.; Wolff, A.; Gajdusek, D.C. Resistance of scrapie infectivity to steam autoclaving after formaldehyde fixation and limited survival after ashing at 360 degrees C: Practical and theoretical implications. J. Infect. Dis. 1990, 161, 467–472. [Google Scholar] [CrossRef]
- Taylor, D.M. Inactivation of prions by physical and chemical means. J. Hosp. Infect. 1999, 43, S69–S76. [Google Scholar] [CrossRef]
- Woerman, A.L.; Kazmi, S.A.; Patel, S.; Freyman, Y.; Oehler, A.; Aoyagi, A.; Mordes, D.A.; Halliday, G.M.; Middleton, L.T.; Gentleman, S.M.; et al. MSA prions exhibit remarkable stability and resistance to inactivation. Acta Neuropathol. 2018, 135, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J. Mammalian prions and their wider relevance in neurodegenerative diseases. Nature 2016, 539, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Browning, S.; Mahal, S.P.; Oelschlegel, A.M.; Weissmann, C. Darwinian Evolution of Prions in Cell Culture. Science 2010, 327, 869–872. [Google Scholar] [CrossRef]
- Courte, J.; Bousset, L.; Boxberg, Y.V.; Villard, C.; Melki, R.; Peyrin, J.M. The expression level of alpha-synuclein in different neuronal populations is the primary determinant of its prion-like seeding. Sci. Rep. 2020, 10, 4895. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | f/m | Age | PMI | Brain wt | NFT | Aβ | α-Syn | Diagnoses | |
|---|---|---|---|---|---|---|---|---|---|
| Control | 1 | ♀ | 44 | 18 | 1365 | 0 | 0 | 0 | malignant neoplasm |
| 2 | ♂ | 46 | na | 1480 | 0 | 0 | 0 | sepsis (abscess) | |
| 3 | ♂ | 52 | 41 | 1332 | I | 0 | 0 | malignant neoplasm | |
| 4 | ♀ | 54 | 22 | na | I | 0 | 0 | malignant neoplasm | |
| 5 | ♂ | 56 | 18 | 1352 | I | 0 | 0 | coronary artery disease | |
| 6 | ♀ | 59 | 20 | 1401 | I | 0 | 0 | sepsis, cardiac failure | |
| 7 | ♀ | 62 | 32 | 1364 | I | 0 | 0 | malignant neoplasm | |
| 8 | ♂ | 62 | 7 | 1410 | I | 0 | 0 | cardiac disease | |
| 9 | ♀ | 67 | 4 | 1180 | I | 0 | 0 | acute myeloid leukemia | |
| Lewy Body Disease | 1 | ♂ | 46 | 71 | 1500 | 0 | 0 | 2 | ILBD, pericardial tamponade |
| 2 | ♂ | 50 | 6 | 1250 | 0 | 0 | 2 | ILBD | |
| 3 | ♂ | 58 | 39 | 1438 | I | 0 | 2 | ILBD, myocardial infarction | |
| 4 | ♀ | 59 | 24 | 1100 | 0 | 0 | 1 | ILBD, malignant lymphoma | |
| 5 | ♀ | 63 | 28 | 1150 | I | 0 | 3 | ILBD | |
| 6 | ♂ | 44 | 26 | 1400 | II | 0 | 1 | ILBD, aspiration pneumonia | |
| 7 | ♂ | 82 | 16 | 1192 | III | 1 | 3 | ILBD, malignant neoplasm | |
| 8 | ♂ | 80 | na | 1400 | III | 0 | 3 | ILBD, coronary artery disease | |
| 9 | ♂ | 60 | 32 | 1470 | I | 0 | 5 | PD, bronchopneumonia | |
| 10 | ♀ | 64 | 96 | 1318 | II | 1 | 4 | PD, acute myeloid leukemia | |
| 11 | ♀ | 65 | 8 | 1175 | II | 2 | 4 | PD, pulmonary embolism | |
| 12 | ♂ | 65 | 34 | 1500 | I | 0 | 3 | PD, myocardial infarction | |
| 13 | ♀ | 68 | 10 | 1145 | III | 3 | 5 | PD, myocardial infarction | |
| 14 | ♀ | 72 | 12 | 1214 | I | 2 | 5 | PD, bronchopneumonia | |
| 15 | ♀ | 74 | 42 | 1294 | II | 2 | 5 | PD, AGD, myocardial infarction | |
| 16 | ♀ | 78 | 21 | 1130 | III | 1 | 3 | PD, pneumonia | |
| 17 | ♂ | 84 | 28 | 1200 | II | 2 | 4 | PD, aspiration pneumonia | |
| 18 | ♀ | 84 | 30 | 1045 | V | 3 | 5 | PD, AD, AGD | |
| 19 | ♂ | 77 | na | 1232 | II | 0 | 5 | PD | |
| 20 | ♂ | 74 | 93 | 1344 | V | 4 | 5 | PD, AD, absolute arrhythmia |
| Case | f/m | Age | NFT | Aβ | PD | Diagnoses | ENS | BO | dmX | LRG | LC | SN |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | ♂ | 46 | 0 | 0 | 2 | ILBD | 0 | ++ | + | + | + | 0 |
| 2 | ♂ | 50 | 0 | 0 | 2 | ILBD | 0 | ++ | + | + | + | 0 |
| 3 | ♂ | 58 | I | 0 | 2 | ILBD | 0 | + | + | + | + | 0 |
| 4 | ♀ | 59 | 0 | 0 | 1 | ILBD | 0 | + | 0 | 0 | 0 | 0 |
| 5 | ♀ | 63 | I | 0 | 3 | ILBD | + | ++ | + | + | + | + |
| 6 | ♂ | 44 | II | 0 | 1 | ILBD | 0 | + | 0 | 0 | na | 0 |
| 7 | ♂ | 82 | III | 1 | 3 | ILBD | + | + | ++ * | ++ | + | + |
| 8 | ♂ | 80 | III | 0 | 3 | ILBD | + | ++ | ++ * | + | + | ++ |
| 9 | ♂ | 60 | I | 0 | 5 | PD | ++ | ++ | ++ * | + | [++] | [++] |
| 10 | ♀ | 64 | II | 1 | 4 | PD | 0 | + | + | ++ | [++] | + |
| 11 | ♀ | 65 | II | 2 | 4 | PD | + | ++ | + | + | [+] | [++] |
| 12 | ♂ | 65 | I | 0 | 3 | PD | + | ++ | ++ * | ++ | ++ | + |
| 13 | ♀ | 68 | III | 3 | 5 | PD | ++ | ++ | ++ * | ++ | [++] | [++] |
| 14 | ♀ | 72 | I | 2 | 5 | PD | ++ | ++ | ++ * | + | [+] | [++] |
| 15 | ♀ | 74 | II | 2 | 5 | PD, AGD | + | ++ | ++ * | ++ | [++] | [++] |
| 16 | ♀ | 78 | III | 1 | 3 | PD | + | ++ | + | + | + | [+] |
| 17 | ♂ | 84 | II | 2 | 4 | PD | + | ++ | ++ * | ++ | [++] | [++] |
| 18 | ♀ | 84 | V | 3 | 5 | PD, AD, AGD | + | ++ | ++ * | ++ | [++] | [++] |
| 19 | ♂ | 77 | II | 0 | 5 | PD | + | ++ | ++ * | ++ | [++] | [++] |
| 20 | ♂ | 74 | V | 4 | 5 | PD, AD | + | + | + | + | [+] | [+] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fenyi, A.; Duyckaerts, C.; Bousset, L.; Braak, H.; Del Tredici, K.; Melki, R.; on behalf of the Brainbank Neuro-CEB Neuropathology Network. Seeding Propensity and Characteristics of Pathogenic αSyn Assemblies in Formalin-Fixed Human Tissue from the Enteric Nervous System, Olfactory Bulb, and Brainstem in Cases Staged for Parkinson’s Disease. Cells 2021, 10, 139. https://doi.org/10.3390/cells10010139
Fenyi A, Duyckaerts C, Bousset L, Braak H, Del Tredici K, Melki R, on behalf of the Brainbank Neuro-CEB Neuropathology Network. Seeding Propensity and Characteristics of Pathogenic αSyn Assemblies in Formalin-Fixed Human Tissue from the Enteric Nervous System, Olfactory Bulb, and Brainstem in Cases Staged for Parkinson’s Disease. Cells. 2021; 10(1):139. https://doi.org/10.3390/cells10010139
Chicago/Turabian StyleFenyi, Alexis, Charles Duyckaerts, Luc Bousset, Heiko Braak, Kelly Del Tredici, Ronald Melki, and on behalf of the Brainbank Neuro-CEB Neuropathology Network. 2021. "Seeding Propensity and Characteristics of Pathogenic αSyn Assemblies in Formalin-Fixed Human Tissue from the Enteric Nervous System, Olfactory Bulb, and Brainstem in Cases Staged for Parkinson’s Disease" Cells 10, no. 1: 139. https://doi.org/10.3390/cells10010139
APA StyleFenyi, A., Duyckaerts, C., Bousset, L., Braak, H., Del Tredici, K., Melki, R., & on behalf of the Brainbank Neuro-CEB Neuropathology Network. (2021). Seeding Propensity and Characteristics of Pathogenic αSyn Assemblies in Formalin-Fixed Human Tissue from the Enteric Nervous System, Olfactory Bulb, and Brainstem in Cases Staged for Parkinson’s Disease. Cells, 10(1), 139. https://doi.org/10.3390/cells10010139