
Biochemistry, Pathophysiology, and Regulation of Linear Ubiquitination: Intricate Regulation by Coordinated Functions of the Associated Ligase and Deubiquitinase


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
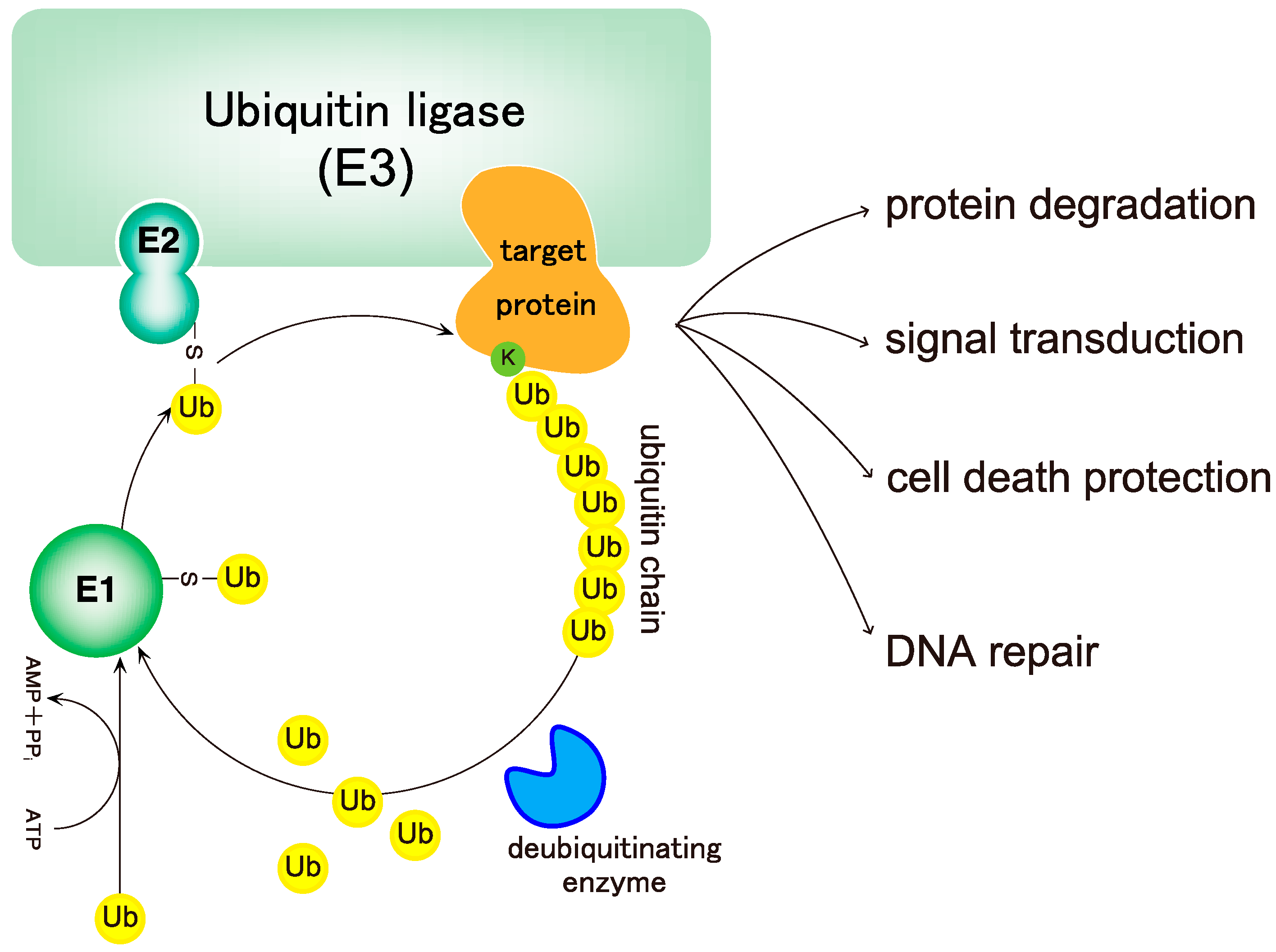
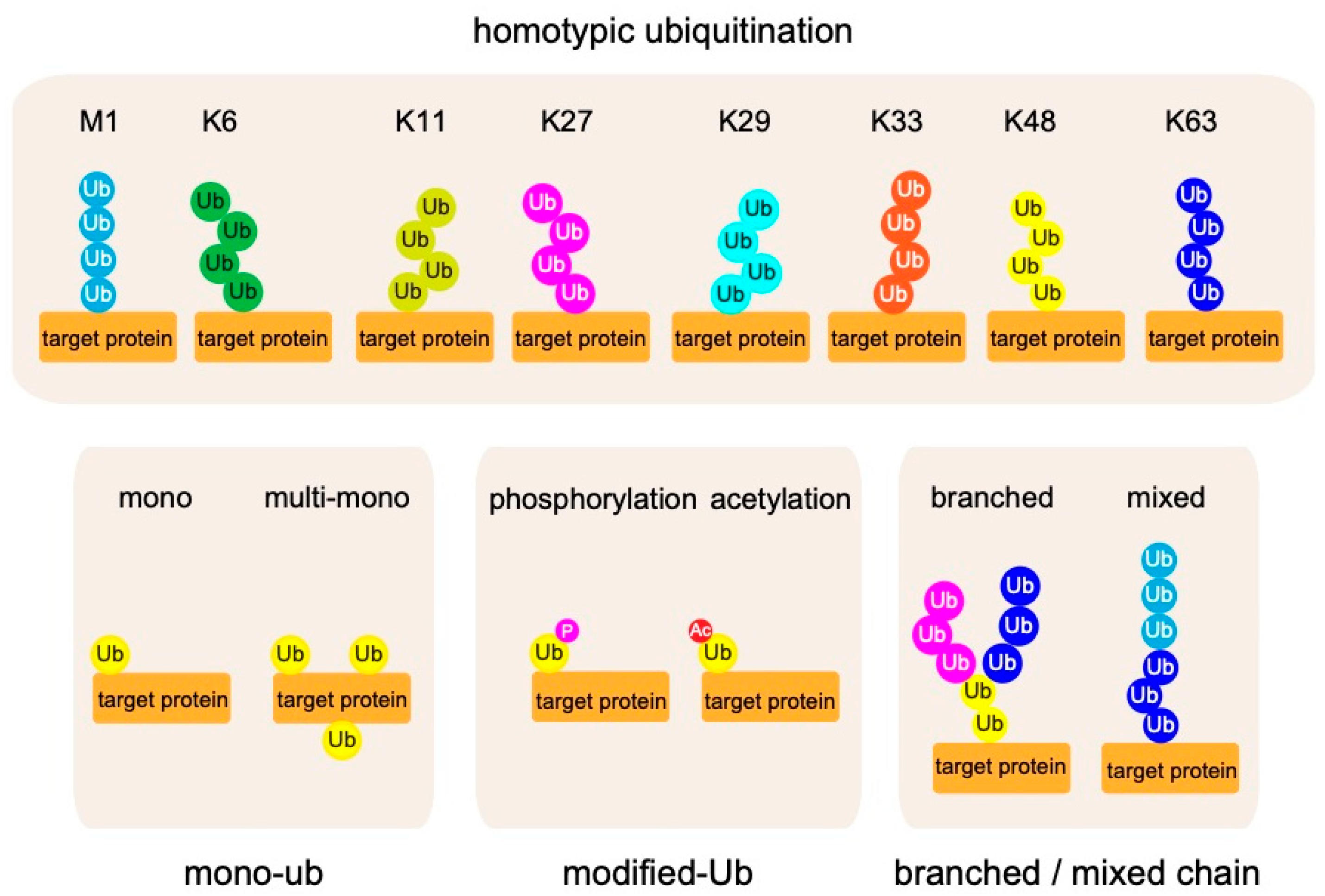
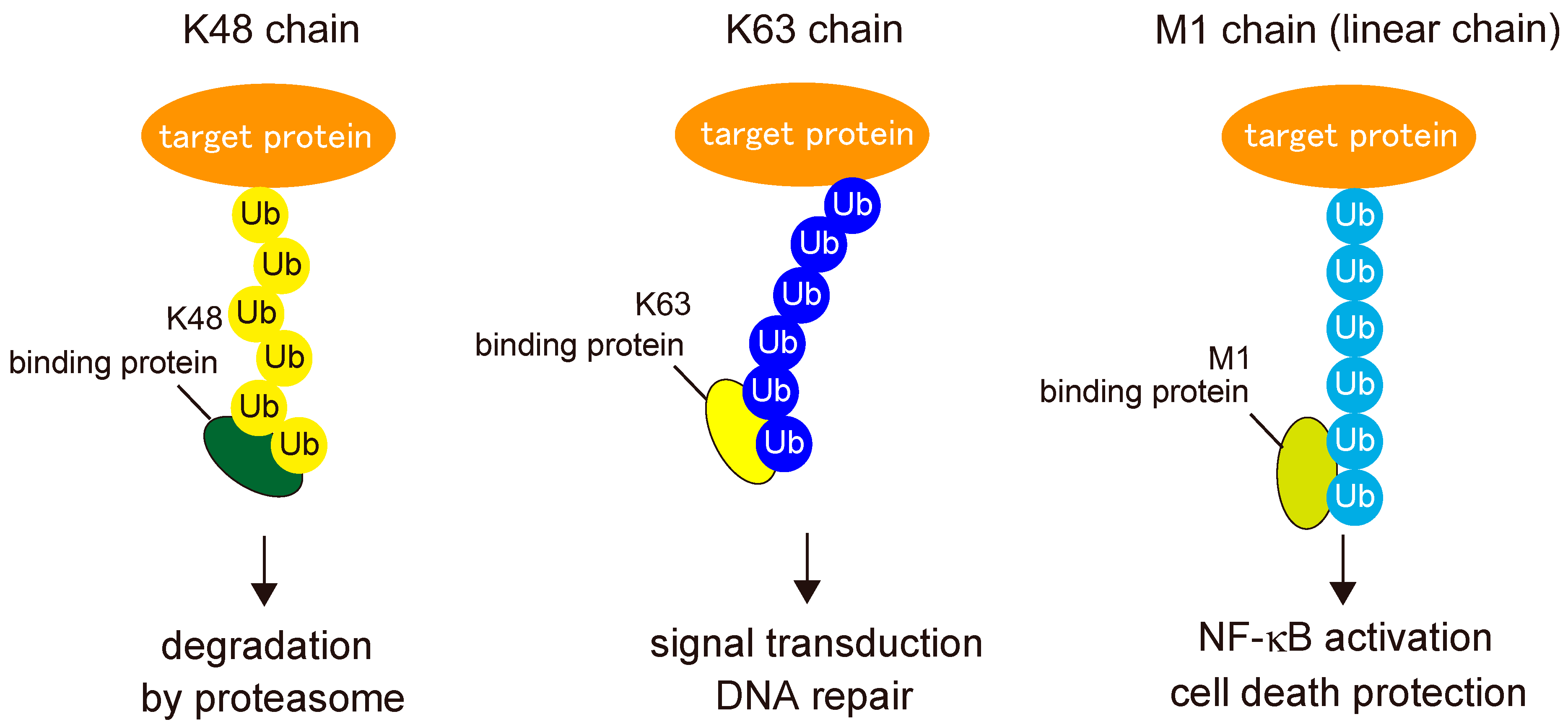
2. Biochemistry of Linear Ubiquitin Chains
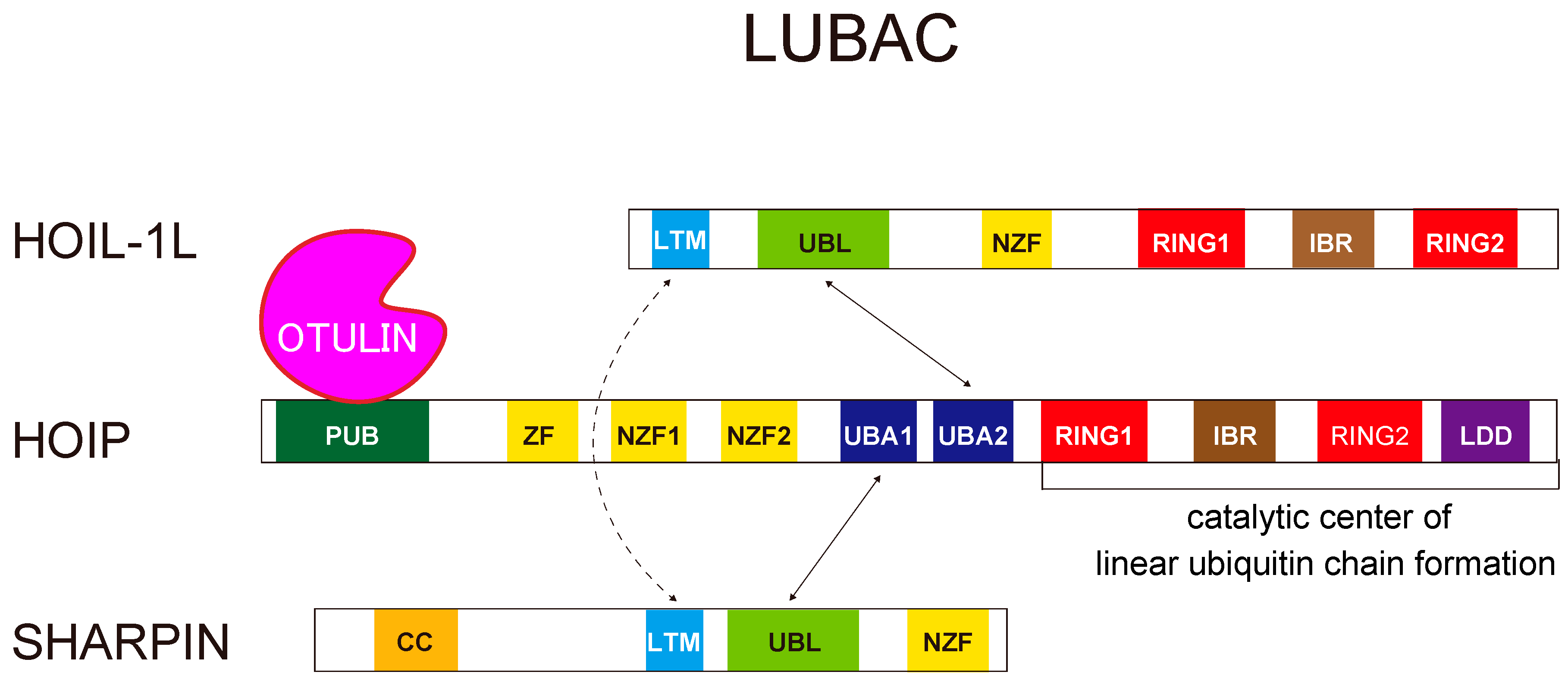
2.1. Linear Ubiquitin Chains Are Generated Specifically by the LUBAC Ligase Complex
2.2. Readers for Linear Ubiquitin Chains
2.3. Deubiquitinating Enzymes of Linear Ubiquitin Chains
3. Structural Insights Regarding the LUBAC Ligase Complex
4. Physiological Functions of Linear Ubiquitin Chains
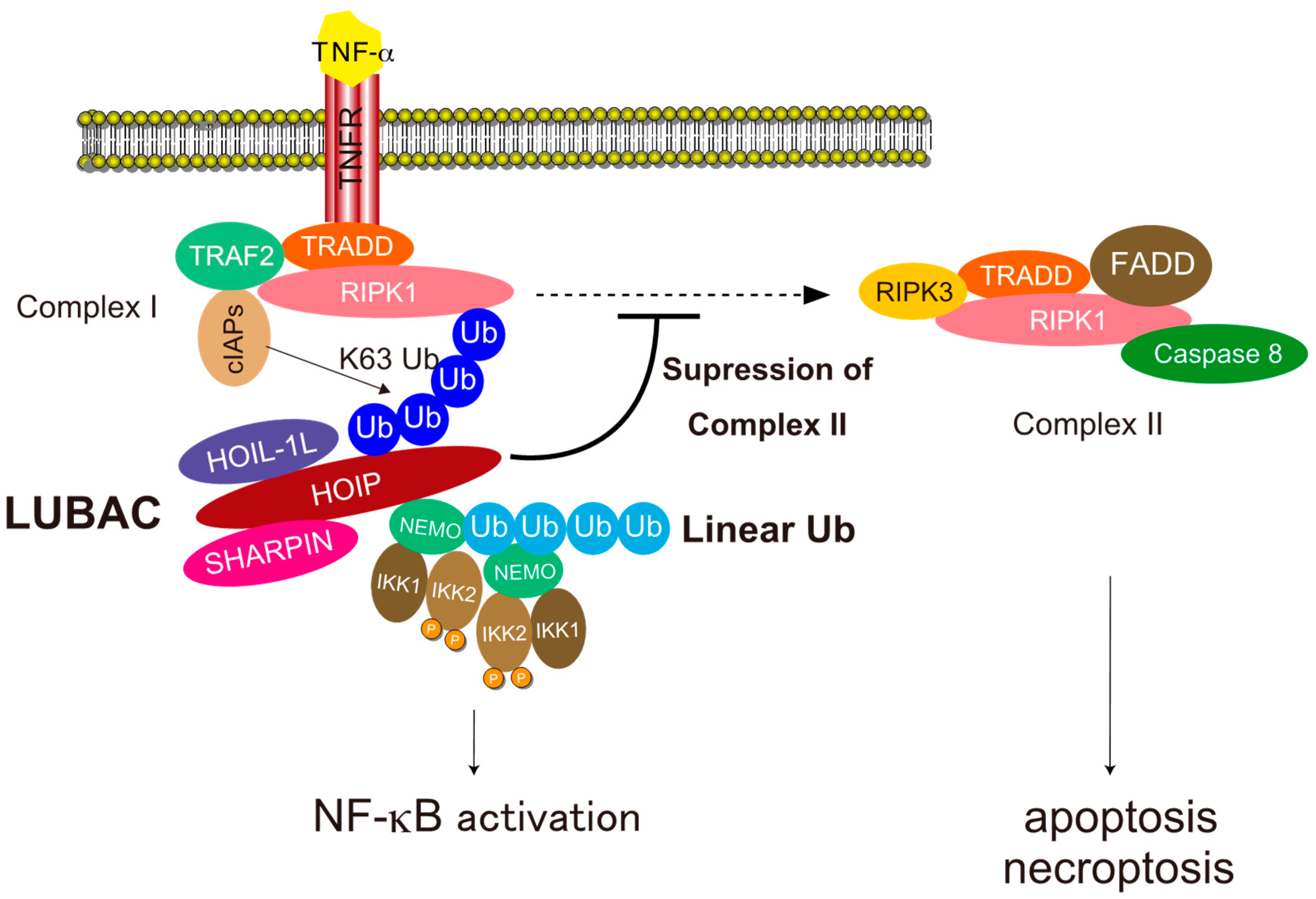
4.1. NF-κB Activation
4.2. Cell Death Protection
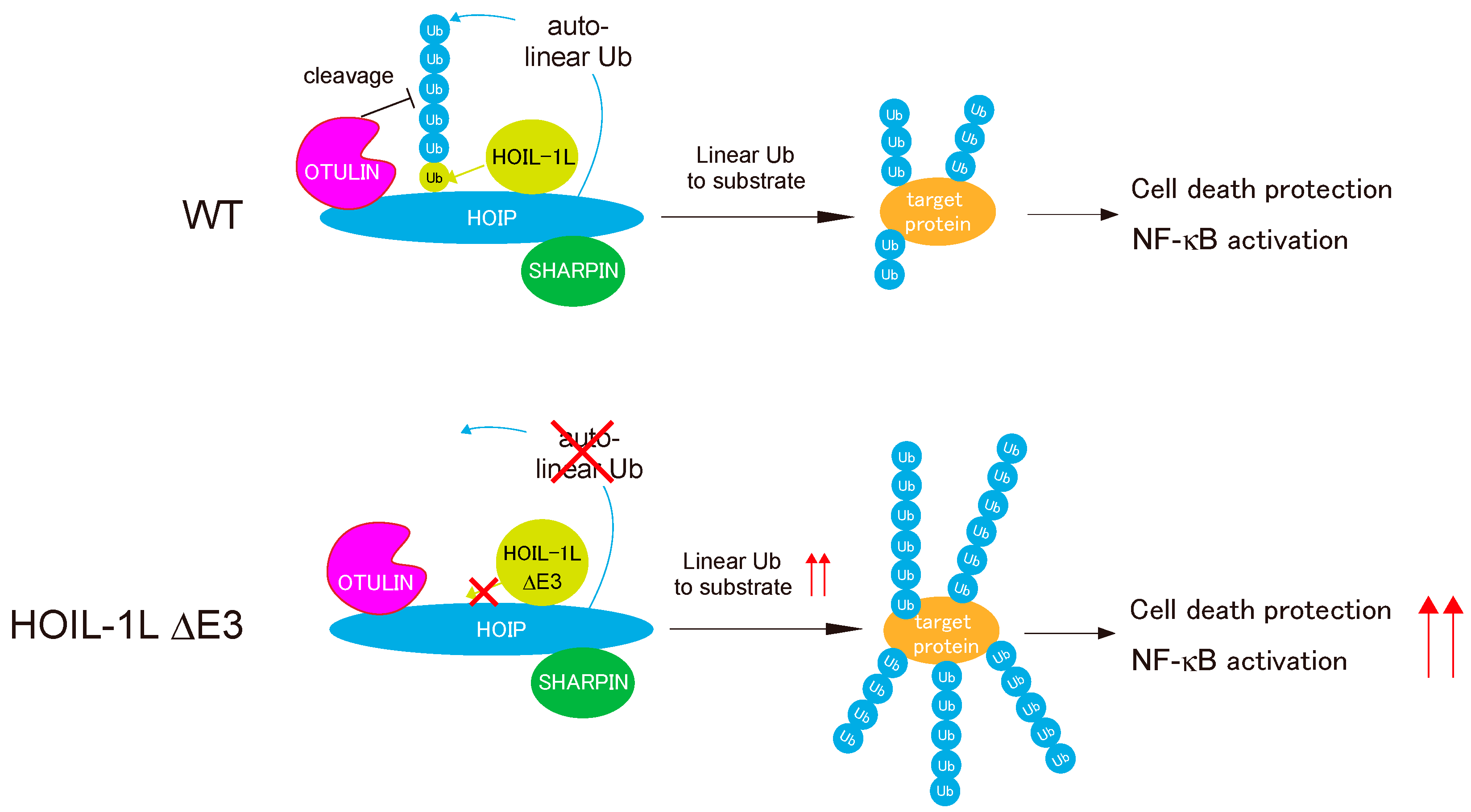
5. Regulation of Linear Ubiquitination Activity of LUBAC
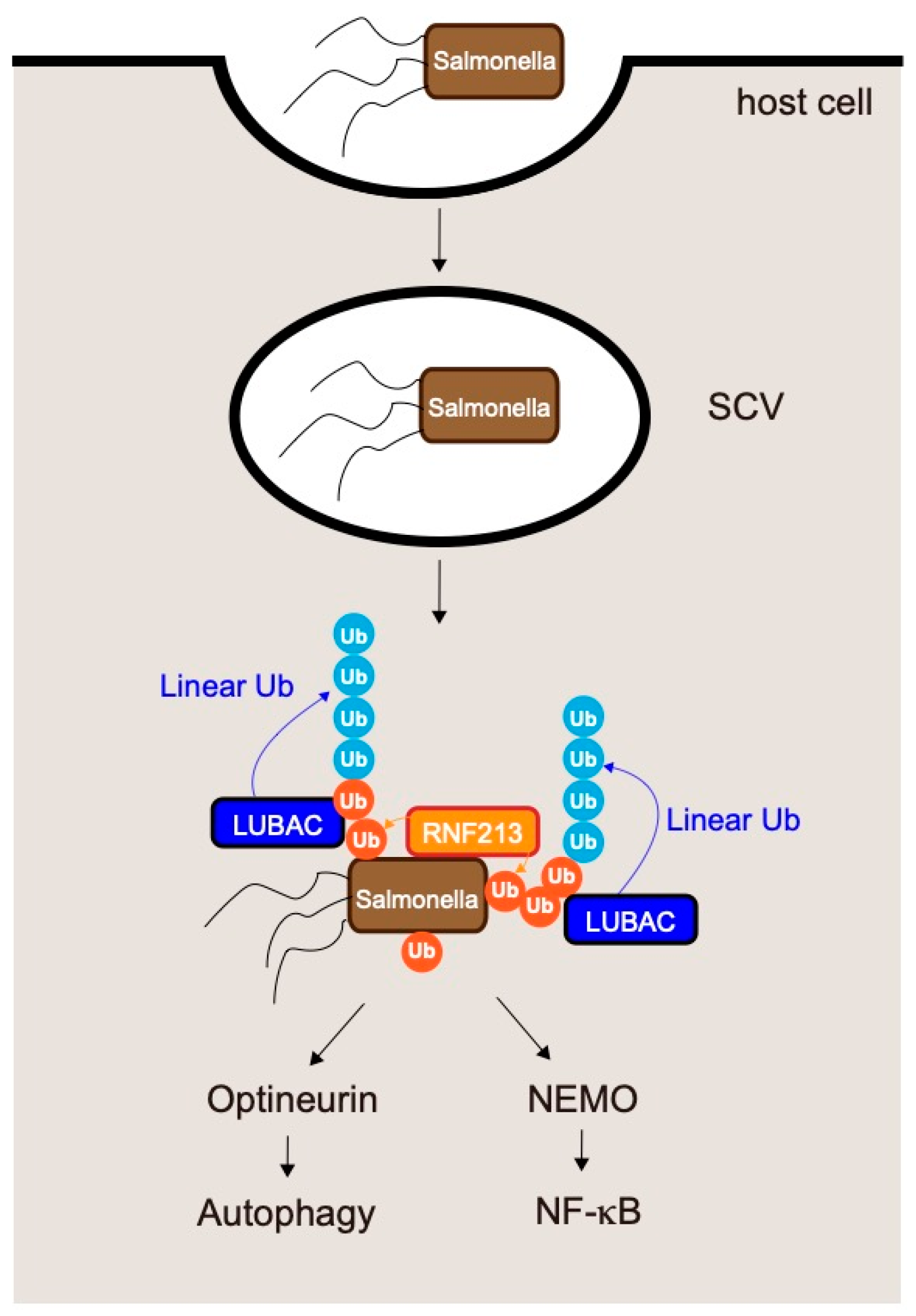
6. LUBAC and Infections
6.1. LUBAC and Salmonella Infections
6.2. Suppression of Linear Ubiquitination by Pathogens
7. Linear Ubiquitination in Diseases
7.1. HOIP Deficiency in Mice and Human
7.2. HOIL-1L Deficiency in Mice and Humans
7.3. SHARPIN Deficiency in Mice and Humans
7.4. OTULIN Deficiency
7.5. Augmentation of LUBAC Activity in Cancer
8. Therapeutic Approaches to Targeting LUBAC
8.1. Cancer Therapy via Attenuation of LUBAC
8.2. Treatment of Infectious Disease via Augmentation of LUBAC
9. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| LUBAC | Linear ubiquitin chain assembly complex |
| Met: | methionine |
| NF-κB | Nuclear factor-κB |
| HOIP | HOIL-1L interacting protein |
| HOIL-1L | large isoform of heme-oxidized iron regulatory protein2 (IRP2) ubiquitin ligase 1 L |
| IRP2: | iron regulatory protein2 |
| SHARPIN | SHANK-associated RH domain-interacting protein |
| SHANK | SH3 and multiple ankyrin repeat domains protein |
| DUBs | deubiquitinating enzymes |
| OTULIN | OTU deubiquitinase with linear linkage specificity |
| CYLD | cylindromatosis |
| RING | Really interesting new gene |
| RBR | RING-in-between-RING |
| E1 | ubiquitin activating enzyme |
| E2 | ubiquitin-conjugating enzyme |
| E3 | ubiquitin ligase |
| K | lysine |
| Cys | cysteine |
| UBAN | UBD in ABIN proteins and NEMO |
| NEMO | NF-κB-essential modulator |
| OPTN | ABIN: A20-binding inhibitors of NF-κB |
| NZF | Npl4-type zinc finger |
| ZF7 | seventh zinc finger |
| TNFAIP3 | tumor necrosis factor α-induced protein 3 |
| IKK | IκB kinase |
| SNPs | Single-nucleotide polymorphisms |
| SLE | systemic lupus erythematous |
| ALS | amyotrophic lateral sclerosis |
| POAG | primary open-angle glaucoma |
| SPATA2 | spermatogenesis-associated 2 |
| RHD | Rel homology domain |
| TNF-α | tumor necrosis factor α |
| CD40L | CD40 ligand |
| TLRs | Toll-like receptors |
| TNFR | TNF-receptor |
| DD | death domain |
| TRADD | TNFR-associated death domain |
| RIPK1 | receptor interacting serine/threonine-protein kinase 1 |
| TRAF2 | TNF-receptor associated factor 2 |
| cIAP1/2 | cellular inhibitor of apoptosis proteins 1 and 2 |
| IκBα | inhibitor of κBα |
| FADD | FAS-associated death domain protein |
| LDD | Linear ubiquitin chain determining domain |
| UBL | ubiquitin-like |
| UBA | ubiquitin-associated |
| LTMs | LUBAC-tethering motifs |
| PUB | PNGase/UBA or UBX |
| Ser | Serine |
| Thr | Threonine |
| RNF213 | ring finger protein 213 |
| MycBP2 | MYC Binding Protein 2 |
| LPS | ipopolysaccharide |
| MALT1 | mucosa-associated lymphoid tissue lymphoma translocation gene 1 |
| Arg | arginine |
| Gly | glycine |
| DLBCL | diffuse large B-cell lymphoma |
| ABC-DLBCL | activated B-cell–like DLBCL |
| SCVs: | Salmonella-containing vacuoles |
| cpdm | chronic proliferative dermatitis in mice |
| ORAS | OTULIN-related autoinflammatory syndrome |
| GCB-DLBCL | germinal center B-cell–like DLBCL |
| RNA-seq | RNA sequencing |
References
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system for protein degradation. Annu. Rev. Biochem. 1992, 61, 761–807. [Google Scholar] [CrossRef] [PubMed]
- Ciehanover, A.; Hod, Y.; Hershko, A. A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes. Biochem. Biophys. Res. Commun. 1978, 81, 1100–1105. [Google Scholar] [CrossRef]
- Spence, J.; Sadis, S.; Haas, A.L.; Finley, D. A ubiquitin mutant with specific defects in DNA repair and multiubiquitination. Mol. Cell. Biol. 1995, 15, 1265–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, D.; Riezman, H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 2007, 315, 201–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickliffe, K.; Williamson, A.; Jin, L.; Rape, M. The multiple layers of ubiquitin-dependent cell cycle control. Chem. Rev. 2009, 109, 1537–1548. [Google Scholar] [CrossRef] [Green Version]
- Zinngrebe, J.; Montinaro, A.; Peltzer, N.; Walczak, H. Ubiquitin in the immune system. EMBO Rep. 2014, 15, 28–45. [Google Scholar] [CrossRef]
- Peng, J.; Schwartz, D.; Elias, J.E.; Thoreen, C.C.; Cheng, D.; Marsischky, G.; Roelofs, J.; Finley, D.; Gygi, S.P. A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 2003, 21, 921–926. [Google Scholar] [CrossRef]
- Kirisako, T.; Kamei, K.; Murata, S.; Kato, M.; Fukumoto, H.; Kanie, M.; Sano, S.; Tokunaga, F.; Tanaka, K.; Iwai, K. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 2006, 25, 4877–4887. [Google Scholar] [CrossRef]
- Iwai, K. LUBAC-mediated linear ubiquitination: A crucial regulator of immune signaling. Proc. Jpn. Acad. Ser. B. Phys. Biol. Sci. 2021, 97, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Iwai, K.; Fujita, H.; Sasaki, Y. Linear ubiquitin chains: NF-κB signalling, cell death and beyond. Nat. Rev. Mol. Cell. Biol. 2014, 15, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Iwai, K. Roles of linear ubiquitinylation, a crucial regulator of NF-κB and cell death, in the immune system. Immunol. Rev. 2015, 266, 175–189. [Google Scholar] [CrossRef] [Green Version]
- Haakonsen, D.L.; Rape, M. Branching Out: Improved Signaling by Heterotypic Ubiquitin Chains. Trends Cell Biol. 2019, 29, 704–716. [Google Scholar] [CrossRef]
- Koyano, F.; Matsuda, N. Molecular mechanisms underlying PINK1 and Parkin catalyzed ubiquitylation of substrates on damaged mitochondria. Biochim. Biophys. Acta 2015, 1853, 2791–2796. [Google Scholar] [CrossRef] [Green Version]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res 2016, 26, 399–422. [Google Scholar] [CrossRef] [Green Version]
- Ohtake, F.; Saeki, Y.; Sakamoto, K.; Ohtake, K.; Nishikawa, H.; Tsuchiya, H.; Ohta, T.; Tanaka, K.; Kanno, J. Ubiquitin acetylation inhibits polyubiquitin chain elongation. EMBO Rep. 2015, 16, 192–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtake, F.; Saeki, Y.; Ishido, S.; Kanno, J.; Tanaka, K. The K48-K63 Branched Ubiquitin Chain Regulates NF-κB Signaling. Mol. Cell 2016, 64, 251–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtake, F.; Tsuchiya, H. The emerging complexity of ubiquitin architecture. J. Biochem. 2017, 161, 125–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herhaus, L.; Dikic, I. Expanding the ubiquitin code through post-translational modification. EMBO Rep. 2015, 16, 1071–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwai, K. Discovery of linear ubiquitination, a crucial regulator for immune signaling and cell death. FEBS J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Fuseya, Y.; Fujita, H.; Kim, M.; Ohtake, F.; Nishide, A.; Sasaki, K.; Saeki, Y.; Tanaka, K.; Takahashi, R.; Iwai, K. The HOIL-1L ligase modulates immune signalling and cell death via monoubiquitination of LUBAC. Nat. Cell Biol. 2020, 22, 663–673. [Google Scholar] [CrossRef]
- Tokunaga, F.; Nakagawa, T.; Nakahara, M.; Saeki, Y.; Taniguchi, M.; Sakata, S.; Tanaka, K.; Nakano, H.; Iwai, K. SHARPIN is a component of the NF-κB-activating linear ubiquitin chain assembly complex. Nature 2011, 471, 633–636. [Google Scholar] [CrossRef]
- Ikeda, F.; Deribe, Y.L.; Skanland, S.S.; Stieglitz, B.; Grabbe, C.; Franz-Wachtel, M.; van Wijk, S.J.; Goswami, P.; Nagy, V.; Terzic, J.; et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB activity and apoptosis. Nature 2011, 471, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, B.; Cordier, S.M.; Schmukle, A.C.; Emmerich, C.H.; Rieser, E.; Haas, T.L.; Webb, A.I.; Rickard, J.A.; Anderton, H.; Wong, W.W.; et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 2011, 471, 591–596. [Google Scholar] [CrossRef]
- Reiter, K.H.; Klevit, R.E. Characterization of RING-Between-RING E3 Ubiquitin Transfer Mechanisms. Methods Mol. Biol. 2018, 1844, 3–17. [Google Scholar] [CrossRef]
- Smit, J.J.; Monteferrario, D.; Noordermeer, S.M.; van Dijk, W.J.; van der Reijden, B.A.; Sixma, T.K. The E3 ligase HOIP specifies linear ubiquitin chain assembly through its RING-IBR-RING domain and the unique LDD extension. EMBO J. 2012, 31, 3833–3844. [Google Scholar] [CrossRef] [Green Version]
- Stieglitz, B.; Rana, R.R.; Koliopoulos, M.G.; Morris-Davies, A.C.; Schaeffer, V.; Christodoulou, E.; Howell, S.; Brown, N.R.; Dikic, I.; Rittinger, K. Structural basis for ligase-specific conjugation of linear ubiquitin chains by HOIP. Nature 2013, 503, 422–426. [Google Scholar] [CrossRef]
- Oikawa, D.; Sato, Y.; Ito, H.; Tokunaga, F. Linear Ubiquitin Code: Its Writer, Erasers, Decoders, Inhibitors, and Implications in Disorders. Int. J. Mol. Sci. 2020, 21, 3381. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Fujita, H.; Yoshikawa, A.; Yamashita, M.; Yamagata, A.; Kaiser, S.E.; Iwai, K.; Fukai, S. Specific recognition of linear ubiquitin chains by the Npl4 zinc finger (NZF) domain of the HOIL-1L subunit of the linear ubiquitin chain assembly complex. Proc. Natl. Acad. Sci. USA 2011, 108, 20520–20525. [Google Scholar] [CrossRef] [Green Version]
- Tokunaga, F.; Nishimasu, H.; Ishitani, R.; Goto, E.; Noguchi, T.; Mio, K.; Kamei, K.; Ma, A.; Iwai, K.; Nureki, O. Specific recognition of linear polyubiquitin by A20 zinc finger 7 is involved in NF-κB regulation. EMBO J 2012, 31, 3856–3870. [Google Scholar] [CrossRef] [PubMed]
- Verhelst, K.; Carpentier, I.; Kreike, M.; Meloni, L.; Verstrepen, L.; Kensche, T.; Dikic, I.; Beyaert, R. A20 inhibits LUBAC-mediated NF-κB activation by binding linear polyubiquitin chains via its zinc finger 7. EMBO J 2012, 31, 3845–3855. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.C.; Lin, S.C.; Rospigliosi, C.C.; Conze, D.B.; Wu, C.J.; Ashwell, J.D.; Eliezer, D.; Wu, H. Structural basis for recognition of diubiquitins by NEMO. Mol Cell 2009, 33, 602–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwai, K.; Tokunaga, F. Linear polyubiquitination: A new regulator of NF-κB activation. EMBO Rep. 2009, 10, 706–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, H.; Rahighi, S.; Akita, M.; Kato, R.; Sasaki, Y.; Wakatsuki, S.; Iwai, K. Mechanism underlying IκB kinase activation mediated by the linear ubiquitin chain assembly complex. Mol. Cell. Biol. 2014, 34, 1322–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verstrepen, L.; Carpentier, I.; Beyaert, R. The biology of A20-binding inhibitors of NF-κB activation (ABINs). Adv. Exp. Med. Biol. 2014, 809, 13–31. [Google Scholar] [CrossRef]
- Lin, S.M.; Lin, S.C.; Hong, J.Y.; Su, T.W.; Kuo, B.J.; Chang, W.H.; Tu, Y.F.; Lo, Y.C. Structural Insights into Linear Tri-ubiquitin Recognition by A20-Binding Inhibitor of NF-κB, ABIN-2. Structure 2017, 25, 66–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herhaus, L.; van den Bedem, H.; Tang, S.; Maslennikov, I.; Wakatsuki, S.; Dikic, I.; Rahighi, S. Molecular Recognition of M1-Linked Ubiquitin Chains by Native and Phosphorylated UBAN Domains. J. Mol. Biol. 2019, 431, 3146–3156. [Google Scholar] [CrossRef] [Green Version]
- Shamilov, R.; Aneskievich, B.J. TNIP1 in Autoimmune Diseases: Regulation of Toll-like Receptor Signaling. J. Immunol. Res. 2018, 2018, 3491269. [Google Scholar] [CrossRef]
- Ellinghaus, E.; Stuart, P.E.; Ellinghaus, D.; Nair, R.P.; Debrus, S.; Raelson, J.V.; Belouchi, M.; Tejasvi, T.; Li, Y.; Tsoi, L.C.; et al. Genome-wide meta-analysis of psoriatic arthritis identifies susceptibility locus at REL. J. Investig. Dermatol. 2012, 132, 1133–1140. [Google Scholar] [CrossRef] [Green Version]
- Chandran, V. The genetics of psoriasis and psoriatic arthritis. Clin. Rev. Allergy Immunol. 2013, 44, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Liu, H.; Qu, L.; Fu, X.; Yu, Y.; Yu, G.; Tian, H.; Yu, Y.; Sun, D.; Peng, J.; et al. Investigation of 20 non-HLA (human leucocyte antigen) psoriasis susceptibility loci in Chinese patients with psoriatic arthritis and psoriasis vulgaris. Br. J. Dermatol. 2013, 168, 1060–1065. [Google Scholar] [CrossRef]
- Zhou, J.; Wu, R.; High, A.A.; Slaughter, C.A.; Finkelstein, D.; Rehg, J.E.; Redecke, V.; Hacker, H. A20-binding inhibitor of NF-κB (ABIN1) controls Toll-like receptor-mediated CCAAT/enhancer-binding protein beta activation and protects from inflammatory disease. Proc. Natl. Acad. Sci. USA 2011, 108, E998–E1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanda, S.K.; Venigalla, R.K.; Ordureau, A.; Patterson-Kane, J.C.; Powell, D.W.; Toth, R.; Arthur, J.S.; Cohen, P. Polyubiquitin binding to ABIN1 is required to prevent autoimmunity. J. Exp. Med. 2011, 208, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, A.; Ito, S.; Furukawa, H.; Hayashi, T.; Goto, D.; Matsumoto, I.; Kusaoi, M.; Ohashi, J.; Graham, R.R.; Matsuta, K.; et al. Association of TNFAIP3 interacting protein 1, TNIP1 with systemic lupus erythematosus in a Japanese population: A case-control association study. Arthritis Res. Ther. 2010, 12, R174. [Google Scholar] [CrossRef] [Green Version]
- Gateva, V.; Sandling, J.K.; Hom, G.; Taylor, K.E.; Chung, S.A.; Sun, X.; Ortmann, W.; Kosoy, R.; Ferreira, R.C.; Nordmark, G.; et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat. Genet. 2009, 41, 1228–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.W.; Zheng, H.F.; Cui, Y.; Sun, L.D.; Ye, D.Q.; Hu, Z.; Xu, J.H.; Cai, Z.M.; Huang, W.; Zhao, G.P.; et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat. Genet. 2009, 41, 1234–1237. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef]
- Rezaie, T.; Child, A.; Hitchings, R.; Brice, G.; Miller, L.; Coca-Prados, M.; Heon, E.; Krupin, T.; Ritch, R.; Kreutzer, D.; et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science 2002, 295, 1077–1079. [Google Scholar] [CrossRef]
- Nakazawa, S.; Oikawa, D.; Ishii, R.; Ayaki, T.; Takahashi, H.; Takeda, H.; Ishitani, R.; Kamei, K.; Takeyoshi, I.; Kawakami, H.; et al. Linear ubiquitination is involved in the pathogenesis of optineurin-associated amyotrophic lateral sclerosis. Nat. Commun. 2016, 7, 12547. [Google Scholar] [CrossRef] [Green Version]
- Komander, D.; Clague, M.J.; Urbe, S. Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef]
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef] [Green Version]
- Lork, M.; Verhelst, K.; Beyaert, R. CYLD, A20 and OTULIN deubiquitinases in NF-κB signaling and cell death: So similar, yet so different. Cell Death Differ. 2017, 24, 1172–1183. [Google Scholar] [CrossRef]
- Takiuchi, T.; Nakagawa, T.; Tamiya, H.; Fujita, H.; Sasaki, Y.; Saeki, Y.; Takeda, H.; Sawasaki, T.; Buchberger, A.; Kimura, T.; et al. Suppression of LUBAC-mediated linear ubiquitination by a specific interaction between LUBAC and the deubiquitinases CYLD and OTULIN. Genes Cells 2014, 19, 254–272. [Google Scholar] [CrossRef] [Green Version]
- Elliott, P.R.; Komander, D. Regulation of Met1-linked polyubiquitin signalling by the deubiquitinase OTULIN. FEBS J. 2016, 283, 39–53. [Google Scholar] [CrossRef] [Green Version]
- Schaeffer, V.; Akutsu, M.; Olma, M.H.; Gomes, L.C.; Kawasaki, M.; Dikic, I. Binding of OTULIN to the PUB domain of HOIP controls NF-κB signaling. Mol. Cell 2014, 54, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Elliott, P.R.; Nielsen, S.V.; Marco-Casanova, P.; Fiil, B.K.; Keusekotten, K.; Mailand, N.; Freund, S.M.; Gyrd-Hansen, M.; Komander, D. Molecular basis and regulation of OTULIN-LUBAC interaction. Mol. Cell 2014, 54, 335–348. [Google Scholar] [CrossRef] [Green Version]
- Elliott, P.R.; Leske, D.; Hrdinka, M.; Bagola, K.; Fiil, B.K.; McLaughlin, S.H.; Wagstaff, J.; Volkmar, N.; Christianson, J.C.; Kessler, B.M.; et al. SPATA2 Links CYLD to LUBAC, Activates CYLD, and Controls LUBAC Signaling. Mol. Cell 2016, 63, 990–1005. [Google Scholar] [CrossRef] [Green Version]
- Kupka, S.; De Miguel, D.; Draber, P.; Martino, L.; Surinova, S.; Rittinger, K.; Walczak, H. SPATA2-Mediated Binding of CYLD to HOIP Enables CYLD Recruitment to Signaling Complexes. Cell Rep. 2016, 16, 2271–2280. [Google Scholar] [CrossRef] [Green Version]
- Schlicher, L.; Wissler, M.; Preiss, F.; Brauns-Schubert, P.; Jakob, C.; Dumit, V.; Borner, C.; Dengjel, J.; Maurer, U. SPATA2 promotes CYLD activity and regulates TNF-induced NF-κB signaling and cell death. EMBO Rep. 2016, 17, 1485–1497. [Google Scholar] [CrossRef] [Green Version]
- Wagner, S.A.; Satpathy, S.; Beli, P.; Choudhary, C. SPATA2 links CYLD to the TNF-α receptor signaling complex and modulates the receptor signaling outcomes. EMBO J. 2016, 35, 1868–1884. [Google Scholar] [CrossRef] [Green Version]
- Heger, K.; Wickliffe, K.E.; Ndoja, A.; Zhang, J.; Murthy, A.; Dugger, D.L.; Maltzman, A.; de Sousa, E.M.F.; Hung, J.; Zeng, Y.; et al. OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature 2018, 559, 120–124. [Google Scholar] [CrossRef]
- Bignell, G.R.; Warren, W.; Seal, S.; Takahashi, M.; Rapley, E.; Barfoot, R.; Green, H.; Brown, C.; Biggs, P.J.; Lakhani, S.R.; et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat. Genet. 2000, 25, 160–165. [Google Scholar] [CrossRef]
- Regamey, A.; Hohl, D.; Liu, J.W.; Roger, T.; Kogerman, P.; Toftgard, R.; Huber, M. The tumor suppressor CYLD interacts with TRIP and regulates negatively nuclear factor κB activation by tumor necrosis factor. J. Exp. Med. 2003, 198, 1959–1964. [Google Scholar] [CrossRef] [Green Version]
- Trompouki, E.; Hatzivassiliou, E.; Tsichritzis, T.; Farmer, H.; Ashworth, A.; Mosialos, G. CYLD is a deubiquitinating enzyme that negatively regulates NF-κB activation by TNFR family members. Nature 2003, 424, 793–796. [Google Scholar] [CrossRef]
- Draber, P.; Kupka, S.; Reichert, M.; Draberova, H.; Lafont, E.; de Miguel, D.; Spilgies, L.; Surinova, S.; Taraborrelli, L.; Hartwig, T.; et al. LUBAC-Recruited CYLD and A20 Regulate Gene Activation and Cell Death by Exerting Opposing Effects on Linear Ubiquitin in Signaling Complexes. Cell Rep. 2015, 13, 2258–2272. [Google Scholar] [CrossRef] [Green Version]
- Fujita, H.; Tokunaga, A.; Shimizu, S.; Whiting, A.L.; Aguilar-Alonso, F.; Takagi, K.; Walinda, E.; Sasaki, Y.; Shimokawa, T.; Mizushima, T.; et al. Cooperative Domain Formation by Homologous Motifs in HOIL-1L and SHARPIN Plays A Crucial Role in LUBAC Stabilization. Cell Rep. 2018, 23, 1192–1204. [Google Scholar] [CrossRef]
- Tokunaga, F.; Sakata, S.; Saeki, Y.; Satomi, Y.; Kirisako, T.; Kamei, K.; Nakagawa, T.; Kato, M.; Murata, S.; Yamaoka, S.; et al. Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nat. Cell Biol. 2009, 11, 123–132. [Google Scholar] [CrossRef]
- Iwai, K. Diverse ubiquitin signaling in NF-κB activation. Trends Cell Biol. 2012, 22, 355–364. [Google Scholar] [CrossRef]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [Green Version]
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of multiple genes cause deregulation of NF-κB in diffuse large B-cell lymphoma. Nature 2009, 459, 717–721. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.E.; Brown, K.D.; Siebenlist, U.; Staudt, L.M. Constitutive nuclear factor kaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J. Exp. Med. 2001, 194, 1861–1874. [Google Scholar] [CrossRef]
- Shimizu, S.; Fujita, H.; Sasaki, Y.; Tsuruyama, T.; Fukuda, K.; Iwai, K. Differential Involvement of the Npl4 Zinc Finger Domains of SHARPIN and HOIL-1L in Linear Ubiquitin Chain Assembly Complex-Mediated Cell Death Protection. Mol. Cell. Biol. 2016, 36, 1569–1583. [Google Scholar] [CrossRef] [Green Version]
- Rahighi, S.; Ikeda, F.; Kawasaki, M.; Akutsu, M.; Suzuki, N.; Kato, R.; Kensche, T.; Uejima, T.; Bloor, S.; Komander, D.; et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-κB activation. Cell 2009, 136, 1098–1109. [Google Scholar] [CrossRef] [Green Version]
- Walczak, H. TNF and ubiquitin at the crossroads of gene activation, cell death, inflammation, and cancer. Immunol. Rev. 2011, 244, 9–28. [Google Scholar] [CrossRef]
- Peltzer, N.; Darding, M.; Walczak, H. Holding RIPK1 on the Ubiquitin Leash in TNFR1 Signaling. Trends Cell Biol. 2016, 26, 445–461. [Google Scholar] [CrossRef]
- Emmerich, C.H.; Ordureau, A.; Strickson, S.; Arthur, J.S.; Pedrioli, P.G.; Komander, D.; Cohen, P. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc. Natl. Acad. Sci. USA 2013, 110, 15247–15252. [Google Scholar] [CrossRef] [Green Version]
- Kelsall, I.R.; Zhang, J.; Knebel, A.; Arthur, J.S.C.; Cohen, P. The E3 ligase HOIL-1 catalyses ester bond formation between ubiquitin and components of the Myddosome in mammalian cells. Proc. Natl. Acad. Sci. USA 2019, 116, 13293–13298. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P.; Kelsall, I.R.; Nanda, S.K.; Zhang, J. HOIL-1, an atypical E3 ligase that controls MyD88 signalling by forming ester bonds between ubiquitin and components of the Myddosome. Adv. Biol. Regul. 2020, 75, 100666. [Google Scholar] [CrossRef]
- Otten, E.G.; Werner, E.; Crespillo-Casado, A.; Boyle, K.B.; Dharamdasani, V.; Pathe, C.; Santhanam, B.; Randow, F. Ubiquitylation of lipopolysaccharide by RNF213 during bacterial infection. Nature 2021, 594, 111–116. [Google Scholar] [CrossRef]
- Pao, K.C.; Wood, N.T.; Knebel, A.; Rafie, K.; Stanley, M.; Mabbitt, P.D.; Sundaramoorthy, R.; Hofmann, K.; van Aalten, D.M.F.; Virdee, S. Activity-based E3 ligase profiling uncovers an E3 ligase with esterification activity. Nature 2018, 556, 381–385. [Google Scholar] [CrossRef] [Green Version]
- Elton, L.; Carpentier, I.; Staal, J.; Driege, Y.; Haegman, M.; Beyaert, R. MALT1 cleaves the E3 ubiquitin ligase HOIL-1 in activated T cells, generating a dominant negative inhibitor of LUBAC-induced NF-κB signaling. FEBS J. 2016, 283, 403–412. [Google Scholar] [CrossRef] [Green Version]
- Klein, T.; Fung, S.Y.; Renner, F.; Blank, M.A.; Dufour, A.; Kang, S.; Bolger-Munro, M.; Scurll, J.M.; Priatel, J.J.; Schweigler, P.; et al. The paracaspase MALT1 cleaves HOIL1 reducing linear ubiquitination by LUBAC to dampen lymphocyte NF-κB signalling. Nat. Commun. 2015, 6, 8777. [Google Scholar] [CrossRef] [Green Version]
- Douanne, T.; Gavard, J.; Bidere, N. The paracaspase MALT1 cleaves the LUBAC subunit HOIL1 during antigen receptor signaling. J. Cell. Sci. 2016, 129, 1775–1780. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Schmitz, R.; Mitala, J.; Whiting, A.; Xiao, W.; Ceribelli, M.; Wright, G.W.; Zhao, H.; Yang, Y.; Xu, W.; et al. Essential role of the linear ubiquitin chain assembly complex in lymphoma revealed by rare germline polymorphisms. Cancer Discov. 2014, 4, 480–493. [Google Scholar] [CrossRef] [Green Version]
- Jo, T.; Nishikori, M.; Kogure, Y.; Arima, H.; Sasaki, K.; Sasaki, Y.; Nakagawa, T.; Iwai, F.; Momose, S.; Shiraishi, A.; et al. LUBAC accelerates B-cell lymphomagenesis by conferring resistance to genotoxic stress on B cells. Blood 2020, 136, 684–697. [Google Scholar] [CrossRef]
- Noad, J.; von der Malsburg, A.; Pathe, C.; Michel, M.A.; Komander, D.; Randow, F. LUBAC-synthesized linear ubiquitin chains restrict cytosol-invading bacteria by activating autophagy and NF-κB. Nat. Microbiol. 2017, 2, 17063. [Google Scholar] [CrossRef] [Green Version]
- Van Wijk, S.J.L.; Fricke, F.; Herhaus, L.; Gupta, J.; Hotte, K.; Pampaloni, F.; Grumati, P.; Kaulich, M.; Sou, Y.S.; Komatsu, M.; et al. Linear ubiquitination of cytosolic Salmonella Typhimurium activates NF-κB and restricts bacterial proliferation. Nat. Microbiol. 2017, 2, 17066. [Google Scholar] [CrossRef]
- Herhaus, L.; Dikic, I. Regulation of Salmonella-host cell interactions via the ubiquitin system. Int. J. Med. Microbiol. 2018, 308, 176–184. [Google Scholar] [CrossRef]
- Fiil, B.K.; Gyrd-Hansen, M. The Met1-linked ubiquitin machinery in inflammation and infection. Cell Death Differ. 2021, 28, 557–569. [Google Scholar] [CrossRef]
- Wang, L.; Yan, J.; Niu, H.; Huang, R.; Wu, S. Autophagy and Ubiquitination in Salmonella Infection and the Related Inflammatory Responses. Front. Cell. Infect. Microbiol. 2018, 8, 78. [Google Scholar] [CrossRef]
- Ying, H.; Yue, B.Y. Cellular and molecular biology of optineurin. Int. Rev. Cell Mol. Biol. 2012, 294, 223–258. [Google Scholar] [CrossRef] [Green Version]
- Kamada, F.; Aoki, Y.; Narisawa, A.; Abe, Y.; Komatsuzaki, S.; Kikuchi, A.; Kanno, J.; Niihori, T.; Ono, M.; Ishii, N.; et al. A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J. Hum. Genet. 2011, 56, 34–40. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Morito, D.; Takashima, S.; Mineharu, Y.; Kobayashi, H.; Hitomi, T.; Hashikata, H.; Matsuura, N.; Yamazaki, S.; Toyoda, A.; et al. Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PLoS ONE 2011, 6, e22542. [Google Scholar] [CrossRef] [Green Version]
- Ahel, J.; Lehner, A.; Vogel, A.; Schleiffer, A.; Meinhart, A.; Haselbach, D.; Clausen, T. Moyamoya disease factor RNF213 is a giant E3 ligase with a dynein-like core and a distinct ubiquitin-transfer mechanism. eLife 2020, 9. [Google Scholar] [CrossRef]
- Sakamoto, H.; Egashira, S.; Saito, N.; Kirisako, T.; Miller, S.; Sasaki, Y.; Matsumoto, T.; Shimonishi, M.; Komatsu, T.; Terai, T.; et al. Gliotoxin suppresses NF-κB activation by selectively inhibiting linear ubiquitin chain assembly complex (LUBAC). ACS Chem. Biol. 2015, 10, 675–681. [Google Scholar] [CrossRef]
- de Jong, M.F.; Liu, Z.; Chen, D.; Alto, N.M. Shigella flexneri suppresses NF-kaB activation by inhibiting linear ubiquitin chain ligation. Nat. Microbiol. 2016, 1, 16084. [Google Scholar] [CrossRef] [Green Version]
- Wan, M.; Wang, X.; Huang, C.; Xu, D.; Wang, Z.; Zhou, Y.; Zhu, Y. A bacterial effector deubiquitinase specifically hydrolyses linear ubiquitin chains to inhibit host inflammatory signalling. Nat. Microbiol. 2019, 4, 1282–1293. [Google Scholar] [CrossRef]
- Peltzer, N.; Rieser, E.; Taraborrelli, L.; Draber, P.; Darding, M.; Pernaute, B.; Shimizu, Y.; Sarr, A.; Draberova, H.; Montinaro, A.; et al. HOIP deficiency causes embryonic lethality by aberrant TNFR1-mediated endothelial cell death. Cell Rep. 2014, 9, 153–165. [Google Scholar] [CrossRef] [Green Version]
- Boisson, B.; Laplantine, E.; Dobbs, K.; Cobat, A.; Tarantino, N.; Hazen, M.; Lidov, H.G.; Hopkins, G.; Du, L.; Belkadi, A.; et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J. Exp. Med. 2015, 212, 939–951. [Google Scholar] [CrossRef] [Green Version]
- Oda, H.; Beck, D.B.; Kuehn, H.S.; Sampaio Moura, N.; Hoffmann, P.; Ibarra, M.; Stoddard, J.; Tsai, W.L.; Gutierrez-Cruz, G.; Gadina, M.; et al. Second Case of HOIP Deficiency Expands Clinical Features and Defines Inflammatory Transcriptome Regulated by LUBAC. Front. Immunol. 2019, 10, 479. [Google Scholar] [CrossRef] [Green Version]
- Peltzer, N.; Darding, M.; Montinaro, A.; Draber, P.; Draberova, H.; Kupka, S.; Rieser, E.; Fisher, A.; Hutchinson, C.; Taraborrelli, L.; et al. LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature 2018, 557, 112–117. [Google Scholar] [CrossRef]
- Nilsson, J.; Schoser, B.; Laforet, P.; Kalev, O.; Lindberg, C.; Romero, N.B.; Davila Lopez, M.; Akman, H.O.; Wahbi, K.; Iglseder, S.; et al. Polyglucosan body myopathy caused by defective ubiquitin ligase RBCK1. Ann. Neurol. 2013, 74, 914–919. [Google Scholar] [CrossRef]
- Krenn, M.; Salzer, E.; Simonitsch-Klupp, I.; Rath, J.; Wagner, M.; Haack, T.B.; Strom, T.M.; Schanzer, A.; Kilimann, M.W.; Schmidt, R.L.J.; et al. Mutations outside the N-terminal part of RBCK1 may cause polyglucosan body myopathy with immunological dysfunction: Expanding the genotype-phenotype spectrum. J. Neurol. 2018, 265, 394–401. [Google Scholar] [CrossRef] [Green Version]
- Kumari, S.; Redouane, Y.; Lopez-Mosqueda, J.; Shiraishi, R.; Romanowska, M.; Lutzmayer, S.; Kuiper, J.; Martinez, C.; Dikic, I.; Pasparakis, M.; et al. Sharpin prevents skin inflammation by inhibiting TNFR1-induced keratinocyte apoptosis. Elife 2014, 3. [Google Scholar] [CrossRef]
- Sasaki, K.; Himeno, A.; Nakagawa, T.; Sasaki, Y.; Kiyonari, H.; Iwai, K. Modulation of autoimmune pathogenesis by T cell-triggered inflammatory cell death. Nat. Commun. 2019, 10, 3878. [Google Scholar] [CrossRef]
- Damgaard, R.B.; Walker, J.A.; Marco-Casanova, P.; Morgan, N.V.; Titheradge, H.L.; Elliott, P.R.; McHale, D.; Maher, E.R.; McKenzie, A.N.J.; Komander, D. The Deubiquitinase OTULIN Is an Essential Negative Regulator of Inflammation and Autoimmunity. Cell 2016, 166, 1215–1230.e1220. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Yu, X.; Demirkaya, E.; Deuitch, N.; Stone, D.; Tsai, W.L.; Kuehn, H.S.; Wang, H.; Yang, D.; Park, Y.H.; et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc. Natl. Acad. Sci. USA 2016, 113, 10127–10132. [Google Scholar] [CrossRef] [Green Version]
- Nabavi, M.; Shahrooei, M.; Rokni-Zadeh, H.; Vrancken, J.; Changi-Ashtiani, M.; Darabi, K.; Manian, M.; Seif, F.; Meyts, I.; Voet, A.; et al. Auto-inflammation in a Patient with a Novel Homozygous OTULIN Mutation. J. Clin. Immunol. 2019, 39, 138–141. [Google Scholar] [CrossRef]
- Monti, S.; Savage, K.J.; Kutok, J.L.; Feuerhake, F.; Kurtin, P.; Mihm, M.; Wu, B.; Pasqualucci, L.; Neuberg, D.; Aguiar, R.C.; et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood 2005, 105, 1851–1861. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, A.L., III.; Young, R.M.; Staudt, L.M. Pathogenesis of human B cell lymphomas. Annu. Rev. Immunol. 2012, 30, 565–610. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.; Tan, B.; Rosenwald, A.; Hurt, E.H.; Wiestner, A.; Staudt, L.M. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA 2003, 100, 9991–9996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowakowski, G.S.; LaPlant, B.; Macon, W.R.; Reeder, C.B.; Foran, J.M.; Nelson, G.D.; Thompson, C.A.; Rivera, C.E.; Inwards, D.J.; Micallef, I.N.; et al. Lenalidomide combined with R-CHOP overcomes negative prognostic impact of non-germinal center B-cell phenotype in newly diagnosed diffuse large B-Cell lymphoma: A phase II study. J. Clin. Oncol. 2015, 33, 251–257. [Google Scholar] [CrossRef]
- Tilly, H.; Gomes da Silva, M.; Vitolo, U.; Jack, A.; Meignan, M.; Lopez-Guillermo, A.; Walewski, J.; Andre, M.; Johnson, P.W.; Pfreundschuh, M.; et al. Diffuse large B-cell lymphoma (DLBCL): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015, 26 (Suppl. S5), v116–v125. [Google Scholar] [CrossRef] [PubMed]
- MacKay, C.; Carroll, E.; Ibrahim, A.F.M.; Garg, A.; Inman, G.J.; Hay, R.T.; Alpi, A.F. E3 ubiquitin ligase HOIP attenuates apoptotic cell death induced by cisplatin. Cancer Res. 2014, 74, 2246–2257. [Google Scholar] [CrossRef] [Green Version]
- Niu, J.; Shi, Y.; Iwai, K.; Wu, Z.H. LUBAC regulates NF-κB activation upon genotoxic stress by promoting linear ubiquitination of NEMO. EMBO J. 2011, 30, 3741–3753. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, E.J.; Diefenbacher, M.E.; Nelson, J.K.; Sancho, R.; Pucci, F.; Chakraborty, A.; Moreno, P.; Annibaldi, A.; Liccardi, G.; Encheva, V.; et al. LUBAC determines chemotherapy resistance in squamous cell lung cancer. J. Exp. Med. 2019, 216, 450–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oikawa, D.; Sato, Y.; Ohtake, F.; Komakura, K.; Hanada, K.; Sugawara, K.; Terawaki, S.; Mizukami, Y.; Phuong, H.T.; Iio, K.; et al. Molecular bases for HOIPINs-mediated inhibition of LUBAC and innate immune responses. Commun. Biol. 2020, 3, 163. [Google Scholar] [CrossRef] [PubMed]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Pan, D.; Kobayashi, A.; Jiang, P.; Ferrari de Andrade, L.; Tay, R.E.; Luoma, A.M.; Tsoucas, D.; Qiu, X.; Lim, K.; Rao, P.; et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 2018, 359, 770–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDuff, D.A.; Baldridge, M.T.; Qaqish, A.M.; Nice, T.J.; Darbandi, A.D.; Hartley, V.L.; Peterson, S.T.; Miner, J.J.; Iwai, K.; Virgin, H.W. HOIL1 Is Essential for the Induction of Type I and III Interferons by MDA5 and Regulates Persistent Murine Norovirus Infection. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuseya, Y.; Iwai, K. Biochemistry, Pathophysiology, and Regulation of Linear Ubiquitination: Intricate Regulation by Coordinated Functions of the Associated Ligase and Deubiquitinase. Cells 2021, 10, 2706. https://doi.org/10.3390/cells10102706
Fuseya Y, Iwai K. Biochemistry, Pathophysiology, and Regulation of Linear Ubiquitination: Intricate Regulation by Coordinated Functions of the Associated Ligase and Deubiquitinase. Cells. 2021; 10(10):2706. https://doi.org/10.3390/cells10102706
Chicago/Turabian StyleFuseya, Yasuhiro, and Kazuhiro Iwai. 2021. "Biochemistry, Pathophysiology, and Regulation of Linear Ubiquitination: Intricate Regulation by Coordinated Functions of the Associated Ligase and Deubiquitinase" Cells 10, no. 10: 2706. https://doi.org/10.3390/cells10102706
APA StyleFuseya, Y., & Iwai, K. (2021). Biochemistry, Pathophysiology, and Regulation of Linear Ubiquitination: Intricate Regulation by Coordinated Functions of the Associated Ligase and Deubiquitinase. Cells, 10(10), 2706. https://doi.org/10.3390/cells10102706